





摘要
临床和翻译研究在推进对肺动脉高压(pH)的理解方面发挥了重要作用,包括肺动脉高压和其他形式的pH具有严重血管重塑(例如慢性血栓栓塞pH和肺部静脉闭塞疾病)。然而,pH值仍然是一种不稳定的病情,具有高死亡率,强调需要更好地将新颖的科学知识转移到医疗保健干预中。在此,我们审查了最近的病理结果(通过质疑各种形式的pH值的严格形态分类,进入肺部血管的前或后毛细血管中的毛细血管后或毛细血管后累及)和促进与各种肺血管重塑的发作和进展的细胞机制pH的形式。我们还讨论了改进管理的方法,并在本研究领域的支持和优化药物开发。
摘要
与各种形式的肺动脉高压相关的肺血管重塑的细胞和分子基础和病理学的艺术状态和研究视角和病理学http:///wly/cjwp30mgzmh.
介绍
肺动脉高压(PH)包括一组严重的临床实体,如肺动脉高压(PAH)和慢性血栓栓塞性PH (CTEPH),其中肺血管床的缺失和梗阻性重塑是肺动脉压力和肺血管阻力(PVR)上升的原因,导致进行性右心衰和功能下降。肺动脉高压肺血管重塑的特征不仅是不同血管细胞在肺动脉壁(肺动脉平滑肌细胞(PA-SMCs)、内皮细胞、成纤维细胞、肌成纤维细胞和周细胞)的积累,也会导致毛细血管前动脉的缺失,以及血管周围炎性细胞(B淋巴细胞、t淋巴细胞、肥大细胞、树突状细胞、巨噬细胞、等等。).由于目前PAH治疗不专门针对肺血管重构和炎症,迫切需要更好地识别病理生理机制,逐步缩小基本肺动脉腔内及血管周围炎症和血管,以支持治疗的创新瞄准了损失扭转这些功能和再生正常的肺血管。
在病理学和实验医学的最新进展
一般考虑因素
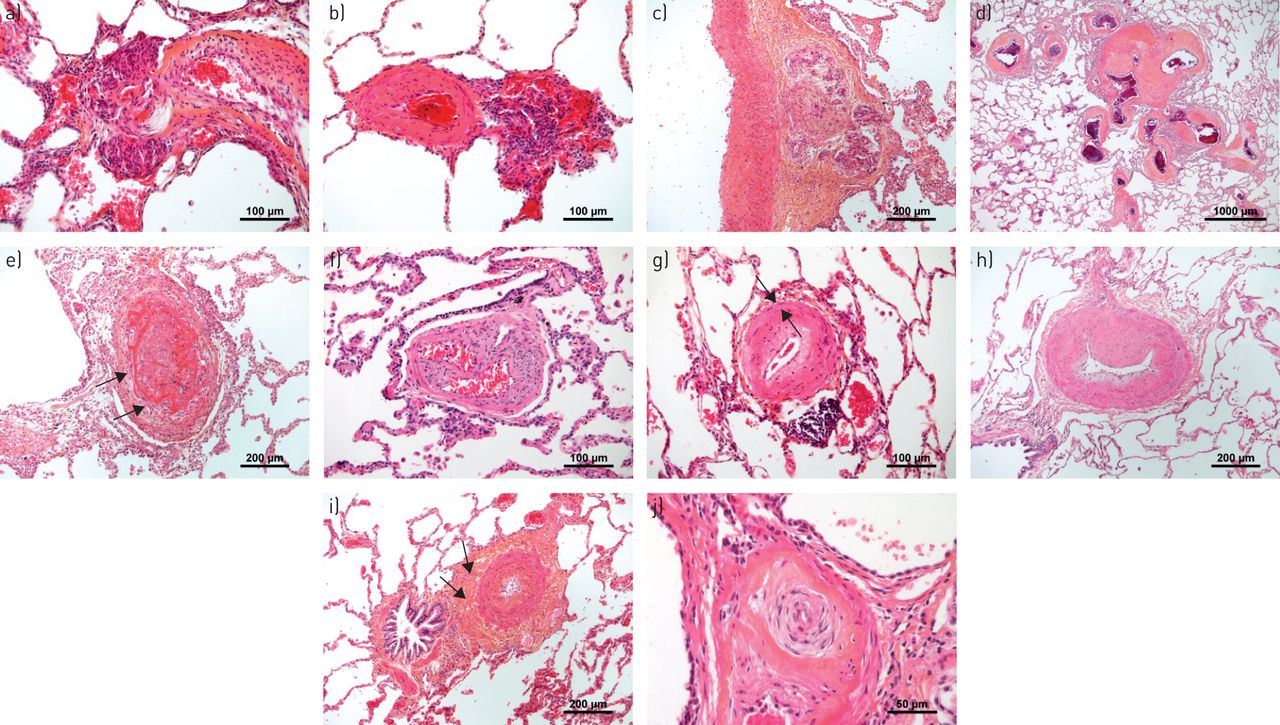
一世n addition to stiffening of large elastic main, lobar and segmental pulmonary arteries, PH can be attributed to lesions mainly occurring in distal muscular-type arteries, ranging in diameter from 500 µm down to 70 µm in humans (medial hypertrophy/hyperplasia, intimal and adventitial fibrosis, and (原位)血栓性病变,网状损伤)(图1).由于肺部内的地形,它们可以清楚地区分肺静脉,因为它们始终由气道(支气管)(支气管)以及它们的微观解剖学以及由内部和内部限定的整齐定义的丘脑介质而邻近。外部弹性薄片。在人体(血细胞)中,直径为70μm至20μm的小毛细血管肺动脉也通过丧失和灭菌,异常肌肉发作和血管内炎症的过程涉及所有人类和实验性pH值(图2).与肌肉型动脉相反,它们只能通过连续切片示踪或(如果可行的话)染色或珠状注射技术间接地与同样大小的毛细血管后小静脉区分。起源于小动脉微血管系统的毛细血管室,代表肺内最大的血管表面,也经常受累。也有越来越多的证据表明,在所有PH组中,毛细血管后肺静脉血管都有不同程度的损伤。在由左心疾病和慢性呼吸系统疾病引起的PH中,毛细血管后累及可能至少部分地解释为肺纤维化和肺气肿患者的实质破坏和炎症。以及左心衰毛细血管后压力的慢性升高,其中PH与整体肺血管重构相关。有趣的是,心力衰竭的PH严重程度与静脉和小的不确定血管内膜增厚相关,类似于肺静脉闭塞性疾病(PVOD)的模式[1];小叶间隔内运行更大的肺静脉,可能会出现“arteriolised”,模仿肌肉型肺动脉的确切显微解剖。在显然纯预毛细管形式PH,诸如PAH和CTEPH,后毛细血管受累的机制并不明显,因为后毛细血管,在理论上,应该从提高预毛细管压力由动脉屏蔽和毛细管舱[2].
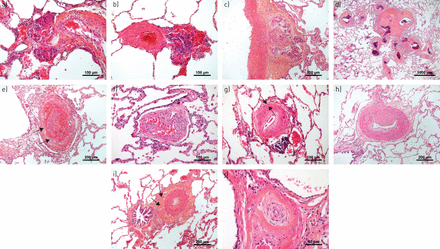
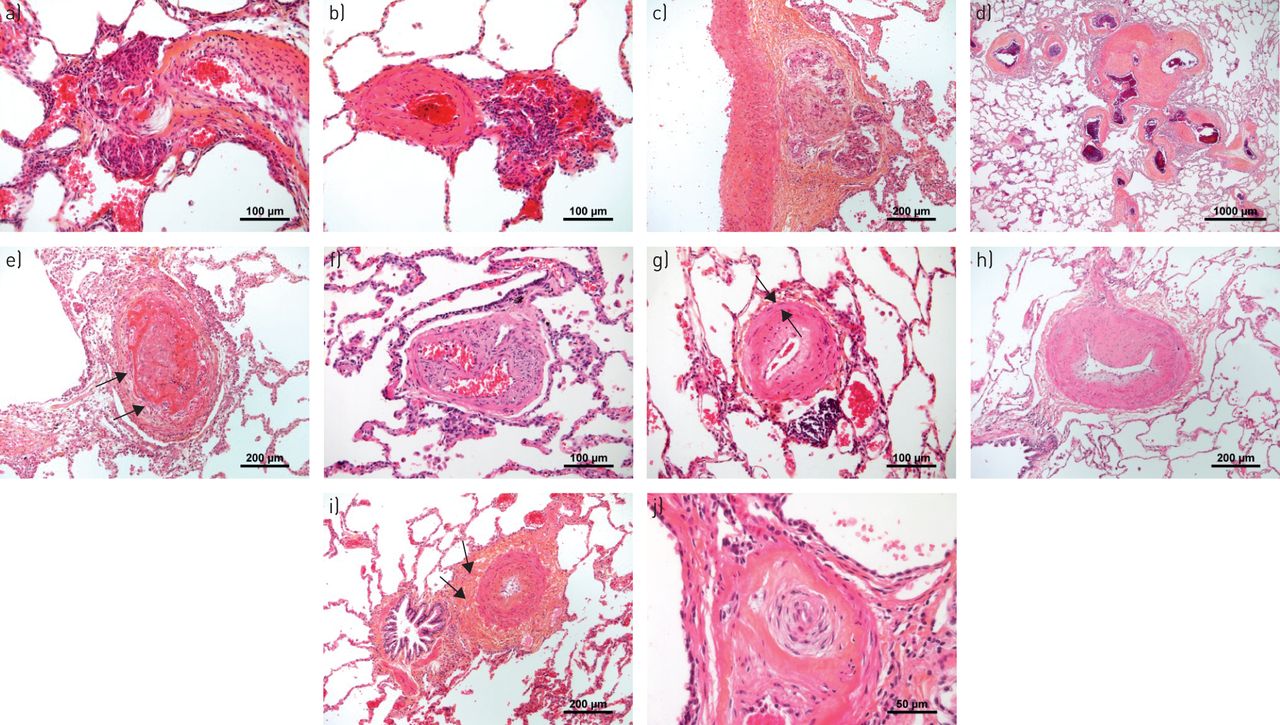
肺动脉高压(PAH)患者肺部典型的血管病变。a - c)丛状的病灶。值得注意的是,丛状核心可能位于a、b)动脉旁位置,似乎与外膜相连,或c)甚至在外膜的周长内,这可能表明累及了系统的血管系统(vasa vasorum)。d)在PAH中发现的毫米大小(注意标尺)的非典型纤维血管病变(也称为SiMFis(单数毫米级纤维血管病变)),主要是遗传性骨形态发生蛋白受体2型相关形式。e)新近出现的血栓性病变,在其核心处显示新鲜的纤维蛋白,并在周围开始组织过程,包括大量成纤维细胞(箭头)。f)组织完整的血栓病变(滤过样病变),有多条再通血管,模糊地使人联想到丛状病变。g)内膜同心、非层状纤维化;中膜(箭头)仅轻微增厚(注意血管下周富含淋巴细胞的浸润)。h)内膜偏心性、垫状纤维化,通常解释为有组织的血栓病变。i)中膜增生和外膜富含胶原的纤维化(箭头)。 j) Concentric laminar fibrosis of the intima (“onion-skin lesion”) due to the concentrically arranged multiple fibrous layers that lead to the progressive obstruction of the pulmonary artery.
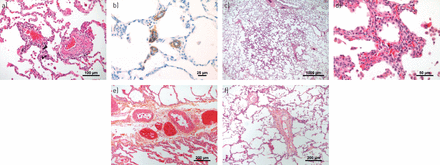
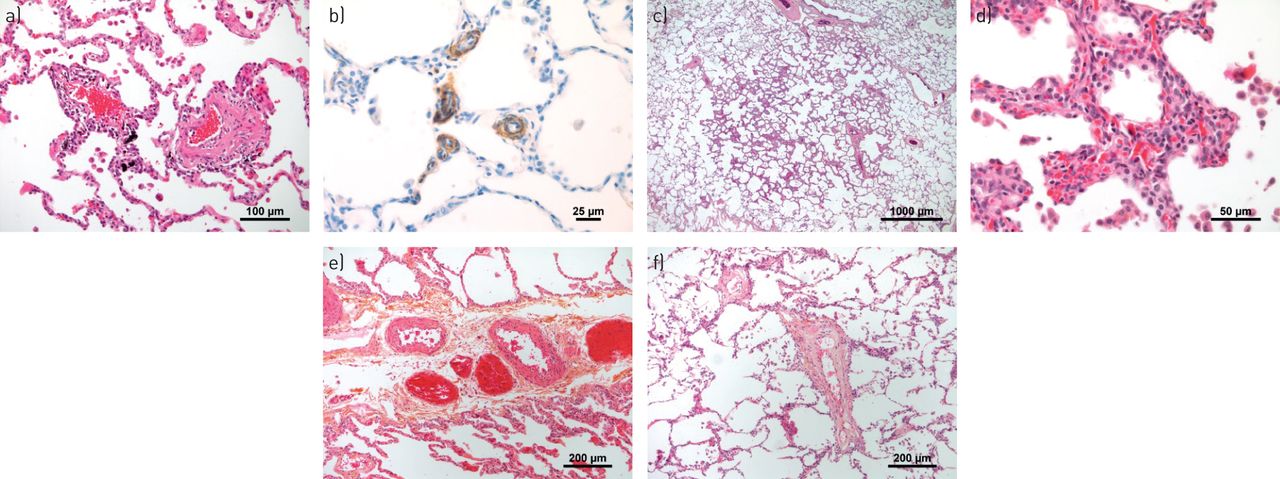
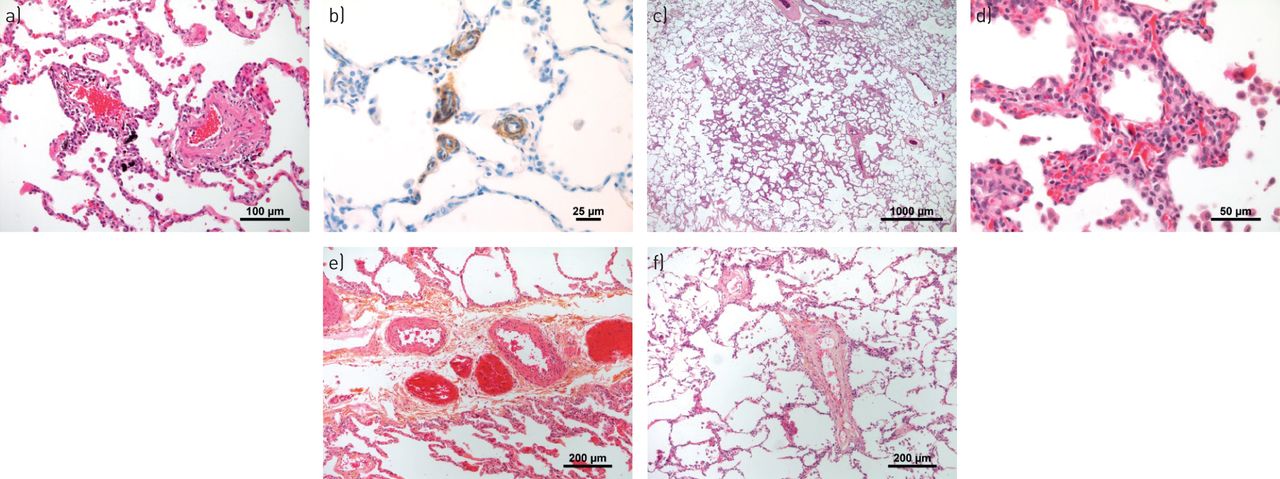
通常在微血管结构,毛细血管和毛细血管后血管(肺静脉)中检测到具有肺动脉高血压(PAH)或肺部静脉闭塞疾病(PVOD)的微血管结构,毛细血管和肺静脉)中的代表性血管病变。a)呈现一些轻度炎症和纤维重塑的微血管(动脉或静脉)(患者用PAH)。b)具有PVOD患者肺部肺部肺部免疫染色的α-平滑肌的代表性图像,突出了肺脉管系统的大量肌肉酶。c)PVOD患者圆形,截然浓度的间质增稠区域;这些区域以PVOD患者的肺整个肺部分发,并且可能对应于计算机断层扫描扫描的典型地玻璃不透明度。d)在更高的放大率下,(c)中的间质增稠是由于肺泡荚膜内形成多层的肺泡毛细血管的局灶性增厚;术语毛细血管血管症状的焦点描述了这种斑驳的间质图案。e)患有PAH的患者的肺部间脉中的肌肉增生和纤维改造。f)用PVOD患者小隔膜静脉的纤维内膜增厚。
特异性肺血管病变的新解释
丛状病变/复杂病变
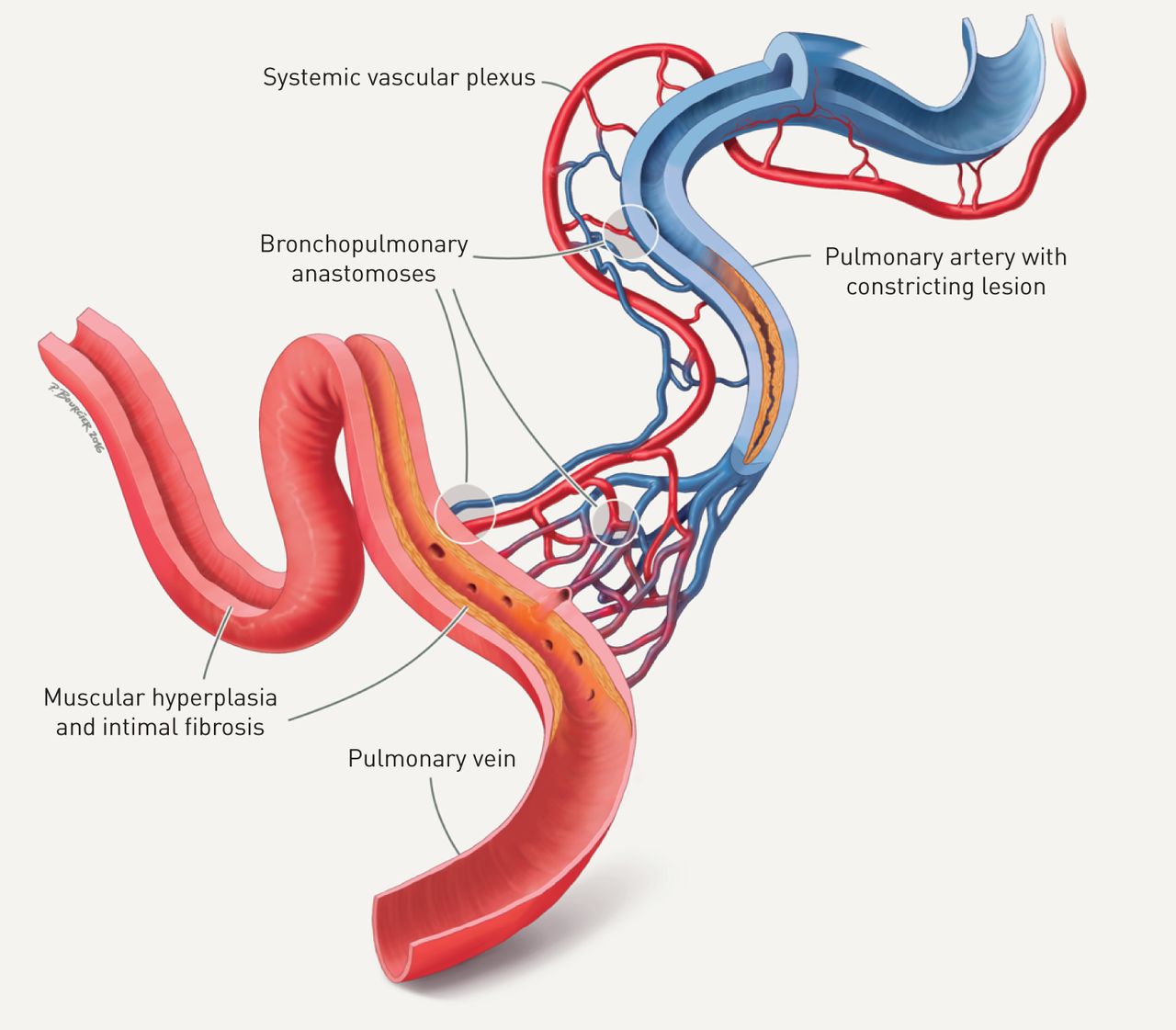
复杂的肺动脉损伤包括不同的元素,例如洋葱皮病变,丛状核心病变和扩张病变,其在近地形关联中通常观察到(图1A-C)。然而,PAH中这些典型血管变化的病理生理学意义尚未阐明。在这方面,最近的报道表明,分别参与了全身血管,例如在肺动脉的外膜内或肺癌结缔组织内的血管血管和支气管动脉,可以参与丛状血管病变(图1C.).数字三维重建对肺动脉高压患者连续切片的分析支持分流假说,其中丛状病变似乎代表支气管微血管与肺动脉和静脉之间的吻合结构[3.].通过对肺动脉高压患者肺组织切片的形态计量学分析,描述了支气管和肺血管系统之间的分流,包括特发性肺动脉高压(IPAH)和遗传性肺动脉高压(HPAH)BMPR2(骨形态发生蛋白受体2型)突变[4.].肥厚和支气管动脉的扩张和增加支气管微血管密度BMPR2突变携带者与肺静脉重塑相关[4.].此外,大型纤维血管结构(“辛比毫升纤维血管病变)似乎将全身脉管系统连接到肺动脉和静脉(图1D).PAH中肥厚系统脉管系统的功能作用,允许短路初级肺动脉阻塞(图3)尚未确认。
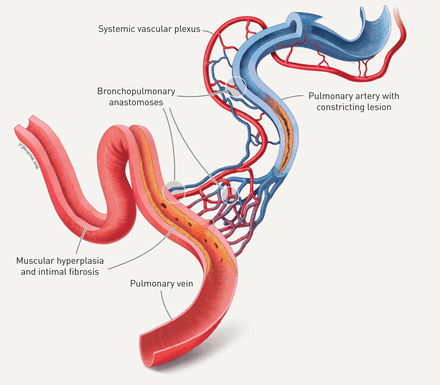
肺动脉高压(PAH)中肥厚性全身血管系统的影响:一种解释方法。肺动脉(上中央,蓝色)被全身血管丛覆盖,包括全身动脉(红色)、静脉(蓝色)血管和微血管。系统神经丛与肺动脉、毛细血管床和肺静脉吻合(左下,红色):这些支气管肺吻合似乎绕过了肺动脉阻塞病变,表现为内侧增厚和内膜纤维化(中间)。最终,进入细动脉、毛细血管和肺静脉的全身血流量增加,导致肺静脉的结构改变:肺静脉内出现肌肉增生和局灶性内膜纤维化。摘自[4.]允许。
静脉和瓣膜病变
大部分PH患者表现为肺静脉和小静脉重塑(图2e) [4.]:来自硬皮病的PAH患者的肺部经常表现出pvod样病理[5.],和肺CTEPH通常显示肺静脉和小静脉异常[2].CTEPH在这方面尤为重要。虽然最主要的侮辱,IE。弹性和肌肉动脉的慢性血栓栓塞性阻塞,发生在肺血管毛细血管前侧,导致PVR增加,微血管重塑也存在,影响毛细血管前小动脉和毛细血管后小静脉[2那6.].重要的是,支气管动脉肥大与CTEPH中的肺部静脉重塑有关,支持与支气管扩孔吻合术相关的全身肺部血管有助于这些变化[2].
在PVOD中,肺血管病变被认为是毛细血管后侧的占主导地位,但是有涉及的动脉[7.].影响室间隔静脉和室间隔前小静脉的毛细血管后病变通常包括松散的纤维性内膜重塑,可完全闭塞管腔。室间隔静脉和室间隔前小静脉壁可表现为平滑肌细胞增生,很难与PVOD肺中直径<70µm的异常肌化小动脉鉴别[7.].毛细血管后重塑经常与肺毛细血管angioectasia和毛细管相关联的血管增生与加倍和可灶性分布在肺泡间隔毛细管层(肺毛细血管haemangiomatosis)的三倍。看图2B.- d和f。
在细胞异常和新兴的治疗靶点的研究进展
肺血管内皮功能障碍
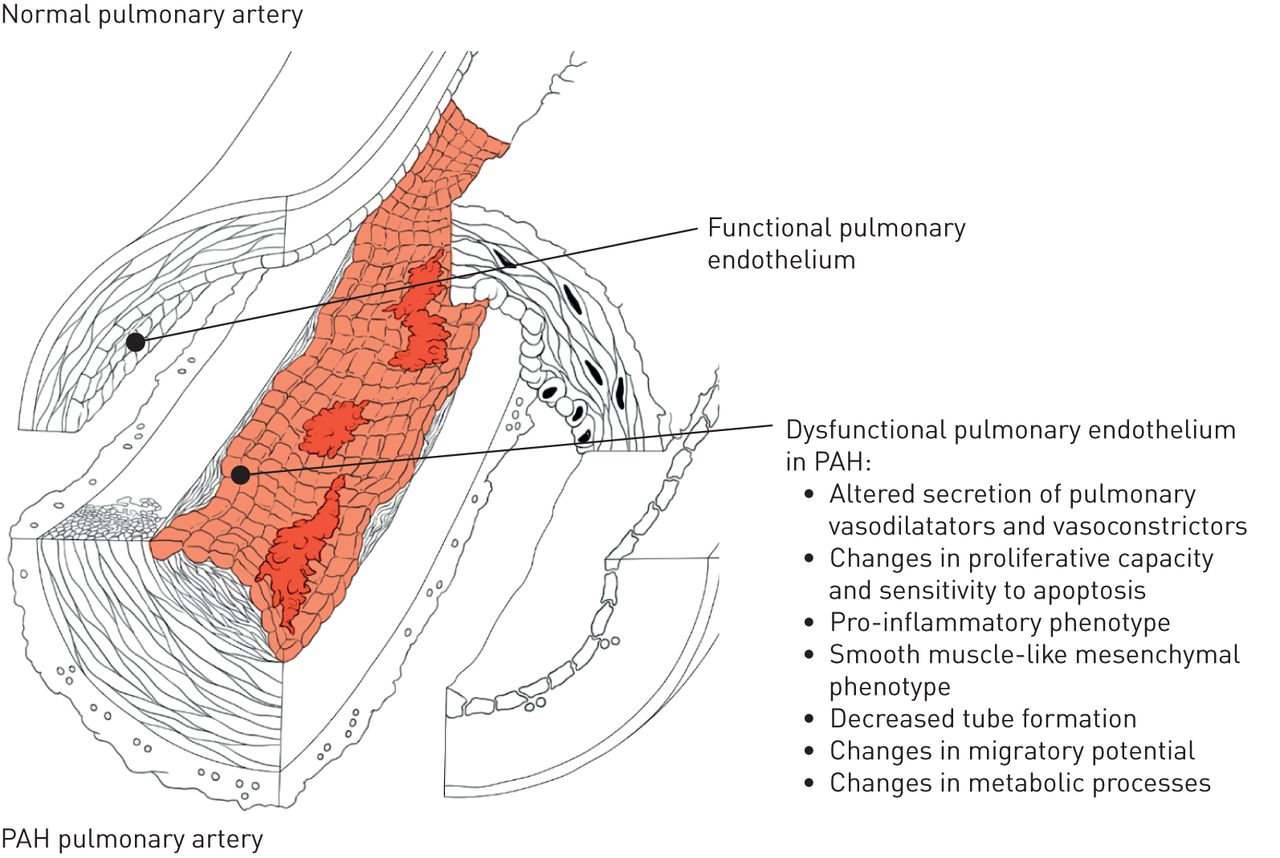
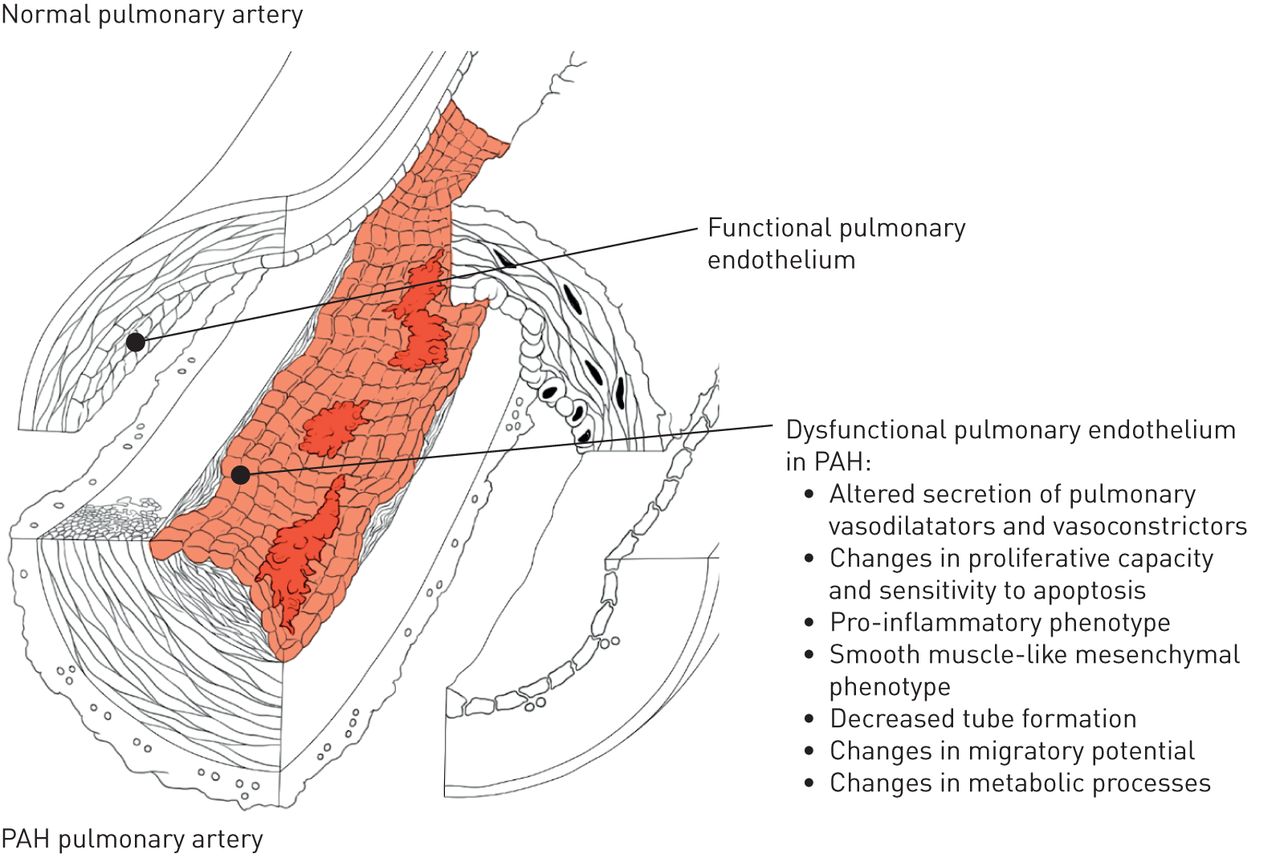
在PAH,术语肺血管内皮功能障碍已被用于有利于血管收缩的内皮依赖性血管舒张的分别表示损害,但它也指降低抗凝血特性,活性代谢变化,活性氧的产生,粘附分子的表达增加(E-选择素,细胞间粘附分子1(ICAM1)和血管细胞粘附分子1(VCAM1)),以及不同的趋化因子,细胞因子和生长因子的局部释放不适应(图4.).后面的这些变化导致在血管生成和修复机制障碍的是发挥肺血管重构主要作用[8.].现在已经证实,培养的肺动脉高压患者的肺内皮细胞可以维持体外几个异常的表型特征或多或少明显,可能反映了不同的亚种群。其中,血管形成能力下降体外,有氧糖醇分析,并且已经描述了一些内皮细胞标记物的丧失并对几种间充质细胞标记物进行了描述[9.-11].在PAH中,据报道,通过e-selectin,ICAM1和Vcam1的表面表达增加,其特征的肺内皮细胞的促炎表型以及过量释放各种关键细胞因子和趋化因子的过度释放12].一些特征可以在由来自同一患者皮肤的诱导多能干细胞(iPSCs)培养的内皮细胞中复制[13那14].各种刺激,如高葡萄糖,胰岛素抵抗,扰乱血流和氧化应激,可导致内皮功能障碍。然而,事业和负责PAH肺动脉内皮功能障碍的潜在机制仍然不完全理解。
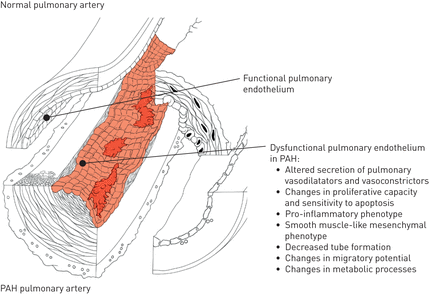
的功能失调性肺血管内皮肺动脉高压(PAH)表型的签名。
最近的研究集中在内皮结构和功能的两种关键调节剂,它们启动并持续与PH/PAH相关的肺血管重构,IE。流体流动引起的高剪切应力以及低氧分压(慢性缺氧)。正常情况下,血管内皮细胞通过失去其鹅卵石的外观和在流动方向上拉伸应对高剪切应力。未能适应这些形态的变化与走向血管重塑增加的趋势相关。有趣的是,压力卸载通过肺动脉绑扎已经报道,以防止甚至逆转闭塞性血管在SUGEN缺氧大鼠模型重塑[15].这一观察结果与最近的研究结果一致,这些研究表明,从肺动脉高压患者分离出来的微血管肺内皮细胞(而非近端肺动脉内皮细胞)表现出对高剪切应力的延迟形态适应体外[16].慢性缺氧也产生了实验和人体PH肺血管结构重塑。最近的研究已经通过显示从患者的PAH网状损伤肺血管内皮细胞具有脯氨酰-4-羟化酶2(PHD2),其促进低氧诱导因子HIF1和HIF2缺氧传感器的降解的酶的表达降低支持了这一概念.此外,与该基因的供PHD2内皮细胞靶向破坏的小鼠(egln1.)发生闭塞性肺血管重塑和复杂病变,如人类PAH [17].安非他命和缺氧的结合可以增加内皮细胞凋亡的倾向,并通过干扰HIF1α转换为糖酵解状态的正常代谢功能而导致DNA损伤[18].达沙替尼可诱导肺动脉高压,并已成为肺内皮功能的调节剂:高剂量达沙替尼可诱导肺内皮细胞功能障碍通过增加的产量的活性氧,从而增加易感性胸腔积液和PH在啮齿类动物中[19-21].更好地理解内皮适应对高剪切应激和慢性缺氧的分子机制将大大提高我们对pH / PAH发病机制的理解,并有助于确定新的治疗策略。与常规血管细胞(PA-SMC,肌纤维素细胞,截瘫)和循环细胞(免疫细胞)进行改变的肺内皮连通的额外见解也是更好地理解pH / PAH发病机制的先决条件。
内皮是也用于功能性血管网络取决于负责协调这个过程中,不同的细胞类型之间交换的信号的发展是至关重要的。在实验和人PH / PAH,血管生成被干扰与预毛细动脉导致肺血管稀疏的图案(“死树”画面),即使若干促血管生成因子是overabundant和/或丢失和进行性闭塞据报道,在PA-平滑肌细胞的Notch3信号的过度活跃和表达。因此,需要进一步的研究,以确定如何内皮微环境,在细胞 - 细胞和细胞 - 基质的接口,也妨碍肺内皮完整性和在PH /其再生血管生成能力PAH。在这种情况下,循环细胞和驻地血管祖细胞的贡献的一个更好的了解,应确定[22-28].实际上,周象不仅是血管成熟和稳定性,而且对于预防内皮芽形成和生产新功能脉管系统所需的内皮增殖也是重要的。在实验性和人pH / PAH中,远端肺动脉肺动脉的总数在疾病进展期间大幅增加[26]和在周细胞功能的缺陷已被证明[28].来自PAH患者的孤立周细胞表现出降低的基因水平,对于WNT /平面细胞极性途径至关重要,并且由于基质管形成测定中评估而导致管形成期间与内皮细胞相关联[28].
现在已经明确证实,异常BMPR2信号会对内皮屏障功能产生不利影响,与受损DNA修复相关的DNA损伤持久性[29]、新陈代谢、线粒体分裂及融合[30.那31],也是炎症及其决议[32-34].因此,应尽快努力以更好地理解BMPR2信号传导系统与肺血管改造过程之间的相互作用。
PA-SMC和Adtoritaial成纤维细胞的积累
在肺动脉高压中,已知肺动脉微环境和一些内在异常和异常信号的存在部分解释了PA-SMCs和外膜成纤维细胞的累积。近年来,临床前和早期临床工作突出了PAH病理生理学的新靶点。在内皮细胞中,DNA损伤反应途径与PAH中的PA-SMC和成纤维细胞存活密切相关。在人类和实验的PH/PAH中,BMPR2下调后BRCA1(乳腺癌1)蛋白的下降与poly(ADP核糖)聚合酶1 (PARP1)的上调有关,以应对DNA损伤的增加[35].PAH papa - smcs中PARP1的上调使其能够通过抑制DNA损伤后果来应对环境压力,使其线粒体功能适应生存模式[36那37].在实验PH模型中抑制PARP1显示出比结合当前标准护理更有效的效果,因此美国食品和药物管理局批准的PARP1抑制剂奥拉帕尼正在对PAH进行临床研究(ClinicalTrials.gov标识符NCT03251872).另一进步来自miRNA-124的下调的观察结果,导致在pH脉管系统中的成纤维细胞和内皮细胞中的RNA剪接因子聚吡啶干燥蛋白1(PTBP1)的上调[38那39].PTBP1与剪接因子的异质核核糖核蛋白家族的其他成员一起,已经被证明可以调节丙酮酸激酶肌肉(PKM)亚型的剪接。PTBP1的增加导致PKM2亚型的积累增加,PKM2亚型在其二聚体(未激活状态)中促进糖酵解、增殖和抗凋亡,即使在有氧环境中。使用药物抑制剂恢复正常的PKM2/PKM1比值可减弱成纤维细胞和内皮细胞的增殖体外和体内.此外,已经发现肿瘤坏死因子-α (TNF-α)抑制BMPR2的表达并促进翻译后切割通过PA-SMCs中的“a disintegrin and metalloproteases”ADAM10和ADAM17,有利于bmp介导的增殖通过备选激活素受体[40].此外,PA-SMCs暴露于高糖会增加SMURF1 (E3泛素蛋白连接酶,抑制BMP信号传导)的表达,并减少磷酸化smad1 /5/8,模拟突变阴性pah衍生的PA-SMCs的信号模式,后者通过阻断葡萄糖摄取而正常化[41].类似地,在PA-SMC和肺的内皮细胞Smad3的耗尽被发现有助于提高的增殖和迁移,这是通过抑制心肌素相关的转录因子衰减[42].
最近在这种情况下鉴定了其他有前途的靶标,其中包括:白酮B4(LTB4),白细胞介素-6(IL-6)和瘦素受体,以及转录核心压缩机C末端结合蛋白1(CTBP1),转化生长因子-β,过氧化物体增殖物激活受体-γ(PPAR-γ),哺乳动物靶标络合物1(MTORC1)和FORKHEAD盒O1(FOXO1)途径[43-47].PAH,也建立了动态和不适应的改造的细胞外基质形成一个宽松的环境,不仅有利于细胞运动性,增殖、凋亡、分化的居民血管细胞和炎症细胞的招聘,但也有相当大的影响船刚度(48-51].由于它们的位置很近,而且有几条线索表明肺动脉细胞与血管周围单核细胞和巨噬细胞之间存在复杂的相互关系[52那53],需要更完全了解这些复杂的相互关系。
先天和自适应免疫系统的失调
在PH实验中,混合炎症细胞的血管周围炎症浸润通常先于结构性肺血管重构,支持炎症和免疫系统的适应不良存在并有助于重构的观点。与这一概念相一致的是,在PAH患者的肺中可以观察到类似于高组织淋巴滤泡的小淋巴细胞聚集到大淋巴细胞聚集。同样,现在已经确定,炎症介质的循环水平与PAH较差的临床结果相关,并且可以观察到循环细胞亚群的改变[54-56].其结果是,直接调节炎症过程已经成为了最近在PAH临床研究的重点治疗。
类固醇或阿司匹林治疗在IPAH和HPAH中没有有效,具有抗炎性质的前列腺素[57,并不能逆转肺血管重构强调了这样一个事实,即进一步了解免疫细胞和关键细胞因子/趋化因子所扮演的角色是发展新的治疗策略的先决条件。最近的研究表明肺血管细胞是肺动脉高压可溶性信号的重要局部来源,有助于肺血管重构。事实上,PAH患者的PA-SMCs、内皮细胞、成纤维细胞和肌成纤维细胞表现出明显的促炎特征,其特征是各种细胞因子和趋化因子以及关键炎症细胞粘附分子(如ICAM1)的表达升高。局部过量分泌IL-1、IL-6、LTB4、巨噬细胞迁移抑制因子、瘦素和TNF-α,以及FoxO1的失活,在介导肺动脉高压肺血管结构和功能改变中发挥了不可或缺的作用[12那40那43-46那58].
受损的T调节细胞的功能,T辅助17细胞免疫极化[59肺动脉血管病变的树突状细胞募集已经在肺动脉高压患者的组织中得到证实,这支持了免疫应答的失调。因此,循环自身抗体通常在PAH患者中检测到,但没有相关自身免疫性疾病的证据。此外,在PH/PAH肺中有淋巴样新生的报道。进一步,更好地理解导致PAH免疫和耐受性平衡改变的机制,可能有助于确定PAH管理的免疫病理学方法。
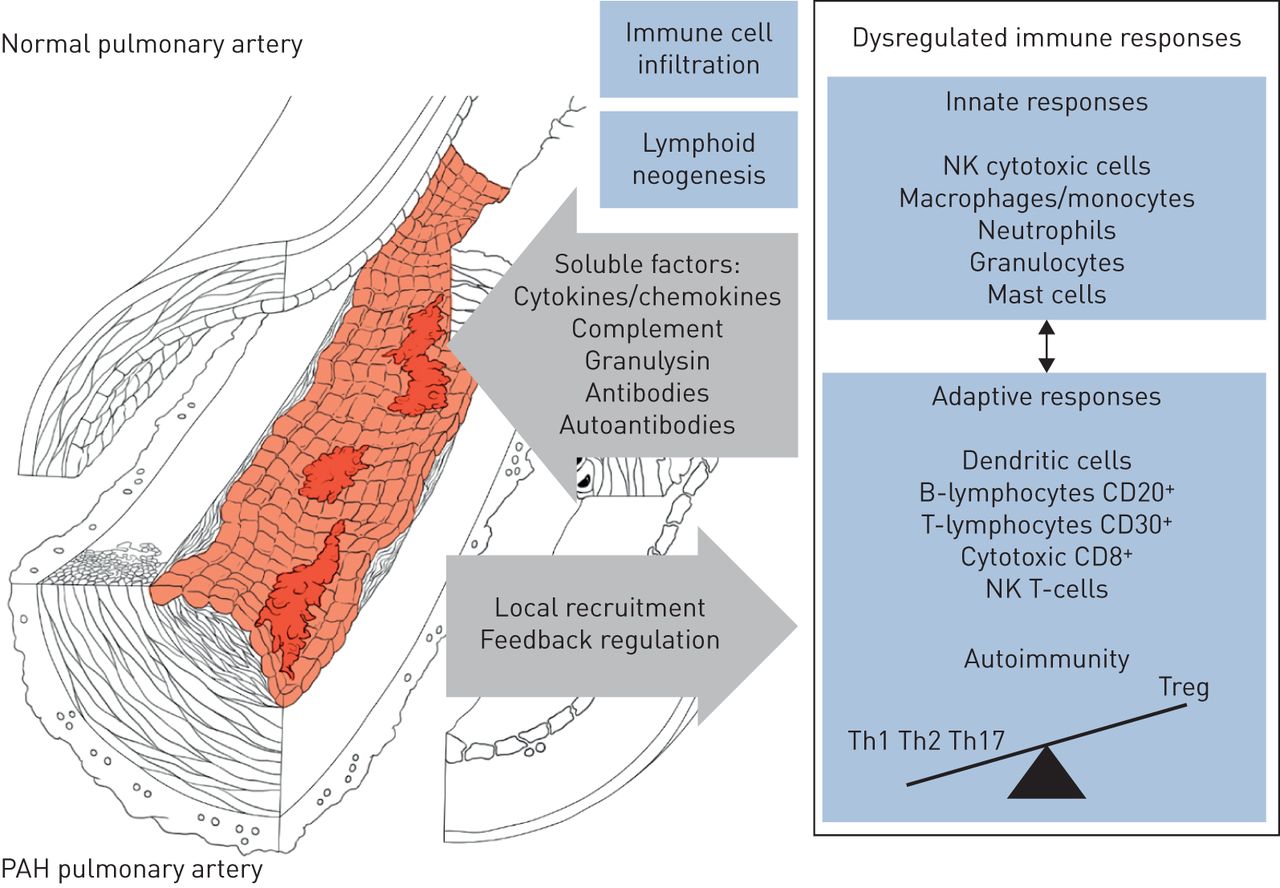
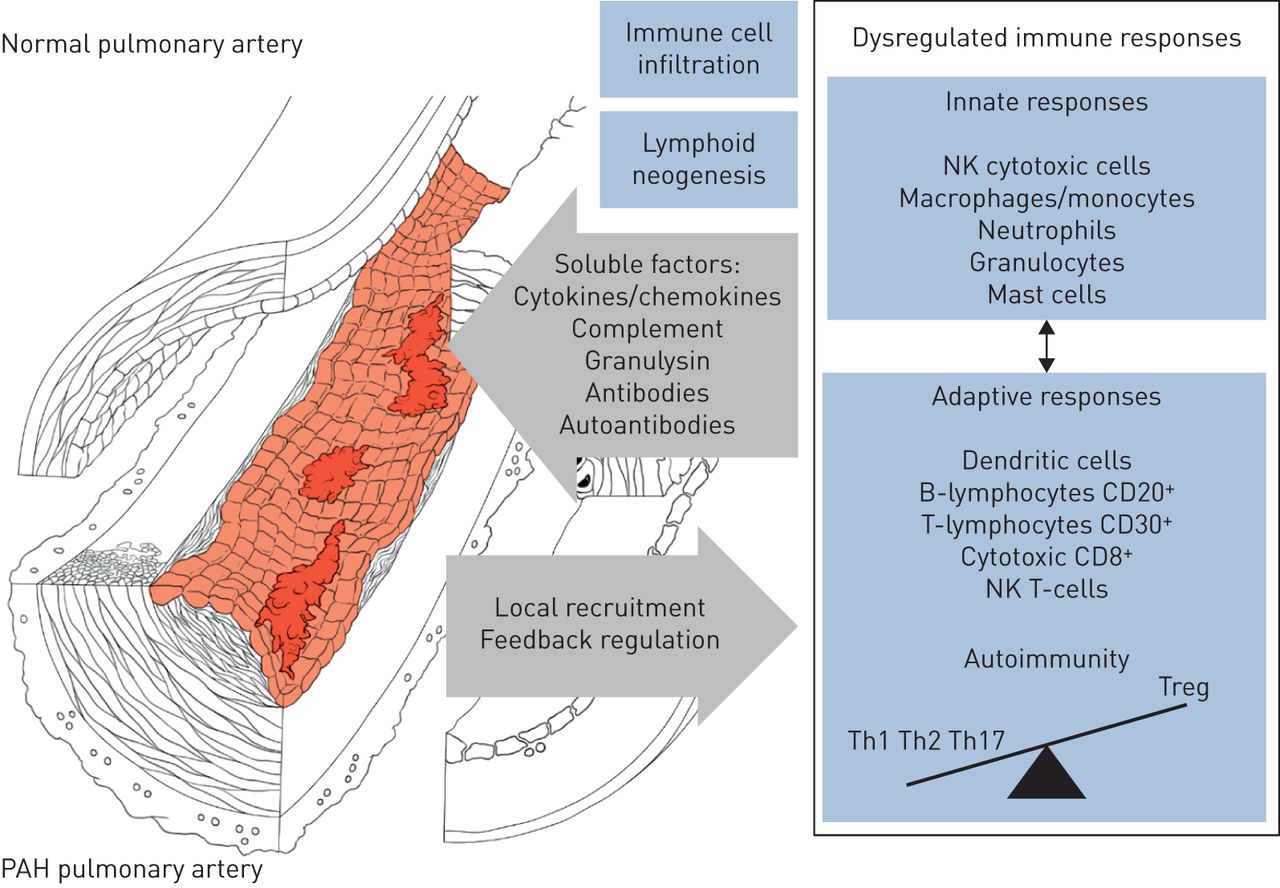
附加起作用的因素在PAH的先天和适应性免疫系统的失调包括,尤其是:剪切应力,长期暴露于缺氧,失调在BMPR2信令,肺循环,代谢紊乱,功能失调或苦恼线粒体的老化,循环自身抗体和免疫复合物。事实上,环境或遗传毒性应力青睐通过血管,炎症和免疫细胞激活肺血管重构。近来,已经发现含抗病毒蛋白SAMHD1(SAM结构域和HD结构域的蛋白1)血管周围的免疫复合物是先天免疫应答,以在人内源性逆转录病毒的序列和蛋白质产物的原因不明的海拔K的PAH观察到血管周围的巨噬细胞和循环单核细胞[60.].这些异常,除其他外,可能延续一个失调的免疫和炎性反应(图5.).不适应免疫和炎症似乎发挥在PH / PAH肺动脉内皮功能障碍和血管重建中发挥积极作用,并且也可能对心脏功能产生不利影响。不过,也有需要进一步调查,以确定是否和抗炎战略将最适合于治疗PAH微妙和复杂性。
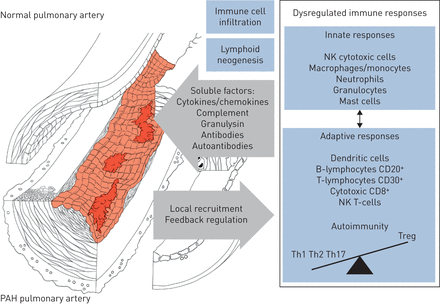
肺动脉高压(PAH)和肺血管重构相关的免疫紊乱的示意图表征NK:自然杀伤;Th:辅助;Treg: t调节。
自身免疫和炎症在PAH发病机制中的突出作用,为未来针对特异性炎症途径的新型生物制剂的临床研究提供了依据。目前,一些临床试验正在探索不同抗炎药物对PAH的疗效和安全性;其中包括:利妥昔单抗,一种嵌合抗人CD20 (ClinicalTrials.gov标识符NCT01086540系统性硬化症相关PAH的患者;tocilizumab,一种人源抗il -6受体抗体(ClinicalTrials.gov标识符NCT02676947.)在PAH患者;FK506,钙调磷酸酶的抑制剂,并且还显示出上调BMPR2表达FKBP12(12-kDa的FK506结合蛋白)的结合配偶;和弹性蛋白酶抑制剂产生作为重组蛋白。
在分子机制和新兴的治疗靶点的研究进展
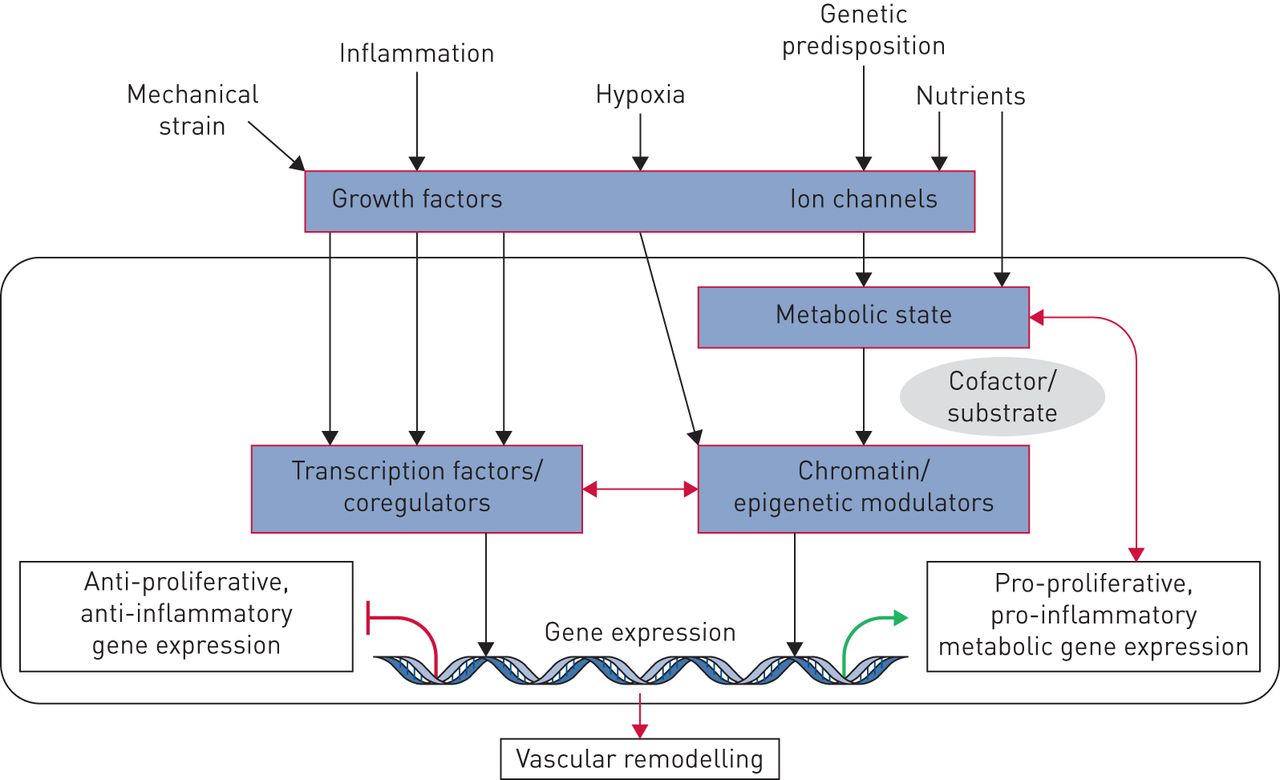
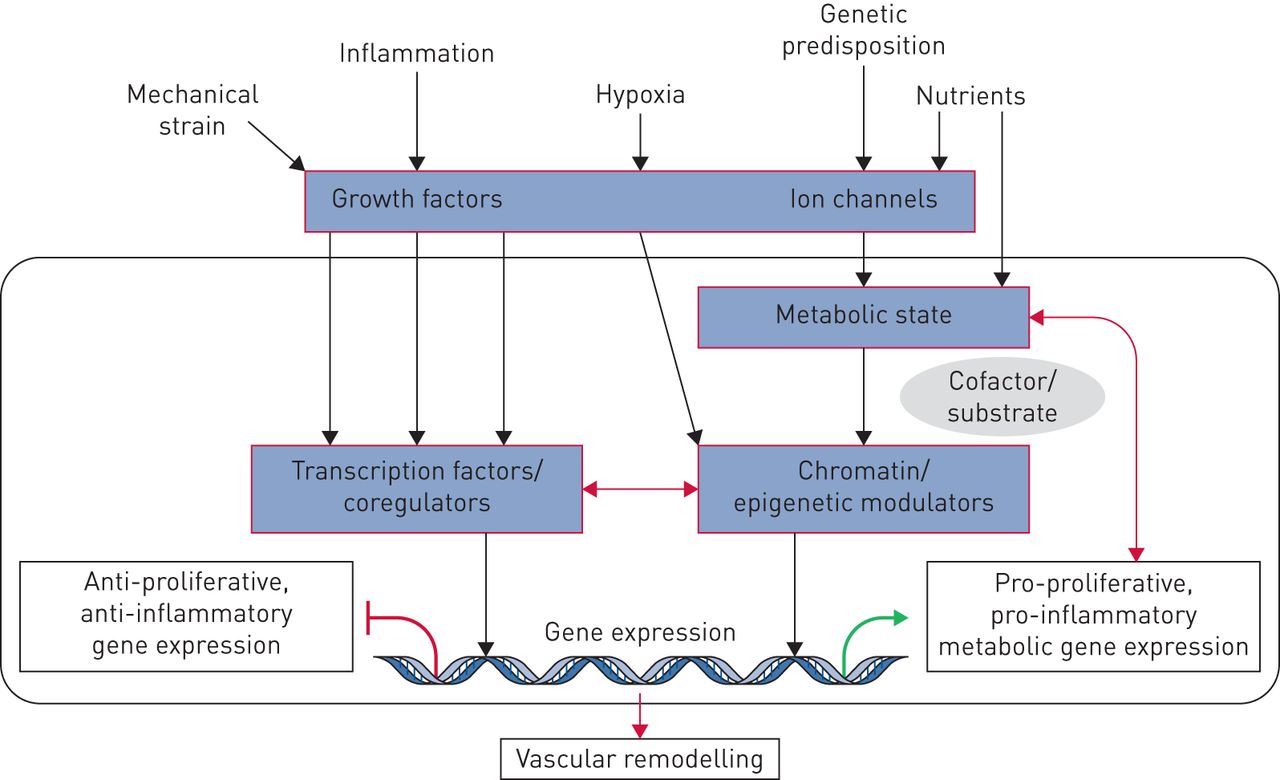
虽然仍有很多遗留情况,但数十年的广泛研究与遗传,表观遗传和环境因素(病毒,药物,毒素,缺氧和炎症)有相关的PAH病理学,这可能导致或加速肺血管床的不可逆重塑。我们建议遗传,表观遗传和环境因素导致沉重生长因子,离子通道,激素和细胞因子的放松管制,其随后激活了血管细胞表型异常的复杂级联,包括增殖,分化/解离和炎症。这表明肺脉管系统中的转录失调可以是一种形状肺血管转录组的早期事件,导致最终导致异常细胞过程的基因产物的耗尽和异位活化,并因此是不良血管重塑[61.].细胞如何感觉和响应导致转录失调的环境触发器仍然是肺血管研究的核心问题。最近的研究提供了新的见解,包括:非受体激酶的作用;离子通道的潜力控制肺动脉调和血管改造过程;基因表达调节因子的活性破坏(IE。转录因子和转录核心特征);表观遗传过程导致染色质重塑蛋白,非编码和微瘤的异常活化;和影响转录,转录后过程和信号通路的严重代谢扰动(图6.).
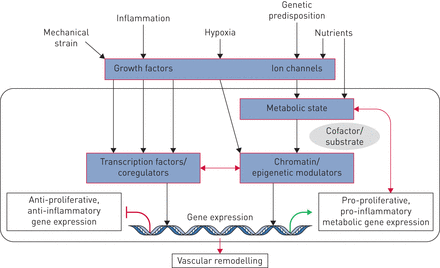
肺动脉高压转录因子,表观遗传学和代谢之间的串扰。生长因子,离子通道,激素和细胞因子激活经典信号通路和下游转录因子,这将募集到局部染色质的染色质调节酶。另一方面,营养水平和细胞代谢将影响代谢物的水平,这是染色质调节酶的所需底物,其使用这些代谢物改变组蛋白和DNA。这些输入中的变化将确定表观簇重塑和转录,随后血管重塑。
受体和非受体激酶信号通路的失调
在PH / PAH,不同的生长因子改变的表达和功能以及它们各自的受体酪氨酸激酶(例如成纤维细胞生长因子2,血管内皮生长因子,血小板衍生的生长因子,表皮生长因子和神经生长因子)与炎症介质一起(例如细胞因子,趋化因子,循环的自身抗体和免疫复合物有助于常规肺血管细胞的表型改变,并涉及远端肺动脉壁的积累。
新兴的离子通道靶点
杂合子功能缺失突变的鉴定KCNK3(钾通道亚家族成员ķ3)基因编码TWIK相关酸敏感的钾通道1(TASK1)作为PAH的原因已经苏醒中离子通道的概念的兴趣[62.].除了电压门控钾通道外,不同类型的瞬态受体电位通道,钙传感器蛋白和钙活化氯化物通道均涉及PAH发病机制[63.-67.].钾通道的调节异常可能在肺血管功能的PAH [立即和长期调节中发挥中心作用68.那69.].这一概念与钾通道功能的恢复可以防止或逆转实验ph值的事实是一致的。体内KCNK3的药理激活在野百合碱诱导的PH中具有有益作用[69.].有趣的是,内皮素、5-羟色胺(5-HT)、氧化应激、BMPR2、二十二碳六烯酸和生长因子(如血小板来源的生长因子)是已知的钾通道活动的调节因子[70].此外,抑制电压门控钾通道可能是一些药物诱导PH的潜在机制[19那71.].目前的挑战是确定小分子或特定策略来恢复这些离子通道在PAH功能失调的肺血管系统中的表达和/或活性。
关键转录因子和转录核心特征
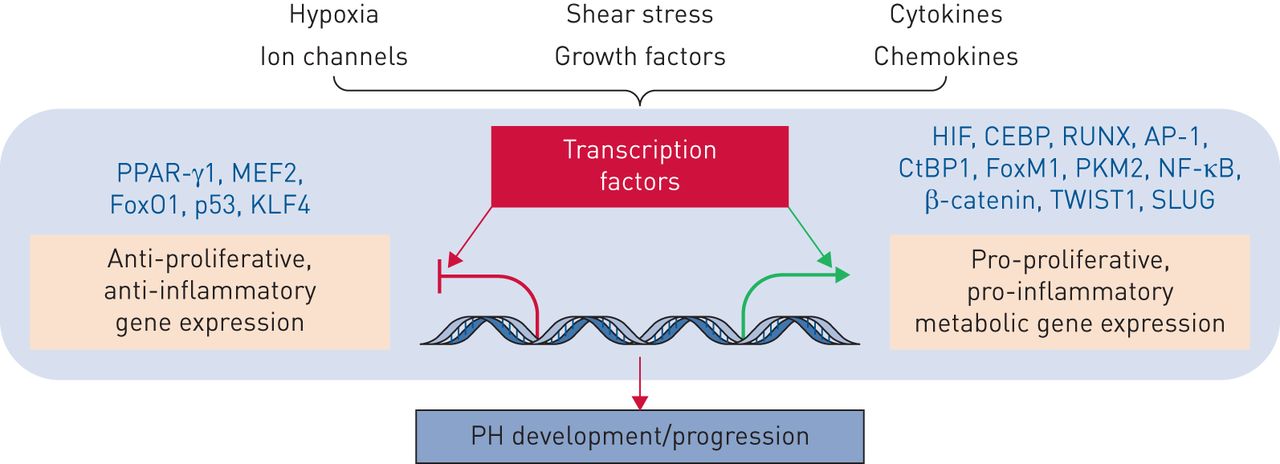
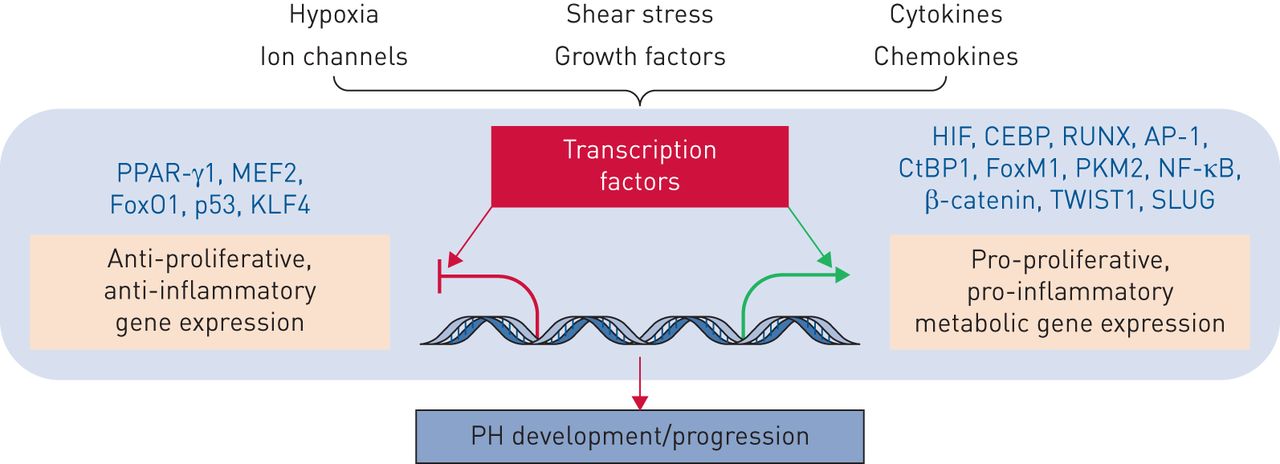
许多转录因子和转录同型(定义为本身没有DNA结合活性的蛋白质,而是可以与增强或抑制转录因子激活基因表达能力的转录因子结合)与pH和右心室相关功能障碍。一些转录因子包括PPAR-γ,肌细胞增强子因子2(MEF2),FOXO,P53,KLF4,HIF,CCAAT-Enhancer结合蛋白(CEBP),RUNT相关转录因子2(RUNX2),活化剂蛋白1(AP-1),CTBP1,FOXM1,PKM2,NF-κB,β-连环蛋白,扭曲家族基本螺旋环 - 螺旋转录因子1(Twist1)和Slug(图7.) [45那51那72.-77.].FoxO1同种型失活涉及PA-SMC的促型和凋亡抗性表型,是不同生长因子和炎症调解员的已知下游介体[45].此外,相关的转录因子FOXM1促进PH PA-SMC积累[77.],建议靶向FOXO-FOXM1轴可能是治疗pH的可行策略。转录因子共膜剂也已达到pH病理生理学。这些包括PKM2和CTBP1 [39那72.].重要的是,正火代谢活动通过代谢抑制剂如2-脱氧葡萄糖或直接降低CTBP1表达衰减pH成纤维细胞增殖和凋亡抗性[72.].
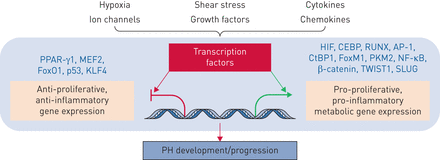
转录因子和转录辅助调节因子在肺动脉高压发病中的作用。有关定义,请参阅正文。多种病理刺激,如缺氧、剪切应激、氧化应激、有丝分裂原和炎症(细胞因子和趋化因子),触发下游信号级联反应,这些级联反应调节转录因子和转录辅助调节因子的招募和激活,这些转录调节因子决定了刺激特异性的PH转录反应。
其他转录因子异常,如Notch3、信号转换器和转录激活因子3 (STAT3)和HIPPO中枢成分大肿瘤抑制因子1 (LAST1) [78.],并且在各种生长因子,肺血管细胞在PAH背后的表型变化。例如,在肺的内皮细胞引线PPAR-γ的损失到有缺陷的复合物与β-catenin和这导致降低Apelin水平,导致受损的肺内皮细胞存活和血管生成[79.].同样,带有pdz结合基元(TAZ)的yes相关蛋白(YAP)/转录共激活因子(TAZ)也作为PAH中细胞生长和迁移的关键调节因子出现,并将机械刺激与血管代谢失调联系起来[80].对慢性缺氧的反应不同的两只大鼠菌株,F344和WKY的比较突出了该基因Slc39a12将锌转运蛋白ZIP12编码为缺氧诱导的肺血管改造的另一个主要调节剂[81.].虽然需要对这些不同策略的总体风险-收益比进行更全面的了解,但这些数据揭示了针对PH/PAH中特定转录因子和/或转录因子共激活物的潜在治疗兴趣。
表观遗传失调的新兴作用
超氧化物的改变的DNA甲基化氧化物歧化酶2倍颗粒溶素的基因,组蛋白H1的水平,组蛋白脱乙酰酶(HDACs)和溴区包含蛋白4的异常表达水平,和失调的微小RNA和长非编码RNA网络一起表明在后生参与的多个级别PAH的发病机制。近日,在HDAC抑制PAH临床意义已经通过发现胞质HDAC6在两个肺动脉重塑和右心室衰竭牵连再生。使用HDAC6特异性抑制剂抑制其可以测试[82.].因此,机构的一个完整的理解参与改变的基因表达在患病细胞[61.对设计新的治疗策略至关重要。缺乏sirtuin 3(一种线粒体去乙酰化酶)的小鼠会自发形成ph。有趣的是,这些小鼠会增加许多线粒体酶和复合物的乙酰化和抑制,从而抑制线粒体功能。此外,功能丧失SIRT3多态与多环芳烃有关[83.].这些研究表明,线粒体的产品,如2-羟基戊二酸,α酮戊二酸,柠檬酸或乙酰辅酶A可调节转录因子和表观遗传机制。此代谢表观遗传学轴功能有助于适应在肺血管和右心室不断变化的环境,提供了潜在的新的治疗靶标。总之,在PH分子,代谢,遗传和表观遗传重新布线之间的相互作用的一个综合理解/ PAH远未完成,但概念上的主题已开始出现。
代谢重塑和线粒体功能障碍
肺血管细胞的异常信号可能是代谢失调的原因或后果,有助于PH/PAH的发展和进展。一些代谢和信号通路可能是PH/PAH治疗的有趣靶点,其中正在研究的包括HIF1和磷酸肌苷3激酶/蛋白激酶B/雷帕霉素途径的哺乳动物靶点,线粒体磷酸酶和紧张素同源物(PTEN)诱导的激酶1 (PINK1),HIPPO和p53信号通路,以及丙酮酸脱氢酶激酶(PDK)的抑制,后者是线粒体丙酮酸脱氢酶(PDH,葡萄糖氧化的守门酶)的抑制剂。在PAH患者中使用二氯乙酸恢复氧化代谢可能对一部分患者有效[84.].缺乏临床反应用的功能性变体的存在相关SIRT3和UCP2(未偶联蛋白2),其预测降低的PDH函数独立于PDK和对二氯乙酸的更大抗性[84.].最近,在PAH患者培养的PA-SMCs中,线粒体热休克蛋白90的积累被发现有助于提高有氧糖酵解和线粒体应激反应[85.].即使需要进一步的研究来更好地了解不同的炎症过程、高剪切应力、慢性缺氧和某些激素在这些代谢失调中的作用,并确定额外的和新的机制靶点,基于代谢的治疗对PH/PAH可能是有希望的。
当前和未来展望
模拟导致PH升高的特定过程的动物模型系统可能对发现新的治疗靶点和研究它们对疾病发病机制的早期和晚期的贡献有价值。然而,同样清楚的是,实验诱导的肺血管病变并不能概括人类疾病的全貌,而且在这些不同的模型中,对促进PH的刺激的反应存在物种、年龄、性别和环境差异。此外,与人类相比,动物的免疫系统在解剖学和功能发育上也存在差异。因此,使用人类组织样本或诱导多能干细胞从肺动脉高压患者获得内皮细胞或SMCs应补充动物研究,为肺动脉高压治疗进入临床提供一条途径。虽然模型PH有明显的限制,但有几个措施应该提高预测效用和转化效率:1)尽可能精确地定义研究问题;2)确定当前可用的每种PH模型的优缺点;3)确定如何最好地使用它们。可以在随机化动物、考虑两性以及与血流动力学和结构终点相关的盲法观察中取得改进[86.-88.].
通过询问大型数据集以寻找新的途径,关键转录因子,MicroRNA,生物标志物和代谢介质,可以帮助开发更好的有针对性疗法的交叉点的重要进展。具有成立数据集的生物信息方法可以生成PAH签名和可用于寻找新颖或重新展示的疗法的反签名,以及提供它们。除了考虑剪切应力水平之外,还需要掺入细胞基质的性质和细胞相互作用的性质。因此,使用所有三层血管壁的工程系统以及模拟这些细胞产生的细胞外基质将是最具信息性的。三维打印等新机会现在可以产生广泛的血管网络。
将激动人心的临床前发现转化为临床检测对像PAH这样的罕见疾病提出了特殊的挑战。首先,由于研究对象的数量有限,从实验室收集的新方法必须作为标准护理疗法的佐剂。研究人员面临的另一个挑战是找到共同的途径或病理生理过程,以安全靶向所有形式的PH/PAH。在这方面的研究中,对公开可用的“多环芳烃组学”数据数据库(最常用的转录组)和多环芳烃注册库的数据进行分析,可能是发现揭示新疗法的常见基因模块的有希望的方法。另一种吸引人的方法可能是从普通途径转向表型患者,将其分为“有反应者”和“无反应者”两组。在这里,挑战是在一个已经很小的群体中发现特别适合辅助治疗的患者群体。
脚注
N. Galiè, V.V. McLaughlin, L.J. Rubin和G. Simonneau编辑的“第六届肺动脉高压世界研讨会论文集”系列第一名
利益冲突:M. Humbert报告来自Actelion和默克的个人费用,以及来自拜耳和葛兰素史克的拨款和个人费用,在提交的工作之外。
利益冲突:C. Guignabert无需披露。
利益冲突:S.邦尼特有没有透露。
利益冲突:P. Dorfmüller报告了来自MSD、拜耳、Actelion和罗氏在提交作品之外的个人费用。
兴趣冲突:J.R. Klinger报告了来自联合治疗的额外细胞囊泡的动物研究,外面提交的工作。
利益冲突:M.R. Nicolls有没有透露。
利益冲突:A.J.Olschewski无需披露。
利益冲突:普拉姆塞蒂没有什么可透露的。
利益冲突:R.T.沉沉有什么可披露的。
利益冲突:K.R.Stenmark报告奖金从辉瑞公司,纽约,指导委员会成员(Enterligence Awards奖)的个人费用的咨询委员会工作的个人费用,以及在外面的詹森研发的科学咨询委员会工作的个人费用提交的工作。
利益冲突:M. Rabinovitch报道了使用FK506治疗PAH。利兰·斯坦福初级大学董事会,受让人。PCT专利/ US2012035793。
- 收到了2018年10月4日。
- 公认2018年10月8日。
- 版权©2019人队
本文是开放式访问,并根据创意公约188滚球软件归因非商业许可证4.0的条款分发。











{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Impact of hypertrophic systemic vasculature in pulmonary arterial hypertension (PAH): an explanatory approach. The pulmonary artery (top centre, blue) is covered by a systemic vascular plexus, comprising systemic arterial (red) and venous (blue) vessels and microvessels. The systemic plexus anastomoses with the pulmonary artery, the capillary bed and the pulmonary vein (bottom left, red): these bronchopulmonary anastomoses appear to bypass an occlusive PAH lesion, represented by medial thickening and intimal fibrosis (centre). Eventually, the increased systemic blood flow into arterioles, capillaries and the pulmonary vein leads to structural changes of the latter: muscular hyperplasia and focal intimal fibrosis within the pulmonary vein are observed. Reproduced and modified from [4] with permission.](http://www.qdcxjkg.com/content/erj/53/1/1801887/F3.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}