





摘要
慢性阻塞性肺病是导致死亡和残疾的主要原因,但直到最近才从细胞和分子的角度进行了广泛的探索。
慢性炎症导致小气道固定狭窄和肺泡壁破坏(肺气肿)。其特征是肺泡巨噬细胞、中性粒细胞和细胞毒性T淋巴细胞数量增加,以及多种炎症介质(脂质、趋化因子、细胞因子、生长因子)的释放。高水平的氧化应激可能会放大这种炎症。弹性溶解也增加,并有证据表明几种弹性溶解酶参与,包括丝氨酸蛋白酶、组织蛋白酶和基质金属蛋白酶。
慢性阻塞性肺疾病的炎症和蛋白水解是对香烟烟雾的正常炎症反应的放大。与哮喘形成鲜明对比的是,这种炎症似乎对皮质类固醇有耐药性,这促使人们寻找新的抗炎疗法,以防止疾病的无情发展。
2002年11月在马耳他举行的“COPD:重要问题”会议由阿斯利康赞助。
这篇综述是2002年11月在马耳他举行的“慢性阻塞性肺病:重要问题”会议的摘要。会议的目的是确定与慢性阻塞性肺病(COPD)所涉及的细胞和分子机制相关的重要问题,并讨论新的研究方法,以更好地了解COPD所涉及的基本机制。
慢性阻塞性肺病是一个日益严重的重大全球健康问题,预计到2020年,它将成为世界上第三大常见死亡原因和第五大常见残疾原因1.虽然在对哮喘的理解和管理方面取得了重大进展,但COPD一直相对被忽视,目前没有任何治疗方法可以减少这种疾病不可避免的进展。然而,由于疾病的巨大负担和不断上升的医疗成本,现在人们对潜在的细胞和分子机制重新产生了兴趣2以及寻找新的治疗方法3.,从而对疾病进行了重新评估4.尽管慢性阻塞性肺病具有巨大的全球重要性,但对它的研究相对较少,就全球疾病负担而言,它是资金最不足的疾病5.
慢性阻塞性肺病的特点是气流限制缓慢进展,可逆性差,与哮喘形成鲜明对比,哮喘有可变气流阻塞,通常是自发或经治疗可逆的。全球阻塞性肺疾病倡议(GOLD)最近采用了COPD的新定义:“以不完全可逆的气流受限为特征的疾病状态。气流限制通常是进行性的,并与肺部对有害颗粒和气体的异常炎症反应有关。”6.这一定义首次包含了COPD是一种慢性炎症性疾病的概念,最近的许多研究都集中在这种炎症反应的性质上。
慢性阻塞性肺病包括伴有纤维化和小气道阻塞的慢性阻塞性细支气管炎,以及伴有气隙增大和肺实质破坏、肺弹性丧失和小气道关闭的肺气肿。相比之下,慢性支气管炎的定义是连续两年以上持续3个月的咳痰;这反映了粘液分泌过多,不一定与气流限制有关。大多数COPD患者同时具有三种病理机制(慢性阻塞性细支气管炎、肺气肿和粘液堵塞),均由吸烟引起,但肺气肿和阻塞性细支气管炎的比例可能有所不同2.在发达国家,吸烟是迄今为止导致COPD的最常见原因,占95%的病例,但还有其他一些危险因素,包括空气污染(特别是燃烧燃料造成的室内空气污染)、不良饮食和职业暴露。COPD的特征是随着年龄的增长,肺功能的正常衰退加速。缓慢进展的气流限制导致残疾和过早死亡,与哮喘的多变气道阻塞和症状有很大不同,哮喘的严重程度很少进展。虽然COPD和哮喘都涉及呼吸道炎症,但炎症过程的性质存在显著差异,包括炎症细胞、介质、对炎症的反应、解剖分布和对抗炎治疗的反应4,7.然而,一些患者似乎具有慢性阻塞性肺病和哮喘的共同特征。而不是代表一个分级的疾病谱系,更有可能的是,这些患者同时患有这两种常见疾病。
与哮喘的区别
慢性阻塞性肺病的组织病理学研究显示主要累及外周气道(细支气管)和肺实质,而哮喘累及所有气道的炎症,但通常不累及肺实质8.细支气管梗阻,纤维化,巨噬细胞和T淋巴细胞浸润。肺实质被破坏,巨噬细胞和T淋巴细胞数量增加,CD8水平明显升高+(细胞毒性)高于CD4+(辅助)细胞9.支气管活检显示类似的变化,巨噬细胞和CD8浸润+细胞和增加的中性粒细胞在严重COPD患者10.支气管肺泡灌洗液和诱导痰中巨噬细胞和中性粒细胞明显增多11,12.与哮喘相反,嗜酸性粒细胞不明显,除非病情加重或伴有哮喘8,13.
气流限制的主要机制是什么?
固定的小气道狭窄、肺气肿和粘液分泌物引起的腔内阻塞都可能导致COPD的气流限制,但关于哪种机制最重要还存在争议。在患者之间和疾病进展的不同阶段,这些过程的作用存在差异,但在患者中进行精确测量的问题使得很难评估每个机制在单个患者中的重要性。
小航空公司
长期以来,人们已经认识到COPD患者的小气道狭窄14- - - - - -17.小气道厚度增加,淋巴滤泡形成增多,外气道壁胶原沉积,可能限制气道开放18.小气道的管腔因含有炎症渗出物的粘膜增厚而减少,这种增厚随着疾病的严重程度而增加。更严重疾病中淋巴滤泡形成的机制尚不清楚,但可能反映了慢性细菌定植和炎症急性加重的反应。气道周围纤维化的机制尚不清楚,但可能代表着修复慢性炎症的一种尝试。特定生长因子的作用,如转化生长因子- β (TGF - β)在外周气道表达增加19,20.和结缔组织生长因子(CTGF)尚不清楚。TGF‐β可能诱导纤维化通过CTGF的释放可能会刺激呼吸道中的胶原沉积21,22.理解小气道阻塞的主要障碍是,由于测量的高可变性和低可重复性,难以使用气流测量来量化患者的小气道阻塞23.
肺气肿
吸烟者可发生全腺性肺气肿和小叶中心性肺气肿24.肺气肿在慢性阻塞性肺病中引起气流阻塞的作用已通过测量切除肺中的宏观肺气肿或与肺功能测试相关的计算机断层扫描(CT)扫描,或通过测量静态跨肺压(Pl)作为肺泡疾病的测量方法。许多研究表明,宏观肺气肿分级与各种肺功能测试之间存在显著(尽管微弱)相关性25,26.然而,宏观肺气肿的评估主要是受损或功能不良的肺,而肺功能测试主要反映存活最好的肺的功能。在非通气性肺气肿大疱被正常肺包围的极端情况下,这两种评估实际上是独立的,肺功能测试仅测量存活肺的缩小体积。在更常见的肺气肿类型中,简单的两室模型不适用,但通常会有更大的疾病异质性,随着疾病的发展,越来越多的单位变得功能不良,导致剩余容积上升和肺活量下降。因此,评估大体肺气肿与肺功能之间的相关性强度将取决于较少受影响的肺中“微观”疾病的严重程度和同质性,而这通常不被测量。
Pl(使用食道导管测量)与不同肺容量下的气流导度或最大呼气流量作对比,以表明肺泡疾病(包括肺气肿)对气流限制的贡献,并假设其余是由于固有气道疾病27- - - - - -29.但是,目前还不能确定下降的幅度Pl准确反映肺气肿的严重程度及其对气道的影响。减少Pl相对均匀的肺气肿可能最大,如全泡性肺气肿(如。α1‐抗胰蛋白酶缺乏),而斑块状小叶中心肺气肿可能接近正常Pl.的确,均匀的“显微”肺气肿可能解释了没有任何CT改变的功能性“假性肺气肿”。
在实践中,电导或最大流量的减少完全可以用流量的减少来解释Pl不常见,除非是轻微疾病。对COPD严重气流阻塞患者切除肺的逆行导管研究均发现,在标准水平下外周阻力大幅增加Pl30.- - - - - -32.但由于肺气肿,气道功能有许多其他可能的变化,这将导致在给定的阻力增加Pl,包括肺周围过度膨胀导致正常气道异常成角或受压,肺气肿破坏导致平行气道丧失,或为肺通风不良区域供气的通畅气道功能丧失。肺气肿的影响不一定会减轻Pl,如。由局部牙槽附着体缺失引起的狭窄。目前对气道形态的分析还不足以揭示外周气流阻力增加这一一致的生理学发现的解剖学基础。假设增加都是由于外周气道的“固有”疾病低估了肺气肿的作用。肺气肿可能在严重疾病中发挥更突出的作用,因为肺功能加速衰退。
粘液分泌过多
黏液分泌过多对COPD气流限制的作用仍不确定。尽管早期的研究支持黏液分泌过多与任何生理缺陷无关的观点33,34但最近的研究表明,粘液分泌过多可能是肺功能加速衰退的潜在危险因素35,36.早期研究检查了COPD的早期阶段,也包括一个职业队列。慢性粘液分泌过多导致COPD进展的最可能机制可能是由于加重的风险增加,这似乎加速了一秒钟内用力呼气量(FEV)的损失1)37.慢性粘液分泌过多可能对COPD早期发作的作用不大,因为急性加重很少发生。慢性粘液分泌过多可能反映粘膜下腺周围的炎症过程38并可能反映更多外周气道的炎症强度。在粘膜下腺体周围也发现中性粒细胞和肥大细胞增多38,39丝氨酸蛋白酶和肥大细胞糜酶是有效的粘液促分泌剂40- - - - - -42.在严重慢性阻塞性肺病中,慢性粘液分泌过多与死亡率相关,这也可能反映了晚期感染风险的增加43- - - - - -45.肺功能正常的吸烟者(GOLD Stage 0)的慢性咳嗽和粘液产生似乎不能预测COPD的后期发展46.
什么是关键的炎症细胞?
COPD与哮喘一样,是一种复杂的炎症性疾病,涉及多种类型的炎症细胞和多种炎症介质。然而,这两种呼吸道疾病的炎症模式和介质谱不同,至少在疾病的稳定状态下是不同的。虽然在COPD中有异常数量的炎症细胞的记录,但这些细胞类型与它们的出现顺序和持久性之间的关系在很大程度上是未知的。大多数研究都是基于选择不同阶段疾病患者的横断面研究,并在没有气流受限的吸烟者(正常吸烟者)和吸烟量相似的COPD患者之间进行比较。没有系列研究,选择偏差(例如从适合肺减容手术的患者中选择组织)可能会给出误导性的结果。肺泡和小气道的细胞谱分析显示,COPD中涉及的所有细胞类型均有所增加,包括巨噬细胞、T淋巴细胞、B淋巴细胞和中性粒细胞47.
中性粒细胞
慢性阻塞性肺病患者的痰液和BAL液中活化中性粒细胞数量增加12,48然而,气道或肺实质只有相对较小的增加49.这可能反映了它们通过气道和实质的快速转运。中性粒细胞分泌丝氨酸蛋白酶,包括中性粒细胞弹性蛋白酶(NE)、组织蛋白酶G和蛋白酶‐3,以及基质金属蛋白酶(MMP)‐8和MMP‐9,这可能有助于肺泡破坏。这些丝氨酸蛋白酶也是有效的粘液兴奋剂。慢性阻塞性肺疾病患者气道中,中性粒细胞向气道和实质的募集涉及到内皮细胞的粘附,E‐选择素在内皮细胞上上调50.然后,粘附的中性粒细胞在中性粒细胞趋化因子(包括白细胞介素(IL)‐8和白三烯B)的引导下迁移到呼吸道4(LTB4).粒细胞-巨噬细胞集落刺激因子(GM - CSF)和粒细胞集落刺激因子(G - CSF)等细胞因子可增加呼吸道中性粒细胞的存活。
中性粒细胞在COPD中的作用尚不清楚。循环中性粒细胞数量与FEV下降有相关性151.支气管活检和诱导痰中中性粒细胞数量与COPD病情严重程度相关10,12随着肺功能下降的速度52.吸烟对骨髓中粒细胞的产生和释放有直接的刺激作用,可能是由肺巨噬细胞释放的GM‐CSF和G‐CSF介导的53.吸烟也可能增加肺中的中性粒细胞潴留54.毫无疑问,随着痰液上清中颗粒蛋白(如髓过氧化物酶和人中性粒细胞脂脂素)浓度的增加,被招募到COPD患者气道的中性粒细胞被激活55- - - - - -57.这些中性粒细胞还表现出与气流限制程度相关的呼吸爆发反应的增加58.
中性粒细胞具有通过释放丝氨酸蛋白酶和氧化剂诱导组织损伤的能力。启动是中性粒细胞脱颗粒和产生超氧阴离子的先决条件59.外周循环中的中性粒细胞显示COPD启动的证据60,但这可能是肺病理生理学的结果,而不是贡献。COPD中有几种趋化信号可能引起中性粒细胞招募,包括LTB4, IL‐8和相关的CXC趋化因子,包括GRO‐α(生长相关癌蛋白)和ENA‐78 (78 kDa的上皮中性粒细胞激活蛋白),它们在COPD气道中升高61,62.这些介质可能来源于肺泡巨噬细胞和上皮细胞,但中性粒细胞本身可能是IL‐8的主要来源63.
中性粒细胞来自肺循环边缘,在进入肺泡间隙前粘附在肺泡壁的内皮细胞上64.中性粒细胞在大气道中迁移的精确路线尚不确定,但更有可能的是它们从气管支气管循环到达气道,并通过毛细血管后小静脉迁移65.在体循环和肺循环中,中性粒细胞粘附和转运的细胞机制不同,这可能赋予来自肺泡或支气管腔室的中性粒细胞不同的性质。肺不同区域的中性粒细胞转运时间可能存在显著差异,这可能导致肺气肿的差异分布,例如小叶中心肺气肿的上叶优势。对于慢性阻塞性肺病气道中中性粒细胞的存活和凋亡知之甚少。理论上,GM‐CSF可以延长中性粒细胞的存活时间,但从痰样本中培养中性粒细胞已被证明是困难的。
然而,虽然中性粒细胞具有引起弹性溶解的能力,但这不是其他肺部疾病的突出特征,其中慢性气道中性粒细胞更为突出,包括囊性纤维化和支气管扩张。这表明其他因素也参与了肺气肿的产生。事实上,慢性阻塞性肺病中中性粒细胞的数量与肺泡破坏的数量之间存在负相关49中性粒细胞并不是COPD实质炎症的显著特征。然而,慢性支气管炎患者气道中性粒细胞增多可能与粘液高分泌有关。来自中性粒细胞的丝氨酸蛋白酶,包括中性粒细胞弹性蛋白酶、组织蛋白酶G和蛋白酶‐3,都是粘膜下腺和上皮中杯状细胞分泌粘液的有效刺激物40,42.
巨噬细胞
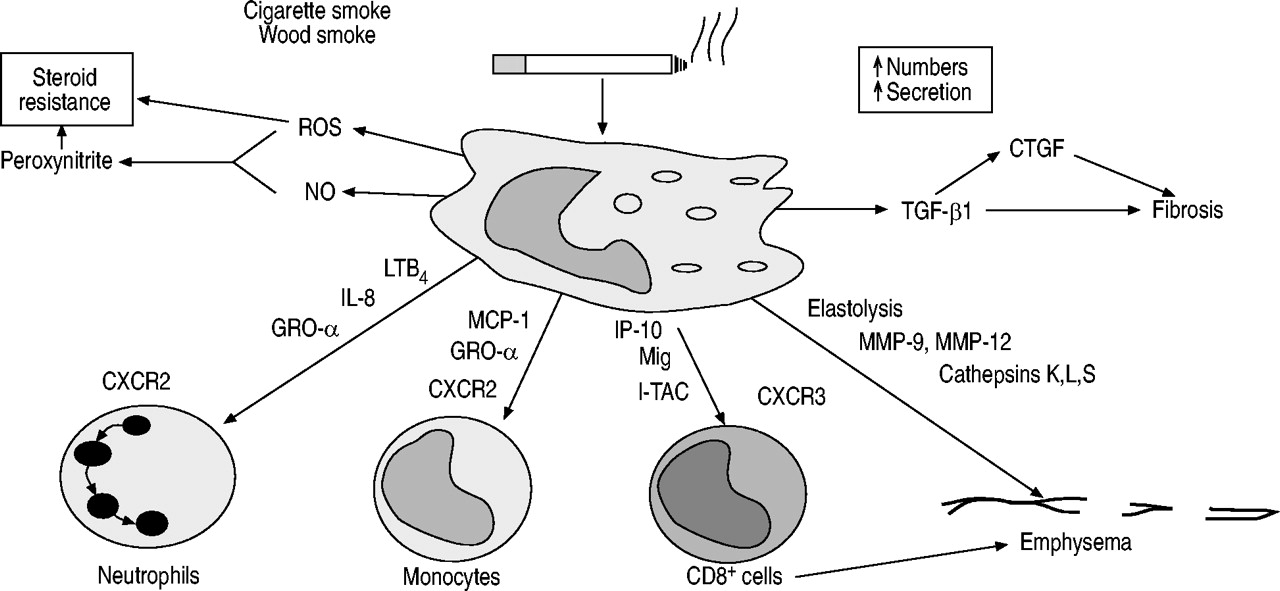
巨噬细胞似乎在COPD的病理生理学中起着关键作用,并可以解释该疾病的大多数已知特征(图1)⇓)66,67.
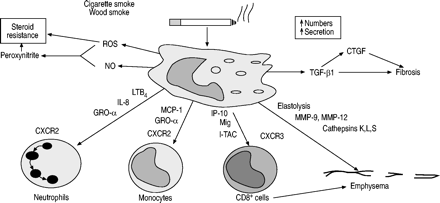
巨噬细胞可能在慢性阻塞性肺疾病(COPD)中发挥关键作用,因为它们被香烟烟雾提取物激活并分泌许多炎症蛋白,这些炎症蛋白可能协调COPD的炎症过程。中性粒细胞可能被白细胞介素(IL)‐8、生长相关癌基因‐α (GRO‐α)和白三烯B所吸引4(LTB4),单核细胞通过巨噬细胞趋化蛋白‐1 (MCP‐1)和CD8+干扰素- γ诱导蛋白(IP - 10)、干扰素- γ诱导的单因子(Mig)和干扰素诱导的T细胞α -趋化剂(I - TAC)。弹性溶解酶包括基质金属蛋白酶(MMP)和组织蛋白酶(cathepins)的释放引起弹性溶解,转化生长因子(TGF‐β)和结缔组织生长因子(CTGF)的释放。巨噬细胞还产生活性氧(ROS)和一氧化氮(NO),它们共同形成过氧亚硝酸盐,可能有助于类固醇抵抗。
COPD患者气道、肺实质、BAL液和痰中巨噬细胞数量明显增加(5-10倍)。对肺气肿患者实质中巨噬细胞数量的仔细形态测量分析显示,与正常吸烟者相比,肺气肿患者组织和肺泡间隙中的巨噬细胞数量增加了25倍47.此外,在肺气肿患者中,巨噬细胞定位于肺泡壁破坏的部位49,68.气道中巨噬细胞数量与COPD严重程度之间存在相关性10.
巨噬细胞可能被香烟烟雾提取物激活,释放炎症介质,包括肿瘤坏死因子(TNF)‐α, IL‐8,其他CXC趋化因子,单核细胞趋化肽(MCP)‐1,LTB4以及活性氧,提供了一种将吸烟与慢性阻塞性肺病炎症联系起来的细胞机制。肺泡巨噬细胞也分泌弹性溶解酶,包括MMP‐2、MMP‐9、MMP‐12、组织蛋白酶K、L和S以及从中性粒细胞中摄取的中性粒细胞弹性酶69,70.COPD患者的肺泡巨噬细胞在基线时比正常吸烟者的肺泡巨噬细胞分泌更多的炎症蛋白,具有更大的弹性溶解活性,暴露于香烟烟雾后进一步增加70- - - - - -72.巨噬细胞即使在培养3天后也表现出这种差异,因此似乎与正常吸烟者和不吸烟的正常对照组的巨噬细胞有本质上的不同70.COPD患者肺泡巨噬细胞分泌的主要弹性溶解酶是MMP‐9。大多数COPD巨噬细胞中上调的炎症蛋白是由转录因子核因子κB (NF‐κB)调控的,它在COPD患者的肺泡巨噬细胞中被激活,特别是在加重期间73,74.
吸烟者和COPD患者中巨噬细胞数量的增加可能是由于单核细胞选择性趋化因子对循环中单核细胞招募的增加。COPD患者痰液和BAL中单核细胞选择性趋化因子MCP‐1升高61,75,在巨噬细胞中表达增加76.CXC趋化因子也是作用于单核细胞的趋化因子通过COPD患者痰液和BAL中CXCR2和CXC趋化因子GRO‐α浓度明显升高61.COPD患者的单核细胞对GRO‐α的趋化反应比正常吸烟者和非吸烟者的细胞更强,但这不能通过CXCR2的增加来解释77.有趣的是,虽然所有单核细胞都表达巨噬细胞炎症蛋白受体CCR2,但只有~ 30%的单核细胞表达CXCR2。这些表达CXCR2的单核细胞可能转化为表现不同的巨噬细胞,如。释放更多炎症蛋白。巨噬细胞还能够释放趋化因子干扰素- γ诱导蛋白(IP - 10)、干扰素诱导T细胞α -趋化因子(I - TAC)和干扰素- γ诱导的单因子(Mig),这些因子对CD8具有趋化作用+Tc1细胞通过与这些细胞上表达的CXCR3受体相互作用78.
COPD中巨噬细胞数量的增加可能是由于单核细胞招募增加,但也可能是由于肺中增殖增加和生存期延长。巨噬细胞在肺中的增殖率非常低,但本文作者已经证明,通过增殖细胞核抗原(PCNA)测量,细胞增殖有一定的增加。79.巨噬细胞的存活时间很长,因此很难直接测量。然而,在吸烟者的巨噬细胞中,抗凋亡蛋白Bcl‐X的表达显著增加lp21表达增加CIP / WAF1在细胞质中79.这表明,巨噬细胞可能在吸烟者和COPD患者中具有延长的生存期。
在COPD患者中,皮质类固醇对抑制炎症无效,包括细胞因子、趋化因子和蛋白酶80,81.在体外来自正常受试者和正常吸烟者的IL - 8、TNF - α和mmp - 9巨噬细胞的释放被糖皮质激素抑制,而糖皮质激素在COPD患者的巨噬细胞中无效82.奇怪的是,这并不适用于GM‐CSF,它在COPD中似乎没有分泌增加,并且被皮质类固醇抑制,尽管其程度低于正常吸烟者的巨噬细胞。慢性阻塞性肺病患者对糖皮质激素以及吸烟者巨噬细胞抵抗的原因可能是组蛋白去乙酰化酶(HDAC)活性的显著降低。83糖皮质激素受体将其招募到激活的炎症基因上,以关闭炎症基因84.巨噬细胞中HDAC活性的降低与TNF - α和IL - 8等细胞因子分泌增加以及对皮质类固醇的反应降低有关。COPD患者HDAC活性的降低可能是通过氧化应激和过氧亚硝酸盐的形成介导的。
虽然皮质类固醇不能有效抑制巨噬细胞分泌细胞因子和蛋白酶,但其他药物可能是有益的。低浓度茶碱增加肺泡巨噬细胞HDAC活性在体外逆转氧化应激引起的类固醇抵抗85.白藜芦醇是红酒中的一种类黄酮,是COPD患者巨噬细胞细胞因子表达的有效抑制剂,但其分子作用机制尚未确定86.
巨噬细胞对细菌具有吞噬作用,并在宿主防御中发挥重要作用。COPD患者巨噬细胞的吞噬潜能尚未被探索,但吞噬功能受损可能导致COPD患者呼吸道细菌负荷增加。巨噬细胞识别凋亡细胞通过与巨噬细胞表面特定受体相互作用的磷脂酰丝氨酸(PS)的表达87.巨噬细胞摄取凋亡粒细胞可诱导TGF‐β1的分泌88.中性粒细胞弹性蛋白酶切割PS受体,因此可能损害巨噬细胞吸收凋亡中性粒细胞的能力,导致气道中凋亡中性粒细胞数量增加89.
地理T淋巴细胞
COPD患者肺实质、外周和中央气道T淋巴细胞总数增加,CD8增加较多+比CD4+细胞47,49,90- - - - - -92.T细胞的数量与肺泡破坏的程度和气流阻塞的严重程度相关。CD4细胞的绝对数量也有所增加+T细胞,但CD4的比例+: CD8+细胞在慢性阻塞性肺病中发生逆转。这主要见于患有COPD的吸烟者,而不是没有气流限制证据的吸烟者90.目前尚不清楚这些细胞是否属于Tc1(产生干扰素γ)亚型或Tc2(产生IL - 4)亚型93但有证据表明,COPD气道中的大多数T细胞属于Tc1亚型78.CD8+和CD4+与循环中的T细胞相比,T细胞激活标志物的表达增加,尽管COPD患者与正常对照组之间没有明显差异94.
CD8的机制+, CD4的影响较小+慢性阻塞性肺病患者的气道和肺部积聚的细胞尚不清楚。然而,T细胞归巢到肺必须依赖于一些初始激活,然后粘附和选择性趋化。COPD患者外周气道中的T细胞CXCR3表达增加,CXCR3是一种由IP‐10、Mig和I‐TAC激活的受体。细支气管上皮细胞中IP‐10的表达增加,这可能有助于CD8的积累+细胞,优先表达CXCR378.
CD8的数量也有增加+不吸烟的COPD患者血液循环中的细胞95,96T -辅助性1型(产生干扰素(IFN)‐γ) CD4的增加+COPD患者的细胞97.这表明可能存在慢性免疫刺激通过抗原呈现通过HLA 1类途径。树突状细胞可能会从气道转移到区域淋巴结,并刺激CD8的增殖+和CD4+T细胞。CD8+细胞通常在气道感染中增加,可能是细菌和病毒病原体对COPD患者下呼吸道的慢性定殖导致了这种炎症反应98.蛋白酶诱导的肺损伤也有可能暴露出先前隔离的自身抗原,或者香烟烟雾本身可能损伤气道上皮细胞并使其产生抗原99.
CD4数量增加的作用+慢性阻塞性肺病中的细胞,特别是在严重疾病中,也是未知的47;它们可能具有免疫记忆,并在不吸烟的情况下在延续炎症过程中发挥作用。自然杀伤因子(NK, CD56+)细胞是抵御病毒感染的第一道防线。COPD患者循环NK细胞减少,吞噬活性降低One hundred.在正常吸烟者身上也发现了类似的结果101虽然在COPD患者肺实质中未发现NK细胞的差异90.吸烟者肺泡中γ/δ T细胞增加,无论有无气道阻塞90.
T细胞在COPD病理生理中的作用尚不确定。CD8+细胞能够通过释放穿孔素、颗粒酶- B和TNF - α引起肺泡上皮细胞的细胞溶解和凋亡102.这与CD8有关+肺气肿中肺泡细胞的细胞与凋亡90.在香烟诱导的肺气肿小鼠模型中,T细胞占主导地位,这与肺气肿的严重程度直接相关103.
嗜酸性粒细胞
嗜酸性粒细胞在COPD中的作用尚不确定。有报道称,稳定期COPD患者气道和灌洗液中非活性嗜酸性粒细胞数量增加,而另一些患者在气道活检、BAL或诱导痰中未发现数量增加104.慢性阻塞性肺病患者中嗜酸性粒细胞的存在预示着对糖皮质激素的反应,并可能表明同时存在哮喘105,106.据报道,慢性支气管炎急性发作时支气管活检和BAL液中嗜酸性粒细胞增多107,108.令人惊讶的是,COPD患者诱导痰中嗜酸性粒细胞基本蛋白的水平与哮喘患者一样升高,尽管没有嗜酸性粒细胞,这表明它们可能已经脱粒,在显微镜下无法识别55.也许这是由于高水平的中性粒细胞弹性蛋白酶,已被证明导致嗜酸性粒细胞脱颗粒109.
树突细胞
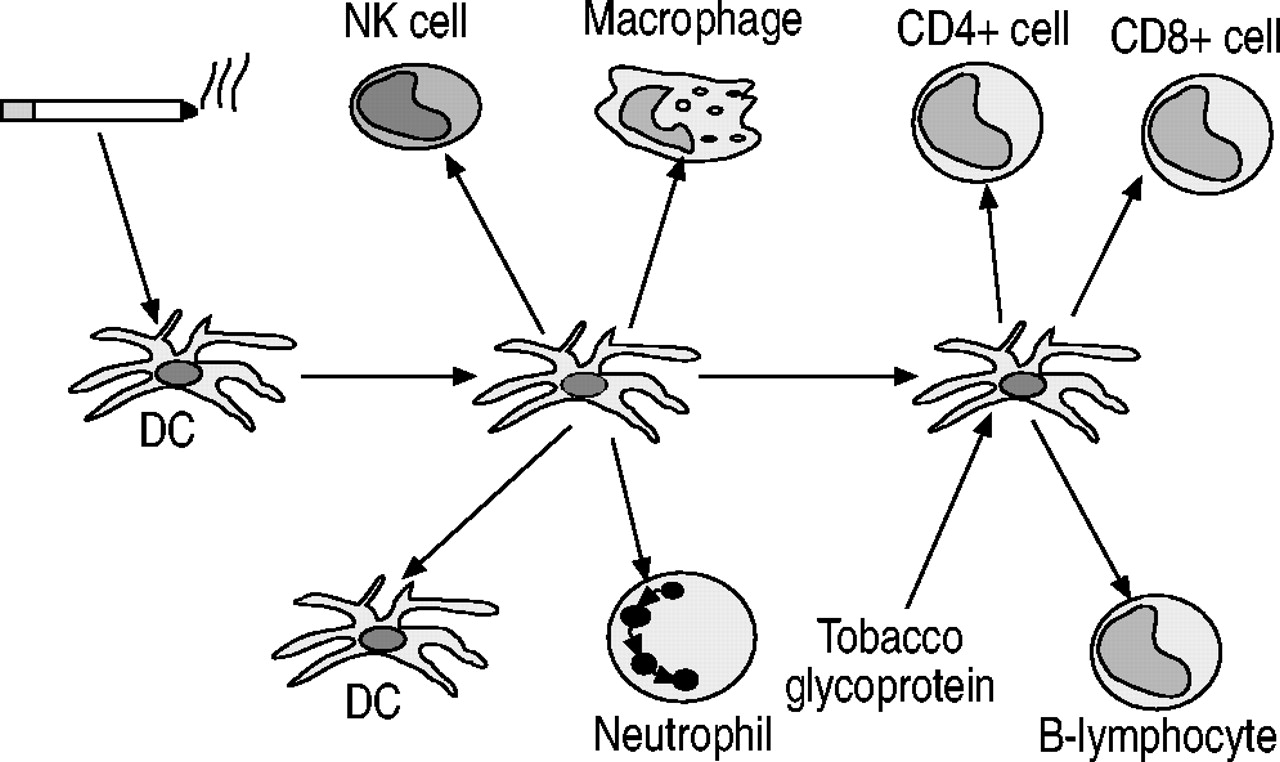
树突状细胞在先天和适应性免疫反应的启动中起着核心作用110.气道和肺部含有丰富的树突细胞网络,这些细胞位于靠近表面的位置,因此它们的理想位置可以发出被吸入的外来物质进入的信号111.树突状细胞可以激活多种其他炎症和免疫细胞,包括巨噬细胞、中性粒细胞、T淋巴细胞和B淋巴细胞112.因此,树突细胞可能在肺部对香烟烟雾和其他吸入有害物质的反应中发挥重要作用,因此可能是COPD的关键细胞成分(图2)⇓).烟草烟雾激活免疫系统的机制尚不清楚,但从烟草中分离出的一种糖蛋白具有强大的免疫刺激作用113.暴露在香烟烟雾中的大鼠肺部树突细胞数量增加114以及吸烟者的气道和肺泡壁115,116.肺组织细胞增多症是一种由肺部树突状细胞肉芽肿引起的疾病,其特征是肺实质的破坏,类似肺气肿117,118.这种疾病的成人形式几乎只发生在吸烟者身上。在长期暴露于香烟烟雾的小鼠中,气道和肺实质中的树突细胞增加119.树突状细胞在COPD中招募其他效应细胞的作用值得进一步研究。
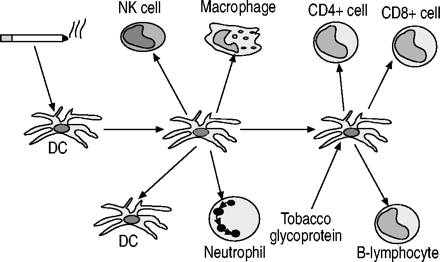
树突状细胞(DC)可能在慢性阻塞性肺疾病的病理生理学中发挥重要作用,因为它们可以被吸烟和烟草糖蛋白激活,导致中性粒细胞、巨噬细胞、自然杀伤细胞(NK)、CD4细胞的招募+和CD8+T淋巴细胞和B淋巴细胞。
上皮细胞
气道和肺泡上皮细胞可能是COPD炎症介质和蛋白酶的重要来源。上皮细胞被香烟烟雾激活产生炎症介质,包括TNF - α、IL - 1β、GM - CSF和IL - 8120- - - - - -122.小气道中的上皮细胞可能是TGF‐β的重要来源,TGF‐β可诱导局部纤维化20..血管内皮生长因子(VEGF)似乎是维持肺泡细胞存活所必需的,而大鼠血管内皮生长因子受体(VEGFR2)的阻断会诱导肺泡细胞凋亡和肺气肿样病理123.然而,VEGF在人类肺气肿发病机制中的作用尚不清楚。
气道上皮细胞在气道防御方面也很重要。杯状细胞产生的黏液可以捕获细菌和吸入的微粒124.上皮细胞分泌防御素和其他具有抗菌作用的阳离子肽,是先天防御系统的一部分,但也参与组织修复过程125.它们还分泌抗氧化剂和抗蛋白酶,如分泌性白蛋白蛋白酶抑制剂(SLPI)。上皮细胞也运输免疫球蛋白A,因此也参与适应性免疫126.香烟烟雾和其他有害物质可能损害气道上皮细胞的这些先天和适应性免疫反应,增加对感染的易感性。
慢性支气管炎和COPD患者气道上皮常表现为鳞状化生,这可能是由于气道上皮细胞增殖增加所致。用PCNA测量的基底气道上皮细胞增殖在一些正常吸烟者中增加,但在慢性支气管炎患者中显著增加,并与鳞状化生的程度相关127.COPD中参与上皮细胞增殖、细胞周期和分化的生长因子的性质尚不清楚。上皮生长因子受体在吸烟者气道上皮细胞中表达增加,并可能促进基底细胞增殖,导致鳞状化生和支气管癌风险增加128.
氧化应激的作用是什么?
当产生的活性氧(ROS)超过抗氧化防御机制并导致有害影响时,就会发生氧化应激,包括对脂质、蛋白质和脱氧核糖核酸(DNA)的损害。越来越多的证据表明氧化应激是COPD的一个重要特征129- - - - - -131.
形成
COPD患者气道中被激活的炎症细胞和结构细胞产生ROS,包括中性粒细胞、嗜酸性粒细胞、巨噬细胞和上皮细胞130.超氧阴离子(O2.-)是由还原的烟胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶产生的,并转化为过氧化氢(H2O2)通过超氧化物歧化酶。H2O2然后被过氧化氢酶分解成水。O2.-和H2O2可在游离铁的存在下相互作用,形成高活性的羟基自由基(.哦)。O2.-也可能与NO结合形成过氧亚硝酸盐,也会生成.哦132.氧化应激导致花生四烯酸氧化,形成一系列新的前列腺素介质,称为异前列腺素,可能发挥重要的功能作用133,包括支气管收缩和血浆渗出134- - - - - -136.
粒细胞过氧化物酶,如中性粒细胞中的髓过氧化物酶,在氧化应激中起重要作用。中性粒细胞H2O2由O生成2-在氯离子存在的情况下髓过氧化物酶代谢为次氯酸,次氯酸是一种强氧化剂。髓过氧化物酶也能硝化酪氨酸残基,过亚硝酸盐也能137- - - - - -139.
抗氧化剂
人体呼吸道中的几种抗氧化机制抵消了氧化剂的正常产生140.呼吸道中主要的细胞内抗氧化剂是过氧化氢酶,超氧化物歧化酶(SOD)和谷胱甘肽,由γ -谷氨酰半胱氨酸合成酶和谷胱甘肽合成酶形成。氧化应激激活可诱导酶血红素加氧酶- 1 (HO‐1),将血红素和血红素转化为胆绿素,形成一氧化碳(CO)。141.胆绿素被转化通过胆红素还原酶是一种潜在的抗氧化剂。HO‐1在人体气道中广泛表达142COPD中CO的产生增加143.在肺中,细胞内抗氧化剂的表达水平相对较低,不受氧化应激诱导,而主要的抗氧化剂是细胞外的144.细胞外抗氧化剂,特别是谷胱甘肽过氧化物酶,在香烟烟雾和氧化应激反应中显著上调。谷胱甘肽系统是呼吸道中的主要抗氧化机制。肺上皮粘膜液中存在高浓度还原性谷胱甘肽140吸烟者体内的浓度也会增加。细胞外谷胱甘肽过氧化物酶(eGPx)是肺中重要的抗氧化剂,可能由上皮细胞和巨噬细胞分泌,特别是对香烟烟雾或氧化应激的反应145.eGPx灭活H2O2和O2-,但也可能阻止活性氮的存在144细胞外抗氧化剂还包括膳食抗氧化剂维生素C(抗坏血酸)和维生素E (α‐生育酚)、尿酸、乳铁蛋白和细胞外SOD3。SOD3在人肺中高度表达,但其在COPD中的作用尚不清楚146.
对呼吸道的影响
活性氧对气道有几种影响,可能会增加炎症反应。这些作用可能是由ROS对气道内靶细胞的直接作用介导的,但也可能是间接介导的通过信号转导通路和转录因子的激活通过氧化介质的形成,如异前列腺烷和羟基壬烯醛。
ROS激活NF‐κB,从而开启多种炎症基因,导致炎症反应的放大147.氧化应激激活NF - κB的分子途径尚未完全阐明,但在激活途径中有几个氧化还原敏感步骤148.许多激活NF‐κB的刺激似乎都是如此通过ROS的形成,特别是H2O2.ROS激活上皮细胞系中的NF‐κB149并增加培养的人气道上皮细胞促炎细胞因子的释放150.氧化应激导致组蛋白乙酰转移酶活性的激活,从而打开染色质结构,并与多种炎症基因的转录增加有关151,152.
另一种激活炎症基因的转录因子是激活蛋白- 1 (AP - 1),一种Fos和Jun蛋白的异二聚体。与NF‐κB一样,活化途径中有几个氧化还原敏感步骤153.外源性氧化剂也可能是气道疾病恶化的重要原因。香烟烟雾、臭氧和二氧化氮(在较小程度上)会对呼吸道造成氧化应激154.
氧化剂也激活有丝分裂原活化蛋白激酶途径。H2O2是一种有效的细胞外调节激酶和p38 MAP激酶通路的激活剂,这些通路调节许多炎症基因的表达和某些细胞的存活,以及巨噬细胞的扩散155.事实上,巨噬细胞功能的许多方面都是由氧化剂通过激活多种激酶途径来调节的156.
氧化应激增加的证据
有相当多的证据表明COPD中氧化应激增加129,130.香烟烟雾本身就含有高浓度的活性氧157.炎症细胞,如活化的巨噬细胞和中性粒细胞,也产生ROS,如上所述。流行病学证据表明,膳食中抗氧化剂摄入量的减少可能是COPD的一个决定因素,人口调查已将膳食中抗氧化剂抗坏血酸(维生素C)摄入量低与肺功能恶化联系起来158,159.
在呼吸中可以检测到氧化应激的几种标记物,一些研究表明,呼出的空气或呼吸冷凝物中氧化剂的产生增加160- - - - - -162.H的浓度增加了2O2在慢性阻塞性肺病患者的呼气冷凝物中,特别是在病情加重期间163,164.
8‐异前列腺素F的浓度也有升高2α(8‐异前列腺素)在呼出气体冷凝物中,即使在戒烟患者中也能发现165并且在急性发作时进一步增加166.正常吸烟者呼吸中的异前列腺素也增加,但增加的幅度小于COPD,这表明COPD中的氧化应激被夸大了。8‐异前列腺素在COPD患者尿液中同样升高,并在加重期进一步升高167.
也有证据表明,COPD患者通过脂质过氧化生化标志物测量的全身氧化应激标志物增加168.免疫细胞化学可以检测到一种特殊的标记物脂质过氧化4‐羟基‐2‐壬烯醛,它与蛋白质中的基本氨基酸残基形成加合物,并已在COPD患者的肺部检测到169.这种氧化应激的特征定位于气道和肺泡上皮细胞、内皮细胞和中性粒细胞。
氧化应激的作用
COPD患者气道氧化应激升高可能通过放大COPD炎症反应在疾病中发挥重要的病理生理作用。这可能反映了NF - κB和AP - 1的激活,然后诱导中性粒细胞性炎症通过IL‐8和其他CXC趋化因子、TNF‐α和MMP‐9的表达增加。NF‐κB在COPD患者的气道和肺泡巨噬细胞中被激活,并在加重期间被进一步激活73,74.在COPD患者中,氧化应激可能是该转录因子的重要激活因子。
氧化应激也可能损害抗蛋白酶如α的功能1‐抗胰蛋白酶和SLPI,从而加速肺实质中弹性蛋白的分解170.
皮质类固醇对慢性阻塞性肺病的疗效远低于对哮喘的疗效,并且不能减少疾病的进展171- - - - - -174.与哮喘患者相比,COPD患者对皮质类固醇没有任何明显的抗炎反应80,81.与正常吸烟者和非吸烟者的细胞相比,COPD患者的肺泡巨噬细胞对皮质类固醇的抗炎作用的反应显著降低82.最近的研究表明,COPD患者氧化应激与皮质类固醇反应不良之间可能存在联系。氧化应激损害糖皮质激素受体与DNA的结合以及这些受体从细胞质到细胞核的易位175,176.皮质类固醇通过将HDAC2招募到活性转录位点,并通过去乙酰化活性转录炎症基因的高乙酰化组蛋白,从而关闭炎症基因,从而抑制炎症84,177.在吸烟者和COPD患者中,HDAC活性显著降低,肺泡巨噬细胞中HDAC2表达降低83外周肺组织中HDAC2表达的减少幅度更大178.HDAC活性的降低与炎症细胞因子抑制的降低和皮质类固醇反应的降低有关。这可能是氧化应激直接或间接的结果,并与H2O2在细胞系中178.
氧化应激也可诱导内皮细胞和上皮细胞凋亡。1型肺细胞的凋亡可能促进肺气肿的发展,这可能是由细胞毒性T淋巴细胞或血管内皮生长因子受体的抑制引起的90,123.ROS可能通过激活NF‐κB通路,直接损伤DNA而诱导细胞凋亡通过活化多聚二磷酸腺苷核糖和通过生成4‐羟基壬烯醛。凋亡信号调节激酶‐1在硫氧还蛋白的作用下处于失活构象,当被ROS氧化时,会触发凋亡通路179.
哪些蛋白酶是重要的?
长期以来,人们一直认为各种蛋白酶可以分解肺实质中的结缔组织成分,特别是弹性蛋白,从而产生肺气肿,并且蛋白酶和内源性抗蛋白酶之间存在不平衡,通常情况下,内源性抗蛋白酶应该能够抵御蛋白酶介导的作用。弹性蛋白可能是这些酶最重要的目标,因为肺气肿患者的肺实质失去弹性,弹性蛋白不能以活性形式再生。COPD中弹性蛋白降解的证据是,与肺功能正常下降的吸烟者相比,肺功能快速下降的吸烟者中,由弹性蛋白交联产生的地石氨酸的排泄增加180.尽管早期的注意力集中在中性粒细胞弹性蛋白酶(NE)上,但许多其他具有降解弹性蛋白能力的蛋白酶现在已经被牵连181.
中性粒细胞弹性蛋白酶
NE的作用一直被特别强调,因为遗传性α1‐抗胰蛋白酶(α1‐AT)缺乏可导致早发型肺气肿。进一步证明了α1‐AT可能因香烟烟雾暴露而失活,这增加了中性粒细胞弹性蛋白酶在血浆α正常的吸烟者中也很重要的可能性1量浓度。气管灌注NE诱导肺气肿和中性粒细胞浸润的动物模型支持这一结论182NE在肺气肿患者肺实质弹性蛋白纤维上的免疫细胞化学定位183.NE (E.C.3.4.21.37)是一种被α抑制的丝氨酸蛋白酶1‐AT位于肺实质。它储存在中性粒细胞和细胞因子启动的细胞中的嗜蓝粒细胞颗粒中,可在细胞表面表达184.
NE随后被证明具有与其在COPD中的潜在作用相关的其他几种作用。它是一种有效的粘膜下腺细胞和杯状细胞的粘液促分泌剂40,185.NE诱导MUC5AC在上皮细胞系中的表达,该机制似乎依赖于活性氧的产生186,187.NE还诱导一些细胞因子的表达,包括气道上皮细胞中的IL‐8188.NE切割巨噬细胞上的磷脂酰丝氨酸受体,从而削弱其清除凋亡细胞的能力89.
另一方面,NE也使细胞表面的脂多糖受体CD14失活,从而减少对内毒素的炎症反应189.东北地区有可能在东道主防御和东北地区发挥作用(- / -)小鼠对压倒性的革兰氏阴性细菌感染的易感性增加,但自发感染似乎没有任何增加190,191.
NE在COPD中的作用只有在NE抑制剂的疗效得到临床研究后才能确定192.在暴露于香烟烟雾的豚鼠中,NE抑制剂显著减少了肺气肿和中性粒细胞炎症反应193.尽管几种NE抑制剂已在人体中进行了测试,但报告的结果很少。目前尚不确定是药物失败还是临床试验设计不当。NE抑制剂MR889对未入选的COPD患者尿中氨脂氨酸没有影响,但在病史相对较短的患者中有小幅降低194.大环内酯类抗生素红霉素和氟红霉素也被证明能抑制NE活性195这可能解释了它们对粘液分泌亢进的有益作用196.
其他丝氨酸蛋白酶
中性粒细胞还在其特定的颗粒中储存另外两种丝氨酸蛋白酶组织蛋白酶G和蛋白酶3。这些其他丝氨酸蛋白酶具有与NE相似的特性,并以类似的方式诱导粘液分泌40,42.蛋白酶3被细胞因子激活后,在中性粒细胞表面强烈表达197.蛋白酶3被α有效抑制1量在198.中性粒细胞弹性蛋白酶抑制剂在发育抑制其他丝氨酸蛋白酶192.
半胱氨酸蛋白酶
溶酶体半胱氨酸蛋白酶(组织蛋白酶)也可能与COPD有关199,200.组织蛋白酶S的表达由干扰素‐γ在几种细胞类型中诱导,包括平滑肌细胞。IFN‐γ过表达诱导小鼠肺气肿,组织蛋白酶B、D、H、L和S表达增加201.组织蛋白酶抑制剂显著减少肺气肿诱导的IL‐13转基因小鼠,表明该组织蛋白酶的弹性溶解潜力202.其他几种组织蛋白酶也具有弹性溶解活性,包括组织蛋白酶B、K和L,它们在肺泡巨噬细胞中表达69,203CD8中的组织蛋白酶W+T细胞应承担的204.组织蛋白酶在COPD中的作用尚不确定。在肺气肿患者的BAL液中检测到组织蛋白酶L浓度升高205COPD患者的肺泡巨噬细胞比正常吸烟者或非吸烟者的巨噬细胞分泌更多的半胱氨酸蛋白酶活性70.组织蛋白酶的内源性抑制剂是胱抑素和stefins,但对它们在COPD中的作用知之甚少。慢性阻塞性肺病患者BAL液中胱抑素C浓度升高205.
基质金属蛋白酶
越来越多的证据表明基质金属蛋白酶在COPD中起作用206.在肺气肿患者中,baln浓度和MMP‐1(胶原酶)和MMP‐9(明胶酶B)的巨噬细胞表达增加81,207,208.肺气肿患者肺实质中MMP‐9活性增加209.肺气肿患者肺部MMP‐1表达也增加,主要集中于II型肺细胞210.正常吸烟者的肺泡巨噬细胞比正常人表达更多的MMP‐972慢性阻塞性肺病患者的细胞也在不断增加71,大大增强了弹性溶解活性70.事实上,使用MMP抑制剂marimastat表明,COPD患者肺泡巨噬细胞在长时间内释放的大部分弹性溶解活性都是由MMP决定的70.
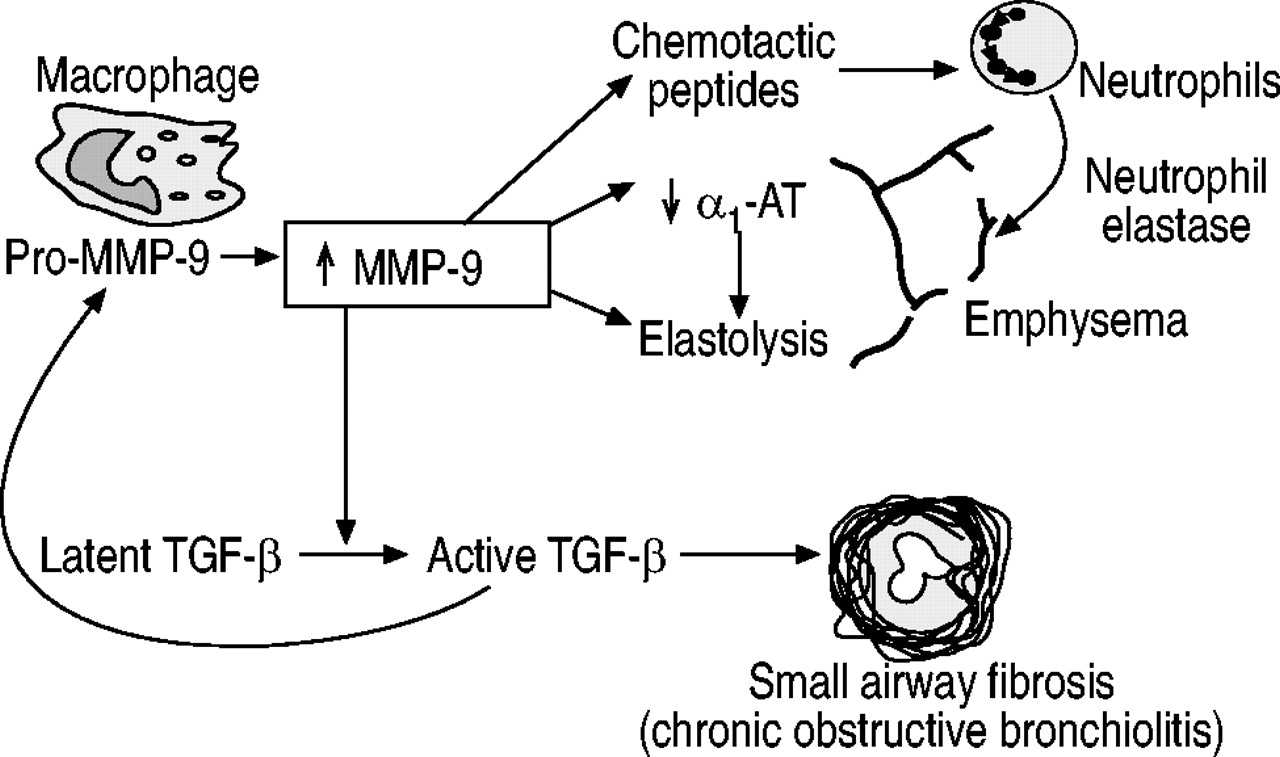
MMP‐12可预防慢性香烟暴露引起的肺气肿,这一研究结果提高了对MMPs的兴趣-/-(巨噬细胞金属弹性蛋白酶)小鼠211.在MMP12-/-IL‐13和IFN‐γ过表达诱导的小鼠肺气肿减轻201,202进入肺的单核细胞数量明显减少。这可能是因为基质蛋白酶产生趋化肽,促进巨噬细胞向实质和气道募集。MMPs可激活潜伏形式的TGF‐β到其活性形式。此外,整合素αvβ6缺失的小鼠(Itgb6‐null小鼠)不能激活TGF‐β,并发生与年龄相关的肺气肿,而MMP12可以预防-/-通过TGF‐β1的过表达来提高小鼠的免疫活性212.这表明TGF‐β1在正常情况下下调MMP‐12,而TGF‐β缺失导致MMP‐12过多和肺气肿。MMP 9应承担的-/-小鼠对香烟烟雾引起的肺气肿没有保护作用,但对小气道纤维化有保护作用213.TGF‐β1被MMP‐9激活214;这可能是被调解的通过MMP‐9诱导潜在TGF‐结合蛋白‐1的蛋白水解裂解,导致TGF‐β1的释放215.因此,这种机制可能是MMP‐9诱导的弹性溶解和TGF‐β1激活同时产生纤维化之间的联系(图3)⇓).因此,MMP‐12在小鼠中是一种突出的MMP,而在人类中似乎不像MMP‐9那么重要。
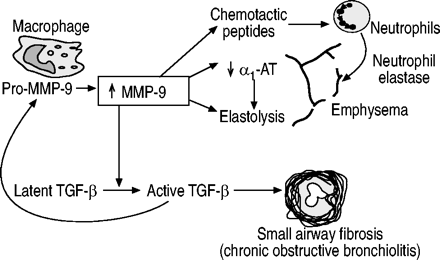
慢性阻塞性肺疾病小气道纤维化与肺气肿的可能相互关系。转化生长因子(TGF)‐β激活基质金属蛋白酶- 9 (MMP - 9),并被MMP - 9激活。α1量:α1抗胰蛋白酶。
Antiproteases
通常蛋白酶会被过量的内源性抗蛋白酶抵消。丝氨酸蛋白酶的主要抑制剂是α1‐AT在肺实质和气道上皮来源的SLPI在气道。其他丝氨酸蛋白酶抑制剂包括弹力蛋白和α1antichymotrypsin。丝氨酸蛋白酶抑制剂灭活NE和其他丝氨酸蛋白酶,如蛋白酶‐3216.α的多种基因变异1‐AT现已被认识到可减少循环活性α1量在浓度217,218.最容易导致早发型肺气肿的缺陷是ZZ型,其中单氨基酸取代(Gly342→Lys)导致α1‐AT结构改变,导致其正常翻译后修饰和肝细胞分泌失败,导致血浆浓度非常低。是否杂合子和其他减少循环α的遗传变异1‐AT浓度在较小程度上比ZZ表型更容易导致肺气肿,这一点更有争议219.ZZα1‐AT缺乏是肺气肿的罕见原因,占患者的1%,但很久以前就有人提出吸烟可能会氧化α1‐AT导致抗蛋白酶功能受损,中性粒细胞弹性蛋白酶活性增加220.其机制似乎是由于氧化应激和氧化蛋氨酸在351或358位削弱抗- NE α活性1量在170.
SLPI是呼吸道中另一种主要的丝氨酸蛋白酶抑制剂221.如α1‐AT, SLPI可能因氧化应激而失活,但也可能因组织蛋白酶L和S通过其活性位点的裂解而失活222在肺气肿患者的BAL液中发现SLPI的蛋白水解片段,这有助于降低这些患者的抗NE活性。SLPI的失活不仅削弱了其抗NE活性,还削弱了其抗菌和抗炎作用。SLPI下调了LPS诱导的单核细胞TNF - α和MMP分泌223,224这可能是通过对IκBα降解的抑制作用介导的,从而抑制NF‐κB225.弹力蛋白和α的作用1‐抗胰凝乳酶在COPD中的作用尚不明确226,227.
四种基质金属蛋白酶的组织抑制剂(TIMP1‐4)可以中和基质金属蛋白酶228.肺泡巨噬细胞的TIMP‐1分泌在炎症刺激下增加,但在COPD患者的细胞中增加减弱,因此有利于增加弹性溶解70,71.在COPD患者中,TIMP‐2功能突变丧失的频率增加229.
放大机制是什么?
慢性阻塞性肺病的炎症改变和蛋白酶失衡也见于无慢性阻塞性肺病的吸烟者,但程度较轻12,47,70,81,82,提示COPD肺功能的加速下降可能是由于正常肺部炎症反应对刺激物的放大。这可能是由于炎症介质和酶的产生增加,或由于内源性抗炎或抗蛋白酶机制的缺陷。这些差异可能是由编码细胞因子、蛋白酶、抗炎蛋白和抗蛋白酶的基因多态性所解释的230,231.
另一种假设是,这些差异是由于潜伏病毒感染造成的232.潜伏腺病毒序列E1A在肺气肿患者的肺中比在匹配的吸烟控制受试者中更常见,并与炎症反应增加相关47.腺病毒感染放大了豚鼠呼吸道对香烟烟雾的炎症反应68.将E1A转染人上皮细胞系后,转录因子NF - κB的激活增加,细胞激活后IL - 8的释放增加,TGF - β1的产生增加,为炎症反应中的扩增提供了分子机制233,234.
COPD炎症放大的另一个分子机制可能涉及肺泡巨噬细胞中HDAC活性受损。在来自吸烟者的巨噬细胞中,HDAC的活性受损,它参与通过逆转与其激活相关的核心组蛋白的乙酰化来关闭炎症基因的转录83,84.COPD患者的外周肺HDAC活性甚至比没有气道阻塞的吸烟者更明显降低178.这可能导致炎症基因表达的扩增,如COPD患者肺泡巨噬细胞中所见71,82.COPD患者的这些细胞中NF‐κB的激活也增加73,74.
虽然吸烟是COPD发病的主要原因,但戒烟似乎并不能缓解气道炎症反应,特别是在疾病晚期47,235,236.这表明,一旦慢性炎症形成,就存在维持慢性炎症过程的持久机制。这可能解释了在首次症状出现前多年停止吸烟的患者出现COPD的原因。疾病持续的机制目前尚不清楚。
为什么对皮质类固醇的反应很差?
吸入糖皮质激素现在是慢性哮喘治疗的主要手段,慢性炎症也存在于慢性阻塞性肺病,这为它们在慢性阻塞性肺病中的使用提供了依据。事实上,吸入糖皮质激素现在被广泛用于治疗慢性阻塞性肺病,几乎和治疗哮喘一样频繁。然而,即使吸入或口服高剂量皮质类固醇也不能抑制慢性阻塞性肺病的炎症80,81,237.这可能反映了这样一个事实,即人体中性粒细胞炎症不能被皮质类固醇所抑制,因为中性粒细胞的存活时间可以被类固醇延长238,239.大约10%的稳定期慢性阻塞性肺病患者在口服糖皮质激素后表现出一些症状和客观改善,这些患者很可能伴有哮喘,因为这两种疾病都很常见。事实上,哮喘的一个特征——气道高反应性,可以预测COPD的下降速度240.对皮质类固醇的反应可以通过痰中嗜酸性粒细胞数量的增加和呼出NO的增加来预测105,106,这都是哮喘的典型特征。
四项大型研究表明,高剂量吸入皮质类固醇不能减少COPD的进展171- - - - - -174.吸入糖皮质激素可能对减少重症患者病情加重有轻微影响241.
COPD患者似乎对皮质类固醇有主动抵抗。高剂量的皮质类固醇不能减少细胞因子和趋化因子,而这些细胞因子和趋化因子应该被皮质类固醇治疗所抑制80,81.皮质类固醇耐药的分子机制尚不清楚,但可能与导致炎症反应放大的机制相同。因此,巨噬细胞HDAC活性降低83可能会阻止皮质类固醇的抗炎作用,而皮质类固醇的抗炎作用依赖于HDACs对炎症基因复合物的募集84.类似的潜伏腺病毒似乎在实验动物中诱导皮质类固醇耐药242.
相比之下,全身性皮质类固醇在治疗COPD急性加重时具有有益的效果,改善了临床结局,缩短了住院时间243,244.急性期类固醇反应差异的原因与慢性COPD可能与炎症反应(嗜酸性粒细胞增加)或加重期气道水肿的差异有关。
急性加重的机制是什么?
尽管慢性阻塞性肺病急性加重(定义为症状加重和肺功能恶化)是住院的常见原因,但其细胞和分子机制尚不清楚245.急性加重期可延长,并对生活质量产生深远影响246并可能加速COPD的进展247.人们一直认为痰量的增加和脓性意味着呼吸道的细菌感染,但现在很明显,COPD的许多恶化(如哮喘)也由于上呼吸道病毒感染(特别是鼻病毒)和环境因素,如空气污染和温度248,249中性粒细胞增多,IL - 6、IL - 8、TNF - α和LTB浓度升高4在病情加重时痰里250,251即使COPD病情稳定,频繁加重的患者IL‐6水平较高,SLPI浓度较低252,253.COPD加重期间肺泡巨噬细胞中NF‐κB的激活也增加,这提供了感染、氧化应激和炎症基因表达增强之间的联系74.化脓性加重与细菌感染有关,其特征是LTB显著增加4痰液浓度254.痰中慢性细菌定植与痰中炎症指数升高相关98更容易发生化脓性恶化255.支气管活检显示轻度慢性支气管炎患者在病情加重期间嗜酸性粒细胞增多107这可能反映了急性加重期间气道上皮细胞中RANTES(由激活、正常T细胞表达和分泌调节)的显著上调39.然而,在严重COPD患者病情加重期间,痰中嗜酸性粒细胞没有增加252.在急性发作期间观察到氧化应激和呼出NO标志物的增加,可能反映了气道炎症的增加163,166,256,257.这可能反映了加重期间NF‐κB活化增加74.因此,COPD的加重似乎是由于炎症过程的进一步放大。这意味着抑制慢性阻塞性肺病炎症的治疗也可以阻止病情恶化。
一些慢性阻塞性肺病患者的下呼吸道被细菌定植的原因,如不可分型的菌株流感嗜血杆菌,链球菌引起的肺炎而且莫拉克斯氏菌属复活是不确定的258.在最近的一项研究中,急性加重期间采集的600个痰标本中有24%含有细菌病原体,而没有加重的住院患者中有18%含有细菌病原体259.大约三分之一的病情恶化与蛋白质组成不同的“新”细菌菌株有关,这表明新获得的感染,而不是现有生物体的突然爆发。然而,这些“新”菌株可能来自持续定殖菌株的抗原变异,这可能使它们不太容易受到局部免疫机制的影响260.慢性阻塞性肺病患者下呼吸道中某些细菌的持续存在可能是由于局限于受保护的部位。流感嗜血杆菌是否局限于气道壁内的部位,如气道壁内细胞之间261当这些生物位于气道上皮细胞之间时,它们对抗生素或抗体介导的防御机制不敏感262.流感嗜血杆菌在COPD患者中,感染与炎症介质(包括IL - 8和TNF - α)浓度升高有关263.炎症过程本身也可能促进某些细菌的存活。例如,人中性粒细胞防御素可增加黏附流感嗜血杆菌影响气道上皮细胞,因而容易受到侵袭264.
接下来的问题是什么?
与对哮喘的了解相比,对COPD所涉及的细胞和分子机制的了解尚处于早期阶段。虽然这两种疾病都涉及呼吸道炎症,但炎症的模式、炎症过程的结果和治疗反应明显不同。这篇综述的重点是目前COPD中正在解决的一些关键问题,但绝不是全面的。
虽然现在已经在COPD中发现了几种炎症细胞的激活,但它们在产生COPD典型病理中的相对重要性和顺序作用仍然知之甚少。然而,香烟烟雾和其他刺激物很可能激活了常驻细胞,包括巨噬细胞、上皮细胞和树突状细胞,然后发出信号,其他炎症细胞,包括中性粒细胞、单核细胞和T淋巴细胞从循环中涌入。这些细胞都释放多种炎症介质,尽管介质的模式与哮喘中的介质不同。病理过程主要位于肺周围,累及小气道和肺实质。然而,炎症与小气道纤维化、肺实质破坏和粘液分泌过多之间的关系尚不确定,这些机制在患者之间和疾病不同阶段的优势存在差异。研究小气道功能需要更好的技术。
在没有气流受限的吸烟者和少数肺功能加速衰退的吸烟者之间,炎症过程似乎有所扩大。这种扩增的分子基础需要进一步研究,但可能的机制与炎症、蛋白水解或保护机制或获得性潜伏病毒感染的遗传差异有关。扩增机制也可能与COPD中发现的相对类固醇耐药有关,一个合理的联系是HDAC激活不足,既放大炎症基因表达,又损害对皮质类固醇的抗炎反应。
现在还需要进一步的研究来回答这些关键问题。然而,从慢性阻塞性肺疾病患者和有类似吸烟暴露而无慢性阻塞性肺疾病的患者身上获得的组织以及新的分子和细胞技术的发展,可能会取得快速进展。这将使预测哪些吸烟者会患上慢性阻塞性肺病成为可能,并可能为开发抑制这种慢性炎症过程的新疗法确定新的靶点。许多治疗慢性阻塞性肺病的新药已经在研发中265其中一些即将进入临床试验。
致谢
“慢性阻塞性肺病:重要问题”2002年11月在马耳他举行的一次会议主席:P. Barnes(英国)、R. Pauwels(比利时)和S. Shapiro(美国);参与者:加拿大: m .科西奥,J.霍格;丹麦: J.维斯博;爱尔兰: G.麦克埃尔瓦尼;意大利: L.法布里,M.路易塞蒂;马耳他: R. Ellul-Micalef;荷兰: L.范·阿尔芬;瑞士: L.Nicod;英国: P.Calverley, E. Chilvers, W. MacNee, N. Pride, W. Wedzicha;美国:查普曼,昆克尔,雷纳德。
- 收到了2003年4月10日。
- 接受2003年6月12日
- ©ERS期刊有限公司
参考文献
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
-
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}