





文摘
自2000年以来已经有了很大的进步在我们理解遗传和基因组学的肺动脉高血压(PAH),但发现其实还有很多。基于现有知识,大约25 - 30%的患者诊断为特发性多环芳烃有一个潜在的孟德尔遗传原因的条件和应归类为可遗传的多环芳烃(HPAH)。在这里,我们总结已知的基因和基因组的司机多环芳烃,这些见解提供病理学,新治疗方法的发展机会。此外,决定不完全外显率的因素中观察到HPAH进行了讨论。基因检测和咨询,目前可用的方法和基因诊断的影响在PAH患者的临床管理。DNA测序技术的进步正在迅速扩大我们的能力进行基因组研究规模庞大的军团。在未来,这样的研究将提供一个更完整的遗传贡献PAH,该疾病的可能,分子分类。
文摘
国家艺术和研究视角的遗传学和基因组学的肺动脉高压和洞察病理学http://ow.ly/dkkq30mgDo2
介绍
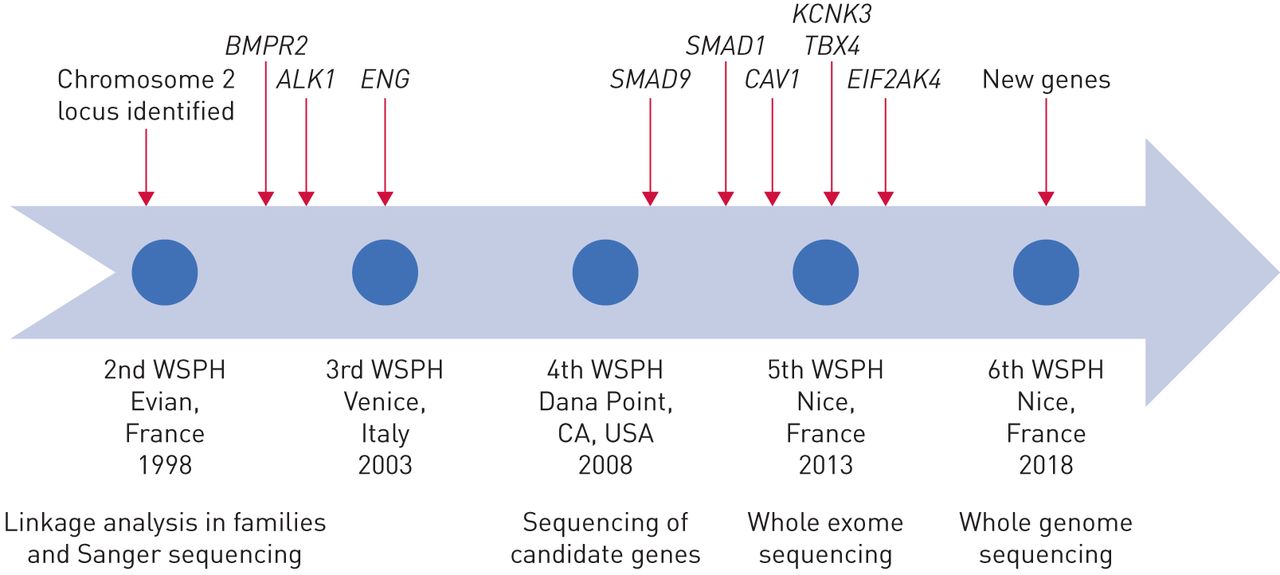
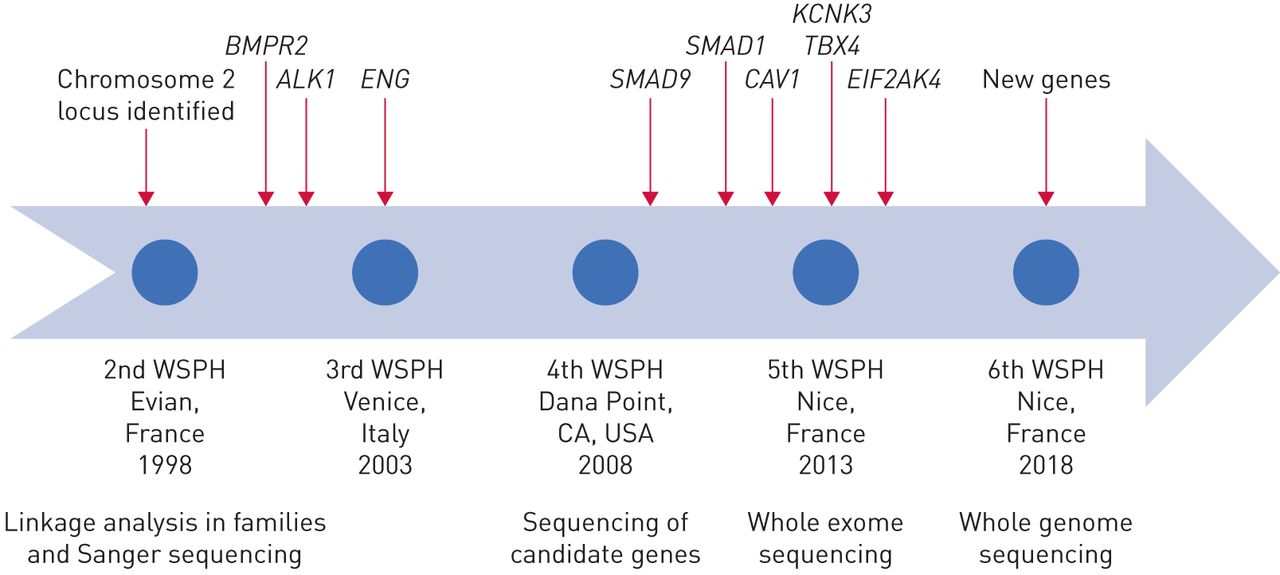
半个世纪后,1900年代中期,当心导管第一次提供临床医师能够安全地测量肺血液动力学、特发性肺动脉高压(IPAH,然后称为原发性肺动脉高压(PPH))被认为是致命的肺血管疾病的神秘起源。附近的这个世纪,新的理解开发特定的遗传倾向的家庭多环芳烃,信息是一个驱动程序修改分类PAH的第三世界在2003年研讨会肺动脉高压。自2000年以来,研究人员的贡献增加遗传学和基因组学的知识多环芳烃。在基因测序技术的进步使得廉价和快速测序的基因组的编码区域(全外显子组测序(韦斯)),或整个基因组(全基因组测序(WGS)),在家庭和大群患者(图1)。见解来自人类遗传学的增加我们的理解多环芳烃的病理学,识别新的潜在的药物靶点,并通知照顾病人和他们的家属。
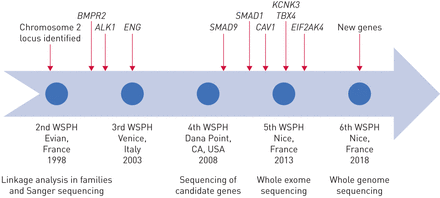
肺动脉高血压的基因发现的历史。WSPH:世界研讨会肺动脉高压。
国家的艺术
孟德尔遗传
有家族病史的多环芳烃在6 - 10%的病人不与其他潜在障碍有关1]。2000年,这样的家庭确定杂合的遗传突变的遗传分析BMPR2,基因编码骨形态形成蛋白受体2型,改变成员增长factor-β(TGF-β)总科2,3]。随后,亦发现了突变IPAH [4]。现在好了,大约70 - 80%的家庭与多环芳烃和10 - 20%的IPAH病例是由突变引起的BMPR2(5]。
测序的基因编码BMP信号受体中介导致罕见的序列变异的识别SMAD1,SMAD4和SMAD9(6,7]。额外的识别SMAD9大军团突变已确认它在多环芳烃中的作用[8]。此外,外显子组测序BMPR2负面个体与多个家庭成员患有PAH突变CAV1编码caveolin-1和函数来身体colocalise骨形态发生蛋白受体(9]。一种罕见的突变CAV1有关与脂肪代谢障碍和多环芳烃在一个年轻的孩子。KCNK3(钾离子通道亚K成员3)突变也被外显子组测序和编码钾通道,导致膜电位确定肺血管张力(10]。在童年和数组比较杂交和测序(很少)成人PAH确认删除和功能丧失的突变TBX4(T-box 4),基因也与小髌骨综合征(11]。突变TBX4是儿童最常见的多环芳烃的遗传原因,建议多环芳烃时至少部分发育肺部疾病呈现在生命早期(12,13]。认识到严重的多环芳烃也可以发生在家庭隔离遗传性出血性毛细血管扩张(HHT)有牵连ACVRL1(激活素受体激酶1 (ALK1))英格(endoglin) PAH突变14- - - - - -16]。
最近,欧洲大型调查是在一个协作的成人患者(> 18年)IPAH,家族多环芳烃(FPAH)和anorexigen-associated PAH (8]。这项研究的1000名患者证实因果变异的存在BMPR2(15.3%),TBX4(1.3%),ACVRL1(0.9%),英格(0.6%),SMAD9(0.4%)和KCNK3(0.4%)。没有致病性编码变异CAV1,SMAD1或SMAD4被确定,可能由于这些基因突变的罕见的成年人。相同的研究发现在新PAH基因突变:ATP13A3(腺苷三磷酸酶13 a3;1.1%),SOX17(SRY-box 17;0.9%),AQP1(水通道蛋白1;0.9%),GDF2(生长分化因子2 / BMP9;0.8%),并建议额外的基因,需要进一步的确认。在所有这些基因突变是常染色体继承和展览居多的外显率降低,这意味着一些人携带突变不清单多环芳烃。在IPAH情况下,突变可能会继承父母或发生的影响新创。在父母的样品,确定突变发生吗新创可以帮助建立致病性。儿科IPAH患者没有已知的危险基因的突变,外显子组测序揭示出2倍浓缩的新创预测有害的变异。新创以新的基因变异可以解释19%的paediatric-onset IPAH情况下(12]。PAH基因的不完全外显率观察表明,额外的遗传,表观遗传或环境因素导致疾病风险/发展。
2014年,biallelic突变EIF2AK4,一个基因编码真核翻译起始因子2α激酶4,被确认为一种遗传原因肺毛细管haemangiomatosis (PCH) [17)和肺静脉阻塞疾病(PVOD) (18]。PVOD和PCH是罕见的和病态的形式的多环芳烃。遗传PVOD和PCH常染色体隐性和几乎完全渗透,与其他形式的多环芳烃。个人被诊断为显然零星PVOD或PCH也可能携带biallelicEIF2AK4变异在25%的情况下(17,18]。检测biallelic致病性EIF2AK4突变建立一个精确和准确的分子诊断PVOD / PCH无需肺活检(19]。
基因迄今报告的摘要HPAH患者所示表1。高水平的证据需要建立在一个特定的基因突变的因果作用之前用于临床筛选和管理。
常见的遗传变异
常见的遗传变异的作用导致PAH的病原学或临床过程是定义良好的。到目前为止只有一个全基因组关联研究已经发表识别变异与多环芳烃(n = 625)。本研究发现一个重要的关联的CBLN2(cerebellin 2前体)轨迹映射到18 q22.3,与风险等位基因赋予PAH的比值比为1.97 (p = 7.47×10−10)[20.]。另一个大型研究评估协会组成内皮素信号通路基因多态性与临床结果在715年欧洲血统的PAH患者步研究(sitaxsentan)。一个协会之间被确认单核苷酸多态性(rs11157866)蛋白γ亚基基因GNG2并结合改善功能类和6分钟步行距离(21]。在较小的一项研究中,血清血管内皮抑制素的水平,一个强有力的抗血管新生蛋白质能力诱导内皮细胞凋亡和抑制内皮细胞增殖,与可怜的功能状态和组1患者的死亡率的重要因素。相比之下,一个错义变体(rs12483377)COL18A1编码血管内皮抑制素,降低蛋白质和循环减少死亡率(22]。线粒体代谢的异常现在承认在多环芳烃。最近的一项研究表明,线粒体基因组的变异可能影响发展中多环芳烃的风险,利率较低的疾病与haplogroup L,最古老的人类祖先haplogroup [23]。最后,在一项对2761名健康成年人,变异雌二醇代谢(CYP1B1(细胞色素P450 1 b1))和雄激素受体基因与右心室功能(24]。鉴于PAH显著性差异和之前的证据CYP1B1变异可能调节疾病外显率BMPR2突变携带者(25),这些变异有关可能的作用值得进一步研究在多环芳烃的右心室重塑。
病理学变异和疾病之间的联系
PAH突变和疾病病理学
BMPR2表面表达细胞的数量,但它特别高表达在肺血管内皮,它形成了一个复杂的I型受体,ALK1或ALK2。ALK1 / BMPR2受体复杂信号循环BMP配体,特别是在应对BMP9 BMP10,利用作为coreceptor ENG (26,27]。ALK1和ENG也高度肺内皮细胞中表达,这要求高水平的BMPR2 / ALK1信号在lung-specific肺动脉内皮细胞可能导致的影响BMPR2突变。BMP9(编码GDF2)函数循环血管静止因子,保护内皮细胞凋亡和过度增殖,抑制血管通透性。的损失BMPR2也有利于内皮功能障碍和促进endothelial-to-mesenchymal过渡。鉴于突变ALK1和英格(通常与遗传性出血性毛细血管扩张症)也会导致多环芳烃,遗传证据强烈表明,肺内皮细胞是一个重要的在多环芳烃病理学启动细胞类型。然而,BMPR2损失函数在其他细胞类型(如。平滑肌细胞,成纤维细胞和免疫细胞)也可能导致疾病的病理学。例如,肺动脉平滑肌细胞BMPR2突变是hyperproliferative耐growth-suppressive每个位置的影响,通过抗增殖Smad1/5信号损失28]。
其它不太常见的PAH突变也与BMP通路有关。Smad8(编码的SMAD9BMP信号下游基因)是一个中介,连同Smad1和5。的杂合突变的SMAD9相对较少影响规范BMP信号通过Smad4,大概由于Smad1/5冗余函数(29日]。然而,相对于Smad4-independent途径促进microRNA成熟(30.),SMAD9突变导致重大损失函数,表明小分子核糖核酸的监管以这种方式可能PAH发病机制中发挥重要作用29日]。也见过类似的结果在一个与先天性心脏病(CHD)相关的多环芳烃,肺动脉内皮细胞进行体细胞杂合的删除SMAD9(31日]。
EIF2AK4,也称为一般控制non-derepressible 2 (GCN2),是一种丝氨酸/苏氨酸蛋白激酶磷酸化的α亚基真核起始因子2,起着关键作用的调节氨基酸代谢,以应对营养不足。EIF2AK4感官氨基酸缺乏通过绑定到无电荷的转移核糖核酸。损失函数的分子和细胞机制的激酶促进PVOD / PCH的发展正在积极调查。在Eif2ak4−−/鼠标,增加压力在肠道炎症反应,二次自噬降低,增加氧化应激及其影响inflammasome激活和炎症(32]。还有待证明,如果类似的机制参与内膜的纤维化和肺血管的内皮细胞增殖观察遗传PVOD。另外,EIF2AK4biallelic损失函数会导致减少TRIB3 (Tribbles-like蛋白质3),可以抑制BMP-mediated差别的对这些细胞反应(33]。在这一假设下,EIF2AK4损失函数将有类似的后果BMPR2或SMAD9通过减少BMP信号突变。
CAV1小窝的主要成分蛋白质(瓶形的质膜内陷)和内皮细胞中高度表达。骨形态发生蛋白受体在小窝本地化,以及其他地区的质膜,并研究表明小窝所需起始BMP信号(34]。CAV1损失减少BMPR2膜本地化和信号35]。相反,BMPR2突变会导致caveolar贩卖缺陷和胞内本地化(36]。最后,值得注意的是,即使没有一个遗传突变,表达的水平BMPR2和CAV1减少多环芳烃肺组织(37,38]。
影响因素疾病外显率
尽管致病性BMPR2PAH突变明显原因,疾病表型的外显率是不完整的。最好的估计是,外显率男性携带者是14%左右,而女性是42%左右(39]。因此,女性性影响的外显率的最重要的因素BMPR2在PAH突变,可能由雌激素代谢(25]。其他因素影响外显率可能是遗传,表观遗传和/或环境。遗传因素可能包括野生型的表达水平BMPR2从影响等位基因(40),基因变异影响TGF-β表达水平的(41)或可变剪接BMPR2(42]。在诱导多能干细胞的研究来源于影响和不受影响BMPR2突变携带者表明,遗传背景,特别是BMP pathway-modifying基因的差异表达,可能导致外显率(43]。额外的大型纵向研究的影响和影响突变携带者需要更系统地搜索外显率的遗传和环境修饰符。
虽然没有研究尚未解决潜在的环境因素影响的外显率BMPR2基因突变在人类群体,研究表明,炎症介质,如。脂多糖(44),肿瘤坏死factor-α(TNF-α)[45]和5-lipoxygenase [46],可以推动发展的PH值在转基因小鼠和patient-derived多能干细胞(47]。TNF-α直接抑制BMPR2信使rna和蛋白质表达。此外,如甲基化的表观遗传机制BMPR2启动子可能扮演一个角色48]。几个小分子核糖核酸也被证明的目标BMPR2信使rna修改蛋白质的表达水平,包括miR-21 [49和mir - 17 - 9250]。
另一个可能导致外显率的因素是体细胞突变在肺血管细胞(51]。几组已经确定了DNA损伤内皮细胞和平滑肌细胞中多环芳烃的肺与控制[相比51- - - - - -53]。的损失BMPR2与缺陷相关的DNA修复(54),但在细胞DNA损伤也明显增加IPAH和相关的多环芳烃情况下,冰毒等环境因素引起的,可以使用[55]。还需要进一步的研究来理解肺内的DNA损伤的作用及其对疾病发病机制的贡献。
遗传咨询/测试和管理健康的突变携带者
遗传咨询
PAH突变基因已经被鉴定IPAH和FPAH anorexigen-associated PAH, PVOD / PCH和儿童IPAH和多环芳烃与冠心病有关。有法医学的义务告知所有患者在这些群体遗传的可能性条件,家庭成员可能会携带突变的风险增加PAH,允许筛查和早期诊断。即使基因检测不执行,家庭成员应该意识到早期的症状和体征,以确保及时和适当的诊断。如果突变中确定一个病人,病人应该重新归类为HPAH症状。
遗传教育和咨询之前应该进行基因检测的多环芳烃,解决复杂的问题不完全外显率,监测高危基因家族成员的问题,生殖问题,基因歧视的担忧,以及心理内疚和责任的问题,可以伴随遗传基础的疾病。检测前的遗传教育影响个人可以通过PAH提供者和/或执行遗传专业人士,并通过集中教育视频(如。www.youtube.com/watch?v=36rlvtj_Qrs)。深入与基因遗传咨询专业人士包括基因顾问或医学遗传学家之前是至关重要的基因测试无症状的家庭成员。家庭也应该被称为遗传顾问和/或临床遗传学家,如果他们希望考虑生育的选择。国家参考中心的遗传咨询经验PAH基因测试显示了潜在的兴趣在无症状的家庭成员56]。
基因检测在家庭中应该首先受影响个体尽可能识别相关的突变的家庭。否则,消极的基因测试结果影响家庭成员不是信息。如果家族突变是已知的和一个突变的影响家庭成员测试为阴性,多环芳烃对那个人的风险是一样的普通人群(大约每1 000 000在北美)。这可以提供很好的心理救援,家庭成员,他们可以放弃评价为PAH屏幕。
基因检测
基因检测可以帮助解释疾病的病因学和其他家庭成员的风险分层和未来的孩子。临床基因检测可以在北美和欧洲,和材料可以发送到美国或欧洲遗传实验室从世界的其他地方。当前的测试成本范围从约1000美元到1500美元来分析的第一个成员的家庭。一旦family-specific突变,测试其他家庭成员在美国成本300 - 500美元。包括基因的确切数字,测试成本和测试因国家的不同而有所不同,保险公司的保险。最经常被引用的原因PAH基因检测是提供信息,关于孩子,和兴趣测试对于paediatric-onset PAH(尤其高57]。
有针对性的测序
随着基因与多环芳烃的数量增加,它已经成为艰苦单独为每个这些基因测试。下一代测序技术的出现使得基因的开发板同时查询多个基因。有几种不同的技术和平台,许多现在提供临床的基础上(gtr www.ncbi.nlm.nih.gov /条件/ C0152171 /)。此外,一些可以分析这些基因的剂量变化检测缺失或重复的一个或多个外显子。这种突变是常见的BMPR2和TBX4。一个适当的方法,比如多路复用ligation-dependent探测器放大,应该使用,因为这些类型的突变是不易被测序。是很重要的检查面板中所包含的基因测试的时候由于成分变化基因的发现。
全外显子组测序和全基因组测序
如果PAH基因的一个最新的小组未能确定一个致病变种在家族PAH患者或一个孩子,韦斯可能是适当的识别潜在的新基因。对于paediatric-onset PAH,测试应该包括孩子和父母允许简单的识别新创变体。这三个方法是用来确定的CAV1和KCNK3基因(9,10]。最大的三个分析表明,19%的儿童IPAH是因为新创突变(12]。外显子组测序比委员会更贵的基因测试,应该下令遗传专业人士可以讨论学习的选择或二次偶然发现的,目前推荐的59个基因组成的美国大学医学遗传学和基因组学,医学上可操作的和潜在的救生,包括基因遗传性癌症和心脏性猝死58]。
在地平线上是WGS的使用,使变异在非编码区域的识别病人的基因组,但是还没有经常用于临床基础。外显子组和基因组测序的优势基因识别迭代再分析随着时间的小说。
测序数据的解释
韦斯和WGS的主要问题是大量的变异在任何个人识别。解释的功能后果错义和非编码变体是特别具有挑战性。因此,识别潜在的致病基因变异在小说需要过滤掉参考基于大型数据库常见变异等位基因频率和生物信息学预测可能的功能效果。隔离研究家庭有时可以澄清不确定意义的变种(VUS开头),特别是如果变体新创或如果它将影响多环芳烃在其他家庭成员(58),而升值,外显率通常是不完整的,航空公司可能不受影响。只分为致病或可能致病的遗传变异(不是VUS开头)可用于基因风险分层家庭成员的影响。
社会心理因素和生殖选择
基因测试结果可能导致一些人弊大于利,因为目前还没有有效的方法来预防或治疗遗传性不同形式的多环芳烃,因为不完全外显率的突变。确定突变可以与家庭的负罪感在父突变传给他们的孩子。也有担忧就业基因歧视和保险。根据国家法律保障反对基因歧视不同。在美国,基因歧视法案保护保险成员反对歧视覆盖或健康保险成本和保护反对就业歧视,但不反对歧视在生活中,长期护理或残疾保险基于遗传倾向。在全民医保的国家,基因歧视的担忧并不伟大和基因检测摄取率更高。
生殖的选择对于那些携带致病突变包括采用,使用捐赠的配子,产前测试,在体外受精(IVF)与胚胎植入前遗传学诊断或不考虑PAH基因状态。可访问性和保险为体外受精和胚胎植入前遗传学诊断不同的国家,但覆盖率通常不是现成的病人由于成本。
遗传学和基因组学的实际应用
在PAH基因诊断患者的效用是什么?
致病性PAH突变与疾病的治疗和预后特征影响。例如,HPAH联系在一起BMPR2或ACVRL1突变的礼物在年轻的时候,有严重的血流动力学异常,和一个非常低的概率急性vasoreactivity,以及减少生存在当前治疗时代(59,60]。患者的遗传形式PVOD也年轻比non-mutation运营商表示,但没有显著差异在风平浪静的生存3年(61年]。
几项研究已经清楚地表明,检测致病性EIF2AK4突变是特别有用的。PVOD / PCH挑战PH值的诊断临床医生、放射科医生和病理学家。识别biallelicEIF2AK4突变可以确认遗传PVOD / PCH没有肺活组织检查和识别多环芳烃的一种独特形式,不仅没有当前PAH药物反应良好,但也易诱发肺部水肿时这些药物管理(61年]。一群个体诊断出患有IPAH或HPAH也可能携带biallelic致病性EIF2AK4突变。岁以下的这些人的特点是展示50年的扩散能力的肺一氧化碳(DLCO)< 50%的预测(62年- - - - - -64年]。因此,我们建议临床医生测试年轻PAH患者低DLCO为EIF2AK4突变,以指导治疗和家庭成员的风险评估(63年]。
从基因组研究发现也开始被用于更好地理解这种疾病过程和治疗反应。H人物et al。(65年)识别患者外周血RNA表达签名钙通道阻滞剂治疗的多环芳烃subphenotype响应,表明非侵入性的方法来预测药物反应。使用韦斯,遗传变异,可能这subphenotype也确认(66年]。正如前面所讨论的那样,常见的基因变异与风险相关的多环芳烃,生存和/或药物基因组学对治疗的反应。最近,这是表明,常见的功能变体SIRT3和UCP2可能预测反应抑制丙酮酸脱氢酶激酶通过二氯醋酸在多环芳烃(2期临床试验67年),为未来铺平道路精密医学研究这种药物。
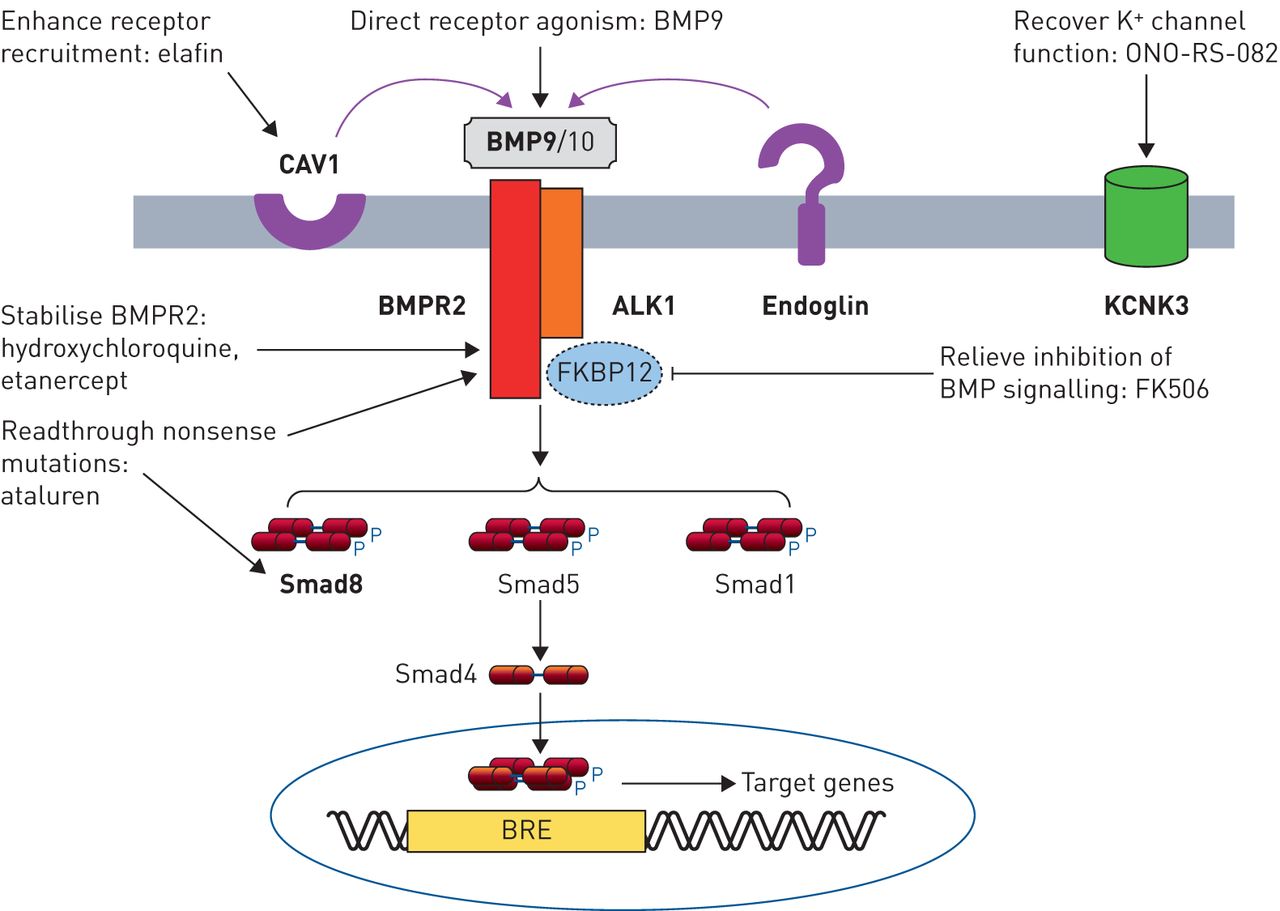
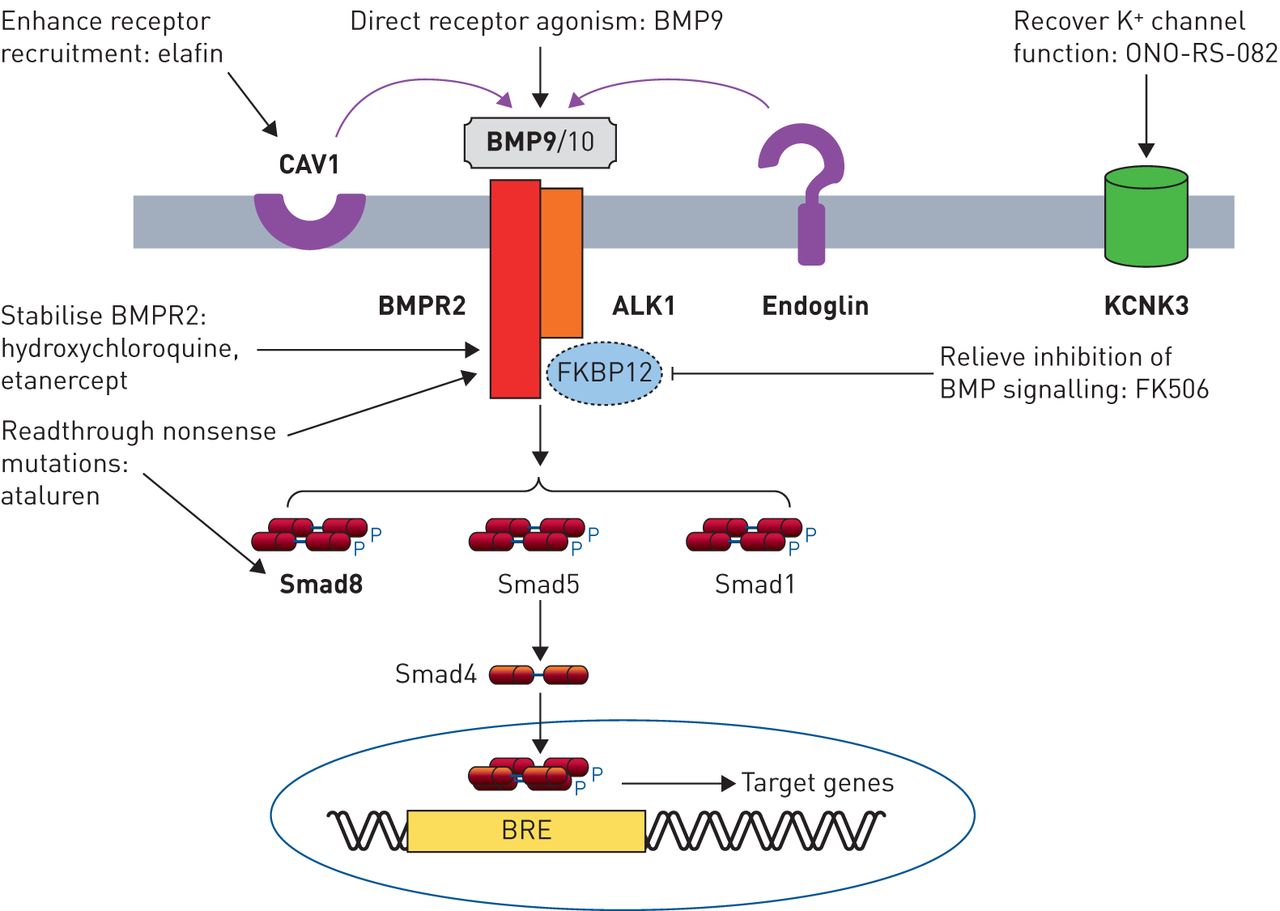
从基因疗法
分子通路基因研究现在的主题强调的几个新的治疗方法(图2)。l昂et al。(68年)报道,在杂合的BMP9政府的有利影响BMPR2基因敲除小鼠,这表明补偿BMPR2haploinsufficiency通过增加剂量的配体可能构成对人类PAH靶向治疗。在受体水平,方法包括平移readthrough无意义突变使用ataluren恢复完整的蛋白质(69年]或减慢溶酶体降解与氯喹BMPR2增加细胞表面的受体密度(70年,71年]。这两种方法有效地恢复BMP信号在体外和溶酶体封锁逆转PH值在实验模型。用依那西普进行抑制TNF-α不仅目标炎症,但可以减少受体脱落和变幻虫的退化BMPR2 [45]。弹力素作用通过CAV1促进受体招聘和增强BMP信号、扭转实验PH值(72年]。下游BMPR2,吸收FK506被发现增加BMP信号和扭转实验PH值,通过绑定FK-binding蛋白12日抑制剂BMP信号(73年]。2期临床试验建立了低剂量的安全性和耐受性吸收FK506在多环芳烃74年]。超出了BMP通路,KCNK3突变导致减少钾通道电导,至少对一些突变,可以恢复的磷脂酶A2抑制剂ono - rs - 082 (10]。这些研究强调了相当大的进步,这在翻译基本遗传研究潜在的治疗方法。而直接修正潜在的突变目前仍然是具有挑战性的,迅速发展的基因编辑技术可能会使肺血管内目标突变校正一个现实的未来前景。
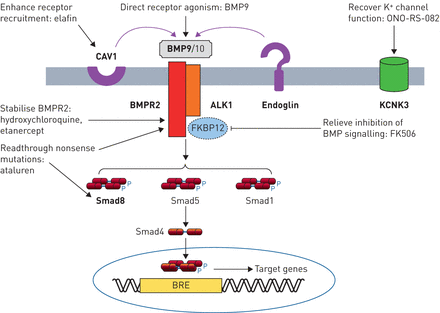
从基因疗法:精密医学方法在肺动脉高血压(PAH)。BMP (R):骨形态形成蛋白(受体);信徒:BMP-responsive元素;CAV1: caveolin-1;FKBP12: 12-kDa FK506-binding蛋白质。在内皮细胞BMP信号是由配体介导的BMP9 BMP10通过ALK1 / BMPR2受体复杂。Endoglin作为辅助受体。受体的信号是由磷酸化Smads (Smad1 5和8),然后与Smad4以及把原子核,调节基因含有启动子想。CAV1促进受体colocalisation,KCNK3编码钾通道,导致肺血管的基调。基因变异在遗传PAH大胆。潜在的治疗方法针对这些途径包括:外生BMP9配体管理,增加可用性的功能性BMPR2受体(羟氯喹,栏),促进readthrough无意义突变恢复功能BMPR2或Smad8蛋白质(ataluren),提高下游信号BMP的缓解FKBP12抑制1型受体(吸收FK506),促进CAV1-mediated受体招聘(选取),或恢复KCNK3通道电导(ono - rs - 082)。
提出未来的发展方向
这些不同的基因组方法强调潜在路径提出利用分子医学改善PAH患者护理。一些大规模的遗传学和基因组学研究目前正在美国和欧洲。值得注意的是,美国国家生物样品和数据存储库的多环芳烃(多环芳烃生物”;www.pahbiobank.org)是产生遗传数据(有针对性的DNA测序,韦斯和全基因组单核苷酸多态性)3000年第1组PAH患者。英国桥项目包括多环芳烃的罕见疾病的WGS数据生成(https://bridgestudy.medschl.cam.ac.uk/pah.shtml)。从欧洲超过1250 IPAH /家族PAH患者被分析调查潜在的遗传变异导致疾病。此外,美国国立卫生研究院PVDOMICS倡议旨在定义新的分子分类在传统世界卫生组织团体相结合深入临床表现型与多个“组学”分析(75年]。只有通过这样的人群和计划,我们将有足够的力量来发现潜在遗传因素的范围多环芳烃,从而提供一个更全面的基因对疾病的理解。理想情况下,这将导致额外的新靶点的鉴定新药。它还可以促进预防策略多环芳烃和预后的预测基于PAH的成因分类。
脚注
2号系列”肺动脉高压学报第六届世界研讨会”由n . Galie编辑教授麦克劳克林,l·j·鲁宾和g . Simonneau
利益冲突:西北莫雷尔报告从Morphogen-IX赠款和个人费用,在提交工作。
利益冲突:硕士Aldred报告国家卫生研究院的资助,在进行这项研究的。
利益冲突:W.K.涌没有披露。
利益冲突:C.G.艾略特报告个人费用从拜耳和柏勒罗丰指导委员会工作,赠款和个人费用从Actelion股价注册表和数据安全监测,和是一个顾问为端点判决肺LLC和费用支付给他的雇主(山间医疗),在提交工作。
利益冲突:厕所尼科尔斯没有披露。
利益冲突:f . Soubrier没有披露。
利益冲突:钢筋Trembath没有披露。
利益冲突:J.E.劳埃德没有披露。
- 收到了2018年10月5日。
- 接受2018年10月9日。
- 版权©2019人队
本文是开放和分布式根据创作共用署188滚球软件名非商业性4.0许可证。











{kind=link}
{kind=link}
{kind=link}
{kind=link}