条文本

文摘
背景:肺动脉高压(PAH)是一个进步的障碍,其特点是引起肺动脉压力与肺小动脉病变。之前的研究表明,大约有70%的家庭多环芳烃和11 - 40%的特发性肺动脉高压(IPAH)病例骨形态形成蛋白受体突变II型(BMPR2)基因。此外,突变激活素受体激酶1 (ALK1)基因在PAH患者已报告。因为BMPR2和ALK1属于转化生长因子(TGF)β总科,使多环芳烃,其他基因突变的TGF -β/ BMP信号通路可能也使多环芳烃。
方法:我们的突变筛查ENDOGLIN(英格),SMAD1、SMAD2 SMAD3 SMAD4、SMAD5 SMAD6和SMAD8基因,参与了TGF -β/ BMP信号,在23个IPAH患者没有突变BMPR2或ALK1。
结果:无义突变的SMAD8指定c。606 C > A, p。C20.2Xwas identified in one patient. The father of this patient was also identified as having the same mutation. Functional analysis showed the truncated form of the SMAD8 C202X protein was not phosphorylated by constitutively active ALK3 and ALK1. The SMAD8 mutant was also unable to interact with SMAD4. The response to BMP was analysed using promoter-reporter activities with SMAD4 and/or ca-ALK3. The transcriptional activation of the SMAD8 mutant was inefficient compared with the SMAD8 wild type.
结论:我们描述了第一次突变SMAD8IPAH患者。我们的研究结果建议的参与SMAD8在多环芳烃的发病机制。
来自Altmetric.com的统计
肺动脉高血压(多环芳烃;MIM 178600)是一种罕见的疾病,发病率以每年每100万在普通人群中1 - 2例。12它的特点是血管内皮和平滑肌细胞的异常增殖肺小动脉,导致持续的海拔平均肺动脉压力⩾25毫米汞柱在休息期间和/或⩾30毫米汞柱运动。13尽管这种疾病可能出现在任何年龄,多环芳烃诊断通常是在人生的第四个十年,女性对男性的比例为2比1。13没有现代治疗疾病进展迅速,导致右心衰后3年内诊断。据报道,在所有PAH患者,家庭多环芳烃占至少6%的情况下,外显率的10 - 20%。13
分类最近的一项研究提出了五个PAH的子组:特发性肺动脉高压(IPAH);家族性多环芳烃(FPAH);多环芳烃与其他相关疾病(APAH)如胶原血管疾病、先天性肺分流术系统,门户高血压、艾滋病病毒感染;多环芳烃与明显的静脉或毛细血管的参与;和新生儿持续肺动脉高压(PPHN)。45
在法国一项研究报道,FPAH和IPAH占4.2%和38.9%的患病率在553年PAH的情况下,分别。6FPAH IPAH共享相同的临床特征,组织病理学和临床过程,FPAH低外显率。因此,真正的发病率和患病率FPAH尚未完全阐明。7
异构在骨形成蛋白(BMP)的种系突变II型受体基因(BMPR2)、转化生长因子受体(TGF) -β总科2号染色体上q33, FPAH已确定。89BMPR2在多达70%的突变已确定FPAH IPAH患者和11 - 40%。1011
突变激活素受体激酶1 (ALK1)基因,TGF-β家族的一员,12号染色体上的问题也被报道在IPAH / FPAH病人1213和患者的遗传性出血性毛细血管扩张(HHT)关联的多环芳烃。1415除了协会BMPR2和ALK1突变,有一个缺乏发表研究报告广泛和系统的突变筛查PAH患者。1216因为两个BMPR2和ALK1基因,属于TGF-β总科,众所周知,使多环芳烃,提出的问题是是否在其他基因突变信号通路可能也使多环芳烃。在这项研究中,23例临床诊断为IPAH,没有突变BMPR2或ALK1在八个基因突变筛查TGF-β/ BMP信号通路-ENDOGLIN (ENG),SMAD1,SMAD2,SMAD3,SMAD4,SMAD5,SMAD6和SMAD8(也被称为SMAD9)——确定是否参与PAH的发病机制。
方法
主题
评估都通过伦理委员会批准东京女子医科大学和东邦大学,东京,日本。患者从东京女子医科大学和东邦大学。书面知情同意了所有研究对象。如果病人是16岁以下,知情同意是由他们的监护人。我们评估每个病人的病史、体格检查和回顾他们的医疗记录。
IPAH / FPAH通过临床的诊断评估、胸片、心电图、超声心动图和心导管基于当前的国际共识标准(威尼斯2003)。56APAH患者,多环芳烃与重要的静脉或毛细血管参与,PPHN和其他肺高血压被排除在本研究由训练有素的心脏病专家。
本研究包含个人临床诊断为IPAH来源于两个群体。第一批12个患者IPAH源自先前的研究13这12个病人没有被证实BMPR2或ALK1突变通过直接测序和多路复用结扎依赖探测器放大(MLPA)分析。11第二组包含21 IPAH或者FPAH,患者和变异分析显示10个病人,其中9个突变包括三个患者其实删除BMPR2,一个有一个突变ALK1(未发表的数据)。这10个患者被排除在本研究之外。剩下的11个患者IPAH第二组和12 IPAH从第一组患者,总共23个病人被证实没有突变BMPR2或ALK1纳入本研究。
总结23例中的基线特征和血液动力学的参数提出了表1。可用的数据特点和23 IPAH患者的血液动力学的参数提供了补充表1。
测序和基因突变分析
基因组DNA提取外周血白细胞的患者或从lymphoblastoid细胞系转化巴尔病毒如前所述。17在23个患者没有突变BMPR2或ALK1,所有编码外显子和邻intronic区域英格,SMAD1,SMAD2,SMAD3,SMAD4,SMAD5,SMAD6和SMAD8放大使用聚合酶链反应(PCR引物的细节可以在补充表2)。PCR扩增产品纯化,直接测序如前所述。18
在所有情况下,任何序列变化发现reamplified和重新测序证实观察到的变化。观察到的变化也证实了放大耐火突变系统(武器)测定。19
生成的序列与野生型相比英格(加入基因库NM_000118数量),SMAD1(基因库NM_005900),SMAD2(基因库NM_005901),SMAD3(基因库NM_005902),SMAD4(基因库NM_005359),SMAD5(基因库NM_005903),SMAD6(基因库NM_005585),SMAD8(基因库NM_005905)。
当检测到突变,我们确认在150年健康控制通过直接测序。
质粒和抗体
人类pcDNA3.0-6xMyc-SMAD8人类pcDNA3.0-Flag-SMAD4,人类pcDNA3.0-ALK1-haemagglutinin (HA),人类pcDNA3.0-ALK3-HA BMP-responsive子记者构造3 gc2-lux, TGF-β响应启动子构建3 tp-lux好心的记者提供的K Miyazono博士(日本东京)。3 gc2-lux包含三个重复的GC-rich序列来自于近端骨形态发生蛋白反应Smad6子元素。20.3 tp-lux TGF-β响应荧光素酶报告基因,包含三个连续tetradecanoylphorbol醋酸(TPA)响应元素和部分纤溶酶原激活物抑制剂1 (PAI-1)启动子区域。21
人类活动(ca) ALK1起来从而ALK3 glu - 201是由突变为天冬氨酸和glu - 233为天冬氨酸突变,分别。
网站定向诱变是由基于PCR的方法。构建质粒被测序验证。抗体使用如下:anti-Flag抗体(F3165,σ,圣路易斯,密苏里州,美国),anti-HA抗体(11867423001,罗氏,曼海姆巴登-符腾堡州,德国),anti-Myc抗体(# 2276,细胞信号技术,丹弗斯,麻萨诸塞州,美国),anti-Myc抗体(# 06 - 549、北部的普莱西德湖,纽约,美国)和anti-phospho-Smad1 / Smad5 / Smad8抗体(# 9511,细胞信号技术)。
转染细胞溶菌作用,免疫印迹和免疫沉淀反应
HEK293和COS1细胞在DMEM / F-12(σ)补充10%胎牛血清(的边后卫)(Gibco,大岛,纽约,美国),100单位/毫升青霉素和链霉素(Gibco)和250 ng / ml两性霉素B(σ)。Lipofectamine 2000执行转染试剂(美国加州表达载体,卡尔斯巴德)根据制造商的指示。转染24小时后,细胞细胞溶解在裂解缓冲(1 M Tris-HCl (pH值8.0)50毫米,0.5 M EDTA 1毫米(pH8.0), 5 M氯化钠120毫米,NP-40 0.25%)。co-immunoprecipitation化验,溶解产物与单克隆抗体anti-Flag孵化(σ)和蛋白质G-Sepharose珠子(通用电气医疗集团,小都,白金汉郡,英国),并与多克隆抗体anti-Myc免疫印迹(北部)。
荧光素酶检测
COS1细胞在Opti-MEM co-transfected(英杰公司)使用Lipofectamine 2000试剂(表达载体)3 gc2-lux或p3TP-Lux野生型和突变体pcDNA3.0-SMAD8和/或pcDNA3.0-SMAD4和/或pcDNA3.0-ca-ALK3(总0.9μg)。转染24小时后,这些细胞被收获。萤火虫和renilla荧光素酶活动测量双荧光素酶报告实验后(Promega,麦迪逊,威斯康辛州,美国)制造商的指示。结果表示为萤火虫荧光素酶活性比renilla荧光素酶的活动。所有分析都是一式三份。
统计数据
所有结果都表示为(SD)。两个样本的统计比较,小动物——一张长有学生t测试使用适用的地方。p < 0.05的值被认为是重要的。
结果
序列分析
确定突变基因在TGF-β/ BMP信号通路,我们筛选突变ENG、SMAD1 SMAD2, SMAD3 SMAD4, SMAD5 SMAD6和SMAD8基因在23个IPAH患者没有突变BMPR2或ALK1。
在这项研究中,未发现突变ENG、SMAD1 SMAD2、SMAD3 SMAD4 SMAD5或SMAD6,而常见的多态性被发现ENG, SMAD3和SMAD6(补充表3)。
然而,一个无义突变SMAD8c。606 C>A, p.C202X was identified in one patient (proband 14) (图1 a, B)。的SMAD8过早终止密码子突变体引入了一个外显子2和结果在一个截断蛋白质缺乏228 carboxy-terminal氨基酸,包括MH2域和SXS的磷酸化网站(图1 c)。
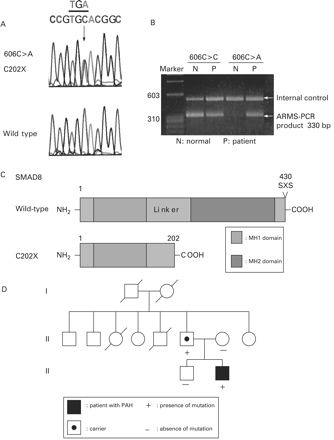
在肺动脉高血压SMAD8突变。(一)DNA序列显示c。606 C >SMAD8。(B)的面板显示确认武器试验使用反向引物特定突变的突变等位基因产生一个产品330个基点,但没有产品在控制个体。上带的内部控制。(C)的示意图表示SMAD8野生型和SMAD8 C202X突变。(D)病人的家族的血统。多环芳烃,肺动脉高血压。
临床特点
病人窝藏的突变SMAD8被诊断出患有PAH基于强调第二心音与肺炎住院期间肺地区8岁。他在8岁时血液动力学的数据显示平均肺动脉压(肺动脉平均)53毫米汞柱,右心房压力(RAP)的5毫米汞柱,心脏指数(CI)的4.31升·分钟−1·米−212.3,总肺阻力(TPR)伍德·单位−1·米−2、肺血管阻力(PVR) 12.4伍德·单位−1·米−2和肺动脉楔压(PAWP) 6毫米汞柱。他的病情发展到世界卫生组织功能的第三类与体育活动期间的气短9岁。他已经收到epoprostenol连续静脉内皮,自从9岁。
他目前的条件是功能类i ii在16岁。他最近的血液动力学的数据显示肺动脉平均49毫米汞柱,说唱的7毫米汞柱,CI的4.98升·分钟−1·米−2,TPR 9.9伍德·单位−1·米−2,PVR 8.5伍德·单位−1·米−2和PAWP 7毫米汞柱。
病人的母亲和他的哥哥没有相同的突变。然而,他58岁的父亲被确认为有相同的PAH突变虽然没有临床症状观察(补充图1)。根据病人的父亲,他是七个孩子的第六位。他的第三个姐姐和五兄弟死于肺部疾病在⩽13岁和2岁,分别为(图1 d)。其他四个兄弟姐妹的父亲没有筛查SMAD8突变,因为他们的血液样本并没有在这个时间点上获得。
免疫印迹分析和co-immunoprecipitation
SMAD8突变被磷酸化和刺激与SMAD4 ALK3和ALK1持续活跃
之前就有报道称,Smad8磷酸化的持续活跃TGF-β/ BMP I型受体,除了ca-ALK5,刺激与Smad4进行交互。2223此外,c端截断Smad8缺乏SXS的磷酸化网站显示是磷酸化在ca-ALK3面前,未能与Smad4交互。22
我们首先检查ca-ALK3和ca-ALK1磷酸化的影响的SMAD8 C202X突变。SMAD8突变不是ca-ALK3和ca-ALK1磷酸化(图2)。我们进一步检查的能力SMAD8突变与co-immunoprecipitation SMAD4的交互。观察SMAD8野生型之间的相互作用和SMAD4的ca-ALK3或ca-ALK1。然而,SMAD8突变未能与SMAD4 (图2 b)。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
SMAD8 C202X突变不是由TGF-β磷酸化/ BMP I型受体,并且不与SMAD4交互。(A)的磷酸化Myc-SMAD8野生型和SMAD8 C202X突变COS1细胞被免疫印迹检测(吸)。Myc-SMAD8野生型或SMAD8 C202X突变与既定的瞬变中的活跃HA-TGF-β/ BMP I型受体ALK3 ALK1。I型受体依赖的磷酸化SMADs anti-phospho-Smad1/5/8显示抗体,也与SMAD8的磷酸化反应。(B)的交互Myc-SMAD8野生型和SMAD8 C202X突变与Flag-SMAD4 HEK293细胞被免疫沉淀反应检查(IP)其次是免疫印迹(吸)。与SMAD8 HEK293细胞转染野生型,SMAD8 C202X突变,ALK3和ALK1 SMAD4以及持续活跃。细胞溶解产物受到免疫沉淀反应anti-Flag抗体和免疫印迹分析anti-Myc抗体。
荧光素酶检测
SMAD8突变并不是能够激活BMP / promoter-reporter TGF-β敏感
据报道,Smad8单独或和Smad4增加BMP中的响应promoter-reporters没有BMP配体或者持续活跃TGF-β/ BMP I型受体。23因此,为了确定SMAD8 C202X突变能够增加BMP和TGF-β响应promoter-reporter活动,我们调查了转录活动由SMAD8野生型和突变体有或没有SMAD4和/或ca-ALK3使用BMP响应promoter-reporter 3 gc2-lux TGF-β响应promoter-reporter, 3 tp-lux COS-1细胞。
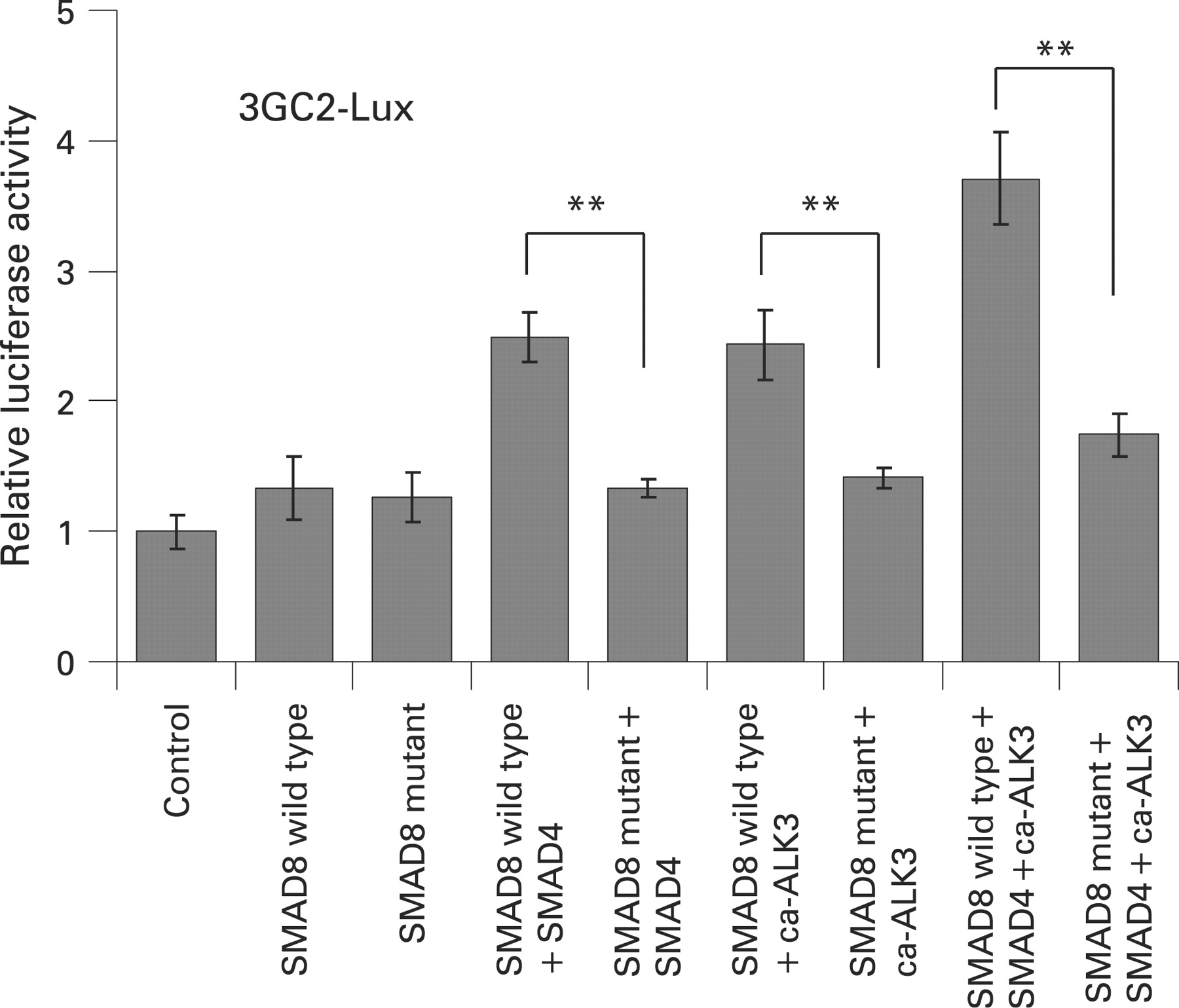
SMAD8野生型BMP诱导响应启动子活动。SMAD8野生型的co-expression SMAD4或者ca-ALK3,诱导活动大约两倍高于SMAD8野生型。SMAD8野生型中的SMAD4和ca-ALK3诱导活动大约3倍高于野生型。然而,SMAD8突变,即使发生SMAD4或/和ca-ALK3低效在激活BMP响应promoter-reporter相比SMAD8野生型(图3)。
{kind=link}

{kind=link}
SMAD8野生型的影响以及SMAD8 C202X突变体的转录激活3 gc2-lux测定存在和SMAD4的缺失和/或ca-ALK3 COS1细胞。值代表的意思(SD)。统计组差异评估学生的t检验。* * p < 0.01。
使用TGF-β响应promoter-reporter类似结果,3 tp-lux(补充图2)。这些结果表明,SMAD8 C202X突变是不能激活BMP / promoter-reporters TGF-β敏感。
讨论
Smad8是受体调节Smads (R-Smads) TGF-β总科的受体。Smad8以及Smad1 Smad5直接基质TGF-β/ BMP I型受体和磷酸化的c端地区SXS的主题。磷酸化Smad8然后用Co-Smad associates和Smad4把进入细胞核,调节靶基因的转录。24
先前的报道表明,信号的损失由SMAD1/5/8扮演着一个重要的角色在肺血管重塑和PAH的发病机制。减少的磷酸化SMAD1媒体和内膜细胞内的小患者的肺动脉IPAH FPAH观察。25的表达BMPR2 IPAH / FPAH肺血管病变的患者,尤其是在那些有BMPR2突变,是减少。26功能研究BMPR2突变表明BMPR2突变监管SMAD1/5/8信号中断或下降。2728ALK3的表达明显降低肺的IPAH病人没有突变BMPR2或ALK1。29日此外,表达磷酸化Smad1/5/8肺明显降低条件基因敲除小鼠缺乏Alk3肺泡上皮细胞。30.遗传性出血性毛细血管扩张症ALK1突变的功能分析表明,ALK1突变破坏或向下调节SMAD1/5/8信号。31日这些观察表明,监管TGF-β/ BMP信号通过SMAD1/5/8 PAH的发病机制中起着重要的作用。
BMPR2已被确定为主要致病基因IPAH / FPAH。8- - - - - -10同时,ALK1突变患者已报告IPAH / FPAH。1213一个英格突变在婴儿IPAH,后来确诊为遗传性出血性毛细血管扩张症患者在8岁。12然而,大约30%的FPAH和60 - 89%的显然IPAH患者没有港口的突变BMPR2。1011大约70%的儿科患者IPAH没有港口的突变BMPR2或ALK1。13因此,在这项研究中,我们调查了其他基因的突变是否参与TGF-β/ BMP信号通路发生在23 IPAH患者。
我们发现了一个无义突变SMAD8c。606 C>A, p.C202X in one patient (图1)。据我们所知,生殖系突变SMAD8尚未报道多环芳烃或其他疾病,而损失SMAD8表达在乳腺癌、结肠癌和前列腺癌已被报道。3233
截断的蛋白质结构预测的SMAD8 C202X突变(图1 c),我们的免疫印迹和co-immunoprecipitation试验表明,它不是由TGF-βphophorylated / BMP I型受体,并没有和SMAD4互动(图2 a, B)。这些结果表明SMAD8突变干扰TGF-β/ BMP的下游信号。
之前就有报道称,Smad8能够把细胞核没有ca-ALKs,尽管其易位效率远低于观察ca-ALKs的存在。23因此,我们调查是否SMAD8突变可以执行一些野生型蛋白的转录功能通过使用BMP响应promoter-reporter, 3 gc2-lux构造,在COS-1细胞。荧光素酶测定表明,SMAD8野生型产生一个高度显著增加BMP响应promoter-reporter活动,特别是当它和SMAD4和ca-ALK3转染。相比之下,SMAD8突变是低效和SMAD4或激活记者表达ca-ALK3 (图3)。这可以解释为截断SMAD8突变的蛋白质结构缺乏MH2域和c端地区SXS的磷酸化的网站。SMAD4交互所需的MH2域不仅也为绑定到其他核DNA结合代数余子式等因素,co-activators和若转录复合体的组装。24
我们的病人的临床表型SMAD8突变没有不同于其他IPAH病人。他没有其他症状以外的多环芳烃在16岁。他的父亲有相同的SMAD8突变,但临床症状或多环芳烃的客观测量诊断并没有被观察到。这种情况并不奇怪,FPAH生殖系BMPR2突变的外显率低。只有10 - 20%BMPR2临床PAH突变携带者将清单。934低外显率也观察到在我们之前的研究;两位父亲的儿科患者FPAH相同ALK1突变作为他们的孩子,但没有多环芳烃或遗传性出血性毛细血管扩张症的症状。13
在这项研究中,一个SMAD8突变被发现只有一个病人的23 IPAH患者(1/23,4.3%)。显然,额外的IPAH / FPAH病人必须筛选来确定的,外显率和精确的角色SMAD8在IPAH / FPAH。
先前的研究已经表明,监管的TGF-β/ BMP信号通过SMAD1/5/8 PAH的发病机制中起着重要的作用。我们的发现SMAD8突变PAH患者提供了一个证据来支持它。
23日的22 IPAH患者(22/23,95.7%)没有发现突变ENG、SMAD1 SMAD2、SMAD3 SMAD4 SMAD5,SMAD6和SMAD8在这项研究中。它表明,仍有不明TGF-β/ BMP信号通路中的基因或其他途径诱发多环芳烃。此外,低外显率的FPAH尽管广泛表达基因突变表明额外的基因和/或环境因素发挥重要作用在多环芳烃的航空公司的发展SMAD8,BMPR2和ALK1突变。据报道,长期缺氧引起肺血管重塑和多环芳烃在老鼠身上。35BMPR2杂合的突变小鼠对炎性应激与右心室收缩压显著增加。36因此,这将是有趣的,以确定Smad8纯合或杂合的突变小鼠发展多环芳烃等刺激反应的慢性缺氧或炎性压力。此外,确定SMAD8在多环芳烃的功能,进一步的调查使用人类肺动脉平滑肌细胞和人类肺动脉内皮细胞是必要的。
确认
我们感谢病人和他们的家庭成员。我们感谢系主任Bernardo nadal - ginard博士他的宝贵意见。我们感谢Miyazono浩平表示博士提供质粒。我们还要感谢博士Shin-ichiro导演今村昌平,Arai Shoichi博士,博士Yoshiyuki Furutani,藤原,玛雅Emiko博士Hayama和美智子女士Furutani优秀的技术援助。
参考
补充材料
-
网络只附录46:5;331 - 7
在这个数据补充文件:
脚注
▸额外的表和数据只在网上发表http://jmg.bmj.com/content/vol46/issue5
资助:这项工作是支持的项目促进建立战略研究中心,专门协调资金促进科技、教育、文化、体育、科学和技术(日本)。
利益冲突:一个也没有。
病人同意:获得的。