





文摘
欧洲呼吸188bet官网地址协会(ERS)研究研讨会题为“肺血管内皮:管弦乐队指挥在呼吸道疾病——突出了从基础研究到疗法”汇集了国际专家在不正常的肺内皮,从基础科学转化医学,讨论在急性和慢性肺部疾病的几个重要方面。本文将简要总结不同主题的讨论从这个在巴黎举行的会议,2016年10月27 - 28日,法国。是很重要的考虑,本文并没有解决所有方面的内皮功能障碍但专注于特定的主题,例如:1)的复杂作用肺内皮编排宿主反应在健康和疾病(急性肺损伤、慢性阻塞性肺疾病、高空肺部水肿和肺动脉高压);和2)肺内皮功能失调的潜在价值目标创新疗法。
文摘
肺内皮功能障碍的核心作用呼吸道疾病http://ow.ly/tVgK30iiZ1S
介绍
肺是由两个截然不同的循环系统,系统性支气管循环系统和小循环系统。从功能的角度来看,肺血管床中循环系统是独一无二的人体,特征是通过高速流,低阻和低压。尽管所有心输出量通过肺循环注入系统,平均肺动脉压力的测量值(肺动脉平均)肺动脉休息,健康的成年人大约14±3毫米汞柱,而全身动脉血压是100±20毫米汞柱。这种独特的小循环系统的功能主要是用肺脉管系统的三个关键特征:1)一个低阻抗血流;2)高合规的肺前毛细管的小动脉的特点是薄的血管壁;和3)高容量招募血管帮助适应流量的增加。事实上,这种循环系统可以容纳流量大约6 L·分钟不等−1在静止的条件下25 L·分钟−1在剧烈运动,以最小的增加肺动脉压力(1]。
由于其战略定位在血液和肺组织之间的界面,肺动脉内皮细胞(ECs)不仅在优化气体交换扮演关键性的角色,在控制屏障的完整性和功能,而且在调节肺血管张力(即。通过一氧化氮(NO)、内皮(PGI2)、内皮素(ET)和血清素(5 -)通路)(2]。此外,肺内皮功能作为一个活跃的和动态receptor-effector组织和响应不同的化学,物理或机械刺激分泌的物质(s),它可能维持血管舒缩性平衡和维管组织内稳态。事实上,肺ECs新陈代谢非常活跃,感知和响应信号从细胞外环境。所有这些交互与相邻细胞(和循环细胞和/或介质)的目标是保持thrombosis-free表面,以控制炎症细胞粘附和贩卖,以确保正常的血管生成和血管壁的完整性(3]。呼吸系统的重要组成部分,改变肺动脉内皮细胞起着核心作用的一些慢性和急性肺疾病的发病机理,常见和罕见。肺内皮改变的主要特点(或功能障碍)是渗透率增加,可以导致血管渗漏和水肿的形成;改变平衡血管收缩和血管舒张;收购的炎性表型招聘增加了炎性细胞粘附分子的表达,增加促炎转录因子的激活,释放炎症介质和氧化应激;pro-thrombotic表型;前导和抗凋亡表型;并与邻近血管细胞壁误解。
虽然已经取得了相当大的进展在肺EC的理解障碍,触发器、机制和后果内皮功能紊乱的急性和慢性肺部疾病仍然没有被完全了解。这些关键方面更好的知识将有助于发现新的疾病生物标记物和/或新药保护体内平衡,以应对损伤和疾病。
遗传和环境因素的重要性
大多数,如果不是全部,急性和慢性肺部疾病源于多个环境因素和许多遗传风险因子之间的相互作用,一个方面,在最近的一次审查从欧洲批判性评估肺白皮书[4]。脉管系统的作用在发展,终身维修和保养肺组织内稳态是至关重要的。实际上,许多疾病包括肺癌、肺气肿、慢性阻塞性肺疾病(COPD) (5,6)和肺动脉高压(PH) [7),显示主要EC异常。更清楚的了解肺ECs有助于肺的正常发展和如何解除这些机制在慢性疾病至关重要的发展新的治疗策略对人肺条件。然而,主题相同的刺激反应的个体差异,在高海拔环境缺氧等良好的文档记录。此外,它已是不争的肺血管加压的人类和动物之间的反应千差万别(甚至不同物种之间),支持未知的遗传影响的重要性。
除了遗传背景和特定的基因突变,其他因素是已知的确定对肺血管结构和功能修改(如外生暴露于药物和毒素,荷尔蒙和老化),导致减少修复能力(8- - - - - -10)和生产不同的受损vasoreactive介质(11]。
另一个潜在的代理能够导致内皮损伤的香烟(烟草制品),在慢性阻塞性肺病扮演了重要的角色,因为“健康的吸烟者”(没有慢性阻塞性肺病)已经显示明显异常的肺动脉包括:船舶改造,炎性细胞浸润,血管内皮功能障碍,减少内皮的表达没有合酶(以挪士),增加生长因子的表达和类似的基因表达谱,观察慢性阻塞性肺病患者(12,13]。在实验模型中也被证实的慢性阻塞性肺病慢性暴露于香烟烟雾中诱发内皮功能障碍在肺动脉14),以及表达的变化以挪士(15)和可溶性鸟苷酸环化酶(国网公司)16]。有趣的是,血管变化先于肺气肿的发展(17]。总之,这些研究结果强烈支持吸烟和肺血管损害的关系在慢性阻塞性肺病。
内皮屏障功能障碍的性质
血管内皮细胞形成最里面的衬里的肺血管的主要障碍是保护空气对血管流体入口空间。渗透率的内皮屏障的完整性是至关重要的维持体内平衡的所有身体的组织和器官,其渗透率的变化是急性和慢性肺部疾病的一个特点。
血管内皮细胞形成一个连续不间断的ECs层由复杂联接的结构,即,粘合连接处并且紧密连接和缝隙连接,这通常被称为管家trans-endothelial运动的液体,蛋白质和细胞。内皮屏障通透性良好,在基底条件下和针对不同的刺激或某些药物,存在着很大的差别在不同血管床(18]。损失的内皮屏障导致肺部水肿、急性肺损伤(ALI)的特征,以及高空肺部水肿(HAPE)。两种情况下的发病机制仍不清楚,没有药物被批准到目前为止这些危及生命的条件。
阿里可以发展为急性呼吸窘迫综合征(ARDS)的多器官衰竭占很高的死亡率和发病率。ARDS可以沉淀直接侮辱肺(即。胃内容物的肺炎或愿望)或间接侮辱(即。脓毒症或多个创伤)。ARDS的脓毒症是最常见的原因人类也是行之有效的肺导致带有相比其他ARDS死亡率最高的目的。ARDS的发病率变化很大,从15到70例,每年每100人000人,代表大约5%的住院,机械通气患者19]。
在血管和肺之间的界面空域,肺内皮是定位在建立免疫反应中扮演重要角色,导致阿里。机械的理解潜在的这些过程仍然不足,被认为是在新发现的光。肺的先天免疫反应包括招聘的免疫细胞在它们迁移到肺血管的空气空间。发生损伤内皮屏障通过旁分泌作用的介质(如花生四烯酸(20.],ATP [21和过氧化22)从相邻的上皮细胞释放、机械应力等血管伸展造成血管压增加(23从低氧红细胞过氧化),释放24)和pro-coagulant蛋白质被血小板(25]。
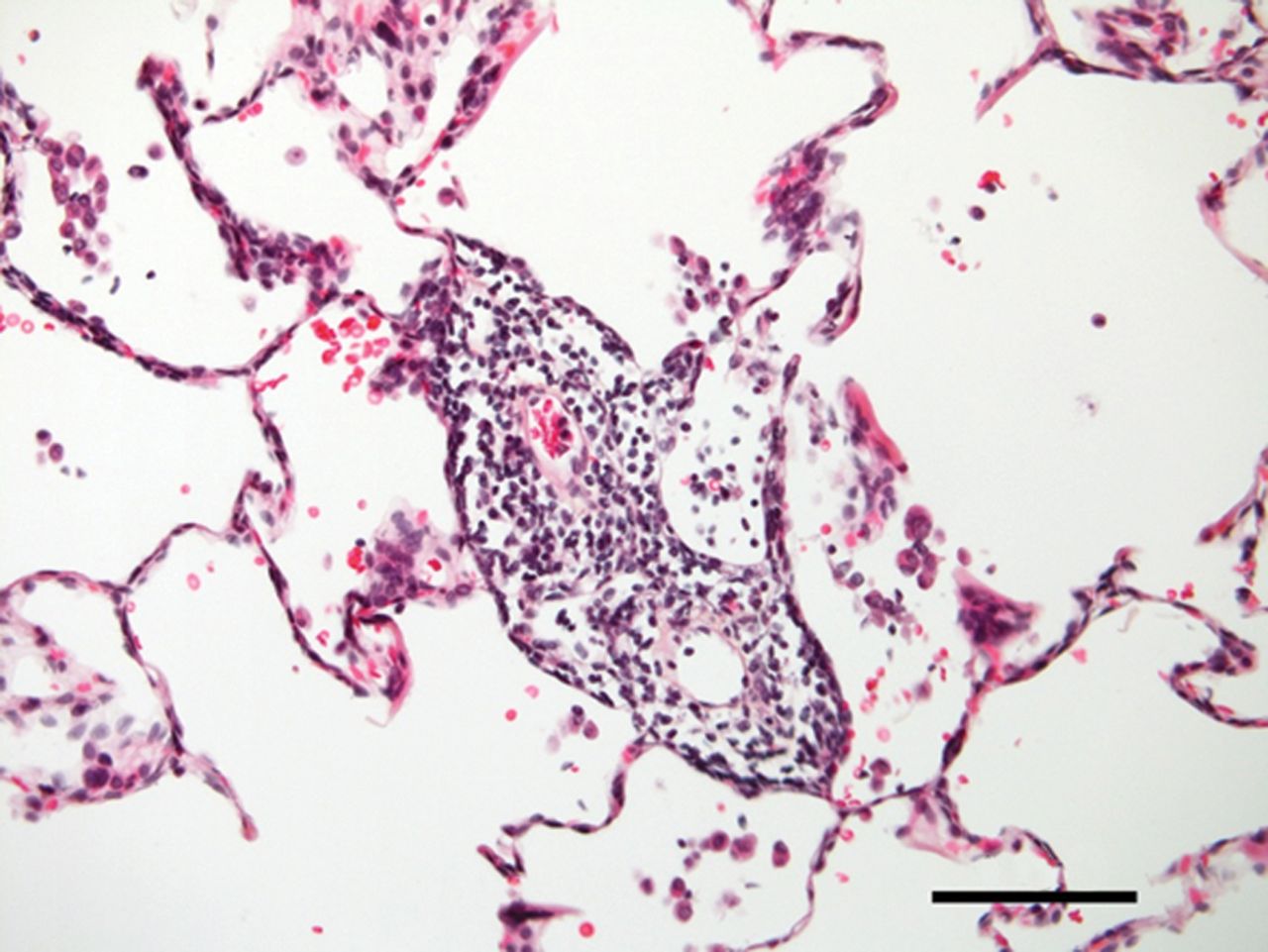
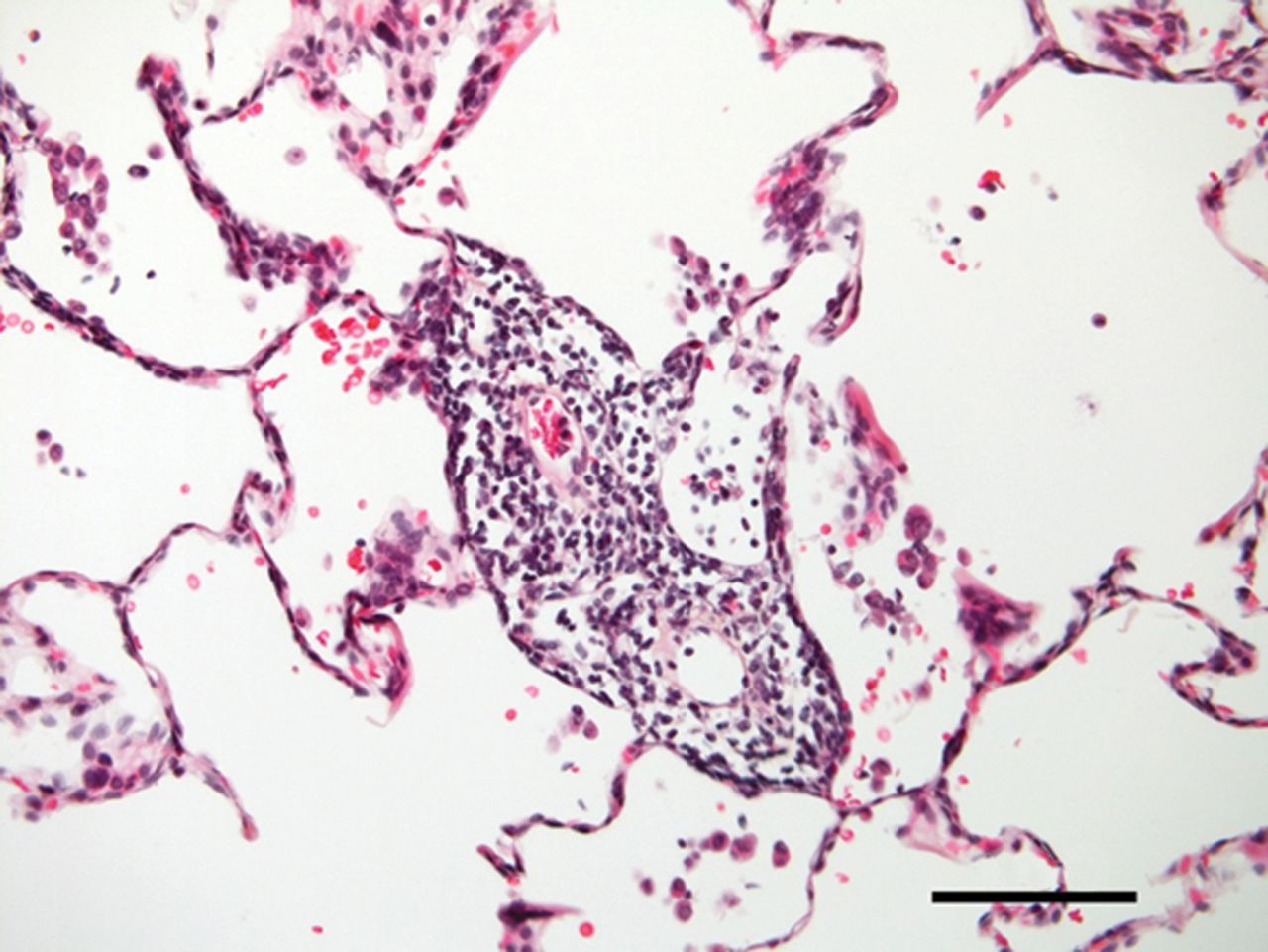
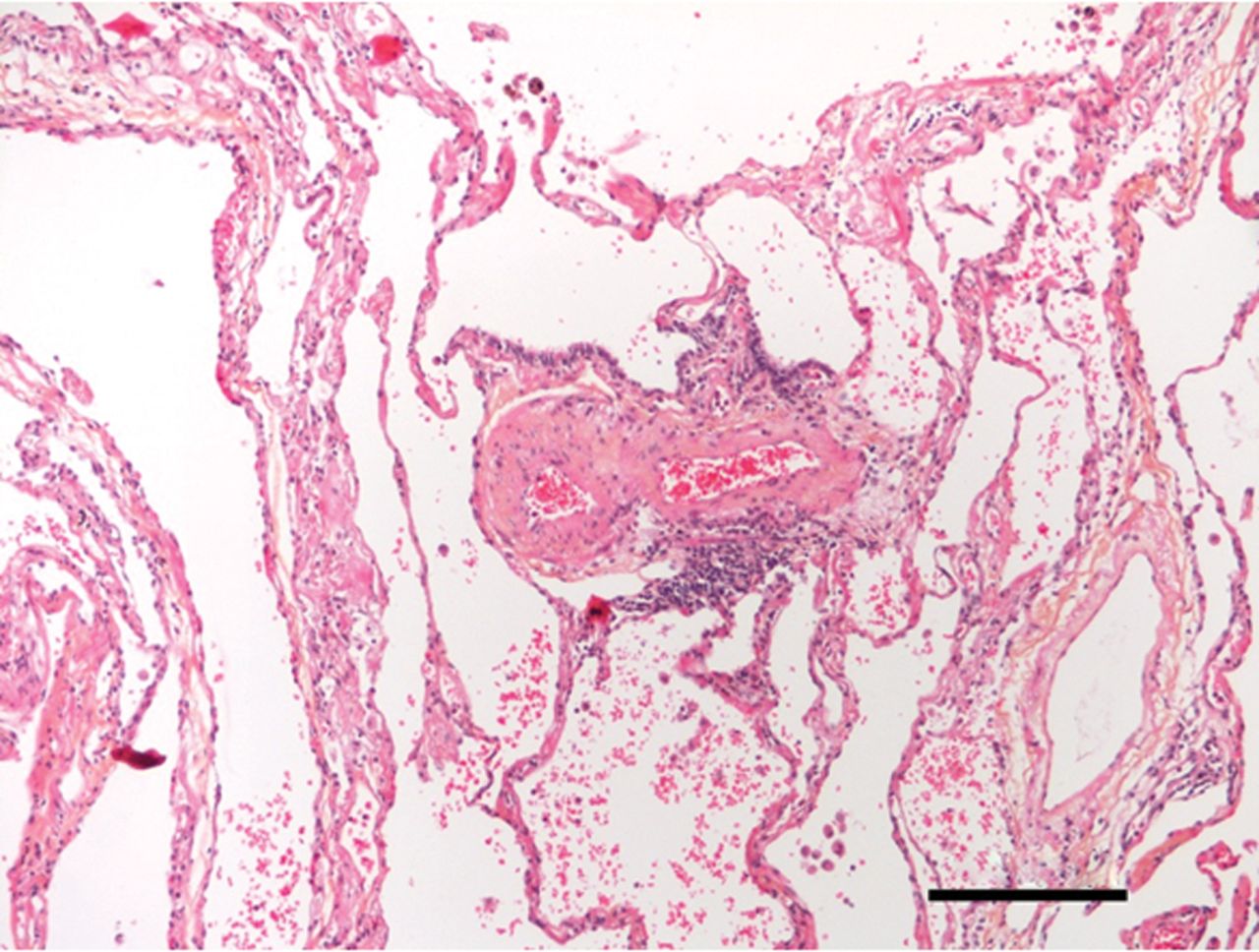
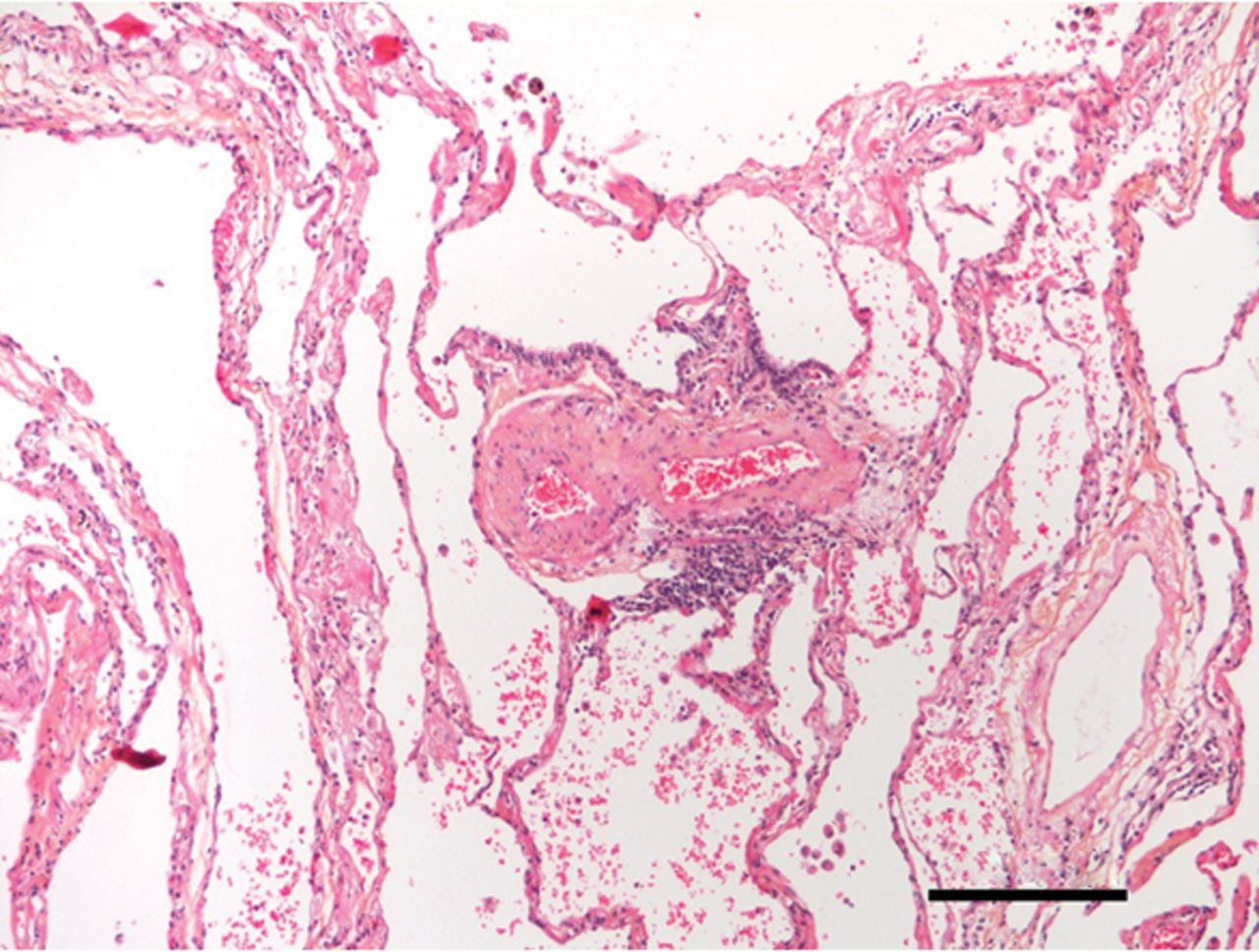
奇怪而强大的角色在肺内皮细胞线粒体的免疫反应越来越明显,尽管线粒体通常被认为是主要的细胞ATP生成和网站对肺内皮细胞线粒体的功能意义的正常或病变的肺。旧的印象,ECs缺乏线粒体,来自培养细胞,消除了最近的实时荧光成像技术(RFI)肺活在研究提供明确的证据肺内皮细胞线粒体的存在(26]。由于增强胞质Ca2 +振荡,要么自发27)或诱导条件与阿里(26,28),线粒体钙振荡2 +增加导致线粒体过氧化氢生产,因此核factor-κB (NF-κB)途径激活26,28]。由此产生的基因转录导致内皮表达P-selectin和白细胞粘附受体E-selectin,定义标记的促炎症激活肺脉管系统。肺动脉内皮细胞,线粒体在毛细管分支点的密度往往是稍高一些比间隔毛细血管(23]。因此,炎症启动毛细管分支点,也是微血管过滤的主要网站,这一过程生成所需的孔隙流体驱动淋巴流。因此,肺内皮细胞线粒体激活先天免疫机制,进而诱发生理反应促进淋巴抗原的淋巴结,促进适应性免疫(图1)。
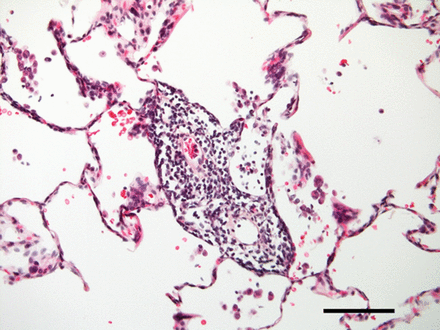
炎症和肺微循环。Inflammation-mediated事件发生在肺小动脉,毛细血管和小静脉毛细血管灌注受损。给出的样本来自患者的肺部慢性血栓栓塞肺动脉高压。注意中度到重度的微血管炎症(没有任何主要的血管壁重塑)。100μm比例尺表示。
内皮细胞线粒体的保护作用也出现了通过RFI研究解决内皮炎性受体的表达,肿瘤坏死因子(TNF)受体1 (TNFR1),这是表示为转移膜蛋白质在细胞腔的膜(28]。过氧化氢造成的线粒体释放肿瘤坏死factor-α(TNF-α)诱导Ca2 +振动激发金属蛋白酶,TNF-α转换酶(TACE),也表达了在腔内内皮细胞膜,导致脱落的TNFR1 ectodomains并限制TNF-α的促炎效应。
新的认识,线粒体是免疫传感器将会超出一般阿里炎症性肺疾病的病理(29日]。需要进一步的研究来理解线粒体损伤肺疾病过程,尤其是在肺内皮和制定治疗策略改善线粒体功能治疗肺部疾病。
肺循环血管内皮屏障属性系统的损失也会导致肺部水肿的一种效果,定义为高山肺水肿,可能发生在以前健康,健康,但不是与会的人迅速提升,在海拔3000 - 4000米[30.,31日]。它是一种多因素疾病,涉及环境(上升的高度和速度)和遗传风险因子(32- - - - - -34]。此外,缺氧引起肺小动脉的血管收缩在缺氧条件下当系统性动脉扩张。这种现象,称为缺氧性肺血管收缩(HPV),更明显的血管直径减少,是一个重要的生理机制肺动脉收缩在缺氧性肺区域血流量重定向的地区更大的氧气供应。现在认识到慢性暴露于低氧(即。慢性阻塞性肺病或住在高海拔地区)导致持续肺泡组织缺氧,导致持续的人乳头状瘤病毒,被认为扮演着重要的角色,让肺毛细血管高压,破坏墙壁,导致肺血管重塑,并可能PH值(35,36]。
异常高在缺氧肺动脉压(PAP)已被证明是一个标志高山肺水肿。高山肺水肿敏感性可以显示在低空短暂缺氧挑战高山肺水肿(历史的人37- - - - - -39]。右心衰catheterisation表现在高海拔显示所有个人发展中肺部水肿毛细管压力泄漏阈值19毫米汞柱以上,表明异常高人民行动党在高山肺水肿导致毛细血管床的40- - - - - -42]。少的地区HPV有更高的灌注引起血流介导增加毛细管压力和泄漏,而区域更活跃的血管收缩低灌注,从而防止水肿的形成。支持这一假设在高山肺水肿,灌注扫描结果表明,肺部水肿发生区域的高血流量(31日),观察水肿在高山肺水肿(片状分布43),通过分析缺氧的肺灌注磁共振成像(MRI) (44,45]。此外,支气管肺泡灌洗(BAL)在个人首先表现在高海拔高山肺水肿报道没有落下帷幕的炎症标记物液(46]。然而,炎症细胞和细胞因子平衡液中发现了更先进的情况下(47与高山肺水肿(在日本)和住院的病人48]。检测不到发炎HAPE因此pressure-induced迹象水肿,可能诱发继发性炎症反应(30.,49]。
最近的一项荟萃分析的高海拔的研究中,人民行动党是由超声心动图测量在人们生活在海拔3600至4350米,得出的结论是,altitude-induced PH值似乎是罕见的(50]。平均收缩压行动党在高海拔是25.3毫米汞柱,这是适度升高而出现在健康个体的一般人群生活在1200米。尽管如此,有一个广泛的分布和一些人在高度不受PH值相比(51]。其他人已经报道,人乳头状瘤病毒在人类的大小可以改变人与人之间的近五倍(52,53]。这有遗传基础和理解的遗传机制提供了重要的见解肺血管张力和结构的分子监管机构可以利用药物靶点。研究极端表型在高海拔种群和老鼠菌株已经开始支持这种方法,开辟新的研究领域。
HAPE-susceptible个人在前臂血流显示内皮功能障碍研究缺氧而不是normoxia,减少呼出没有在高海拔54,55),减少肺没有生物利用度(31日,46]和增加生产活性氧(ROS)在肺(特别是在肺动脉平滑肌细胞缺氧(PA-SMC)水平)(56]。此外,等离子体水平的endothelin-1 (ET-1)增加与增加PAP HAPE-susceptible个体在高海拔(57]。除了这些endothelium-dependent机制占增加HPV HAPE-susceptible个人,必须考虑其他因素,如较低的低氧通气反应(58)和一个更小的毛细血管床所显示低肺容积(59,60],它既可以有助于提高人民行动党在缺氧。
需要更好地理解机制内皮渗透性的差异不同的血管床,不同的信号通路调节inter-endothelial连接和组织液内稳态控制在正常情况下和病理过程。的确,内皮屏障的恢复是至关重要的组织内稳态和复苏后急性炎症事件或损伤。然而,我们在分子水平上了解这一过程的发生仍然是模糊的。应该面向更多的努力研发药物促进inter-endothelial联结的形成或增强的功能屏障完整性的主要介质。
改变平衡血管收缩和血管舒张
已经强调,小循环系统具有独特的血流动力学特性,特点是更放松血管舒缩性的语气在正常生理状态下,与体循环系统。在气体交换发生在胎儿胎盘,人乳头状瘤病毒行为来维持高肺血管阻力和转移远离肺部的血液流动开放性动脉导管。不同的离子通道,包括钾(K+)通道、表达和分布在几种类型的肺血管细胞(包括PA-SMCs)和导致大功能多样性缺氧反应。
肺ECs合成各种各样的旁分泌和内分泌因素,能够控制血管张力,通过有利于放松(即。PGI2也没有)或通过引起收缩(即。ET-1和5)基础条件下,针对不同的刺激或某些代理(61年]。有证据表明,背景通常释放任何有助于低肺血管张力normoxia [61年]。虽然有理论假说,缺氧的合成降低没有理由,缺乏后者似乎并不占急性人乳头状瘤病毒。相反,有证据表明,没有活动是为了增加调节急性肺泡低氧的肺血管收缩反应。受损没有生产,同时降低肺血管放松的能力,也倾向于过度的发生肺血管收缩(5]。缺乏任何合成也可能允许有丝分裂发生和血管壁内的各种细胞类型(扩散62年]。这也证实RhoA / Rho-associated激酶(岩石)通路也调节肺血管张力通过一氧化氮和没有独立机制(63年]。低氧诱导激活的岩石通道增加Ca2 +PA-SMCs收缩作用的敏感性,从而增加血管张力,导致持续的HPV和肺血管阻力增加63年- - - - - -65年]。
在成人,人乳头状瘤病毒至少有两个阶段:初始部分反应在几秒钟内开始,在几分钟内达到最大值,之后经过持续期(30 - 120分钟52,64年,66年]。这些阶段的监管,至少部分由不同的信号通路。第二阶段的人乳头状瘤病毒是影响电子商务功能通过改变释放血管活性的介质,如ET-1 PGI2也没有。在活的有机体内肺,神经体液的调节,红细胞和神经支配也可能影响响应。传感机制的低氧水平细胞一直讨论的话题。线粒体和NAHD / NADPH氧化酶类都被认为是氧传感器。活性氧的水平的变化被认为是重要的但缺乏协议是否信号是活性氧的增加或减少。低氧诱导因子1 (HIF-1)和HIF-2 transactivate共同和不同的下游靶基因的数量显然是候选人解释变异性的肺血管慢性缺氧反应(67年]。除了不同的下游目标,α-subunit表达水平也在细胞和组织的变化而变化。最近,两个老鼠菌株比较,F344和WKY,在他们对慢性缺氧的反应不同,凸显了锌在几个细胞类型的重要性,特别PA-SMCs,在调节肺血管内稳态(68年]。溶质的突变载体家庭39成员12基因(Slc39a12),编码锌转运体ZIP12,预测截断F344大鼠压力和不活跃的蛋白质。引入类似的突变WKY大鼠压力降低缺氧的反应。Upregulation ZIP12表达在肺部的人类生活在高海拔(和特发性肺动脉高血压患者(PAH))已经刺激ZIP12寻找小分子抑制剂,以及感兴趣的细胞内的作用不稳定锌在肺血管疾病。
从动态到不适应的肺血管重塑
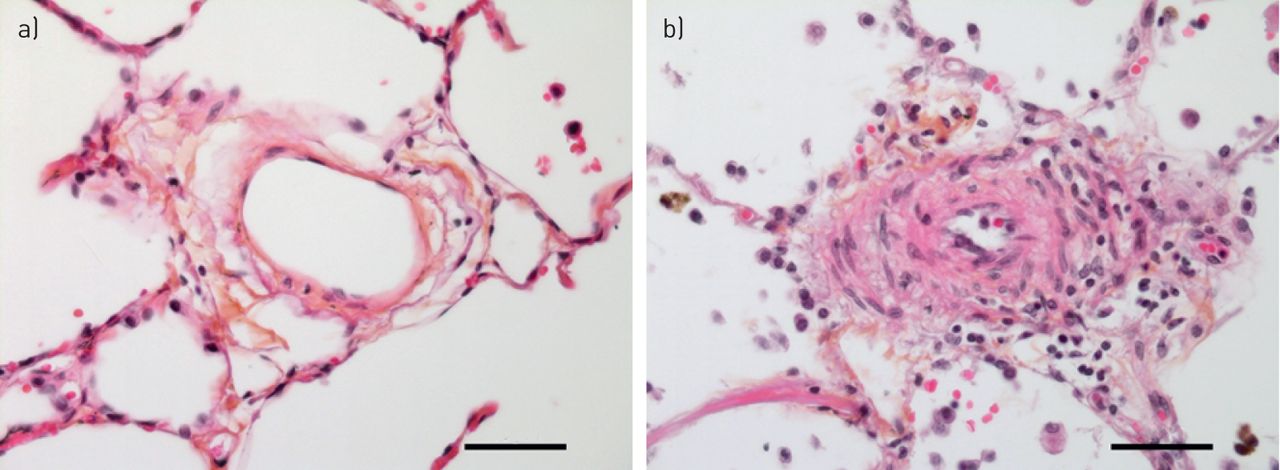
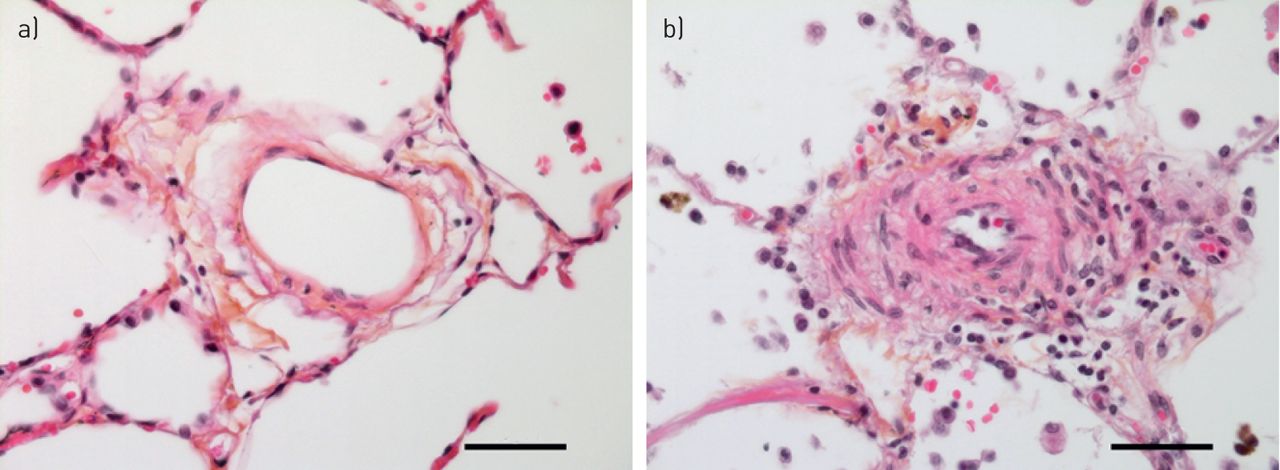
小循环系统响应肺泡低氧通过增加肺血管阻力和HPV负责最初的肺动脉压力上升(4,52,53]。然而,持续暴露于低氧,其他机制驱动的结构性变化阻力血管造成的压力升高,从而解释了为什么2或3周后缺氧呼吸100%的氧气反应。值得注意的是,肺压力随时间逐渐减少他们再次暴露在正常氧环境中,这表明慢性缺氧引起的肺血管重塑是可逆的。从哺乳动物肺组织学检查,包括老鼠,牛和人类暴露慢性缺氧,证明肺小动脉的结构改造69年]。所有层的血管壁参与改造,包括成纤维细胞,但血管慢性缺氧反应的标志是muscularisation增加肌肉的远端血管扩展到以前non-muscularised小动脉(图2)。人乳头状瘤病毒的相对贡献和血管重塑慢性低氧诱导的PH值是辩论的主题70年]。组织学研究老鼠比赛改造的程度缩小血管腔和提供了一个障碍。最近考虑在这场辩论是血管硬化,血管结构的变化导致刚度的增加。这刚度改变脉冲波的传播影响血管及其反射从分支点和右心然后感觉压力负荷增加的推进和反射波。血液动力学的部队和剪切应力的作用在缺氧的启动和维持肺血管重塑最近强调通过对老鼠的研究。肺动脉带不仅可以减少血液动力学的剪切应力和防止血管重塑的发展也改变咬合的病变和血管周的炎症Sugen缺氧模型(71年]。很容易推测,血液动力学的压力和血管闭塞病变可能形成一个恶性循环。如果是这样,这些观察结果表明,减少剪切应力机制,例如在血管舒张药疗法,不应该忽视在寻找治疗的PH值。
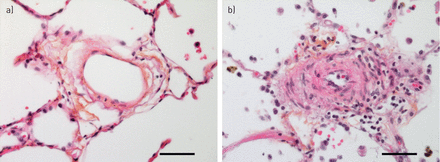
血管慢性缺氧反应。以前nonmuscularised小动脉(一个)显示“muscularisation”招聘的平滑肌细胞和myofibroblasts (b)。注意轻微炎性浸润(主船的右侧),伴随改造过程。规模50μm酒吧代表。
定义为肺动脉平均≥25 mmHg静止评估右心衰catheterisation, PH值可以出现在许多临床场景包括一些高死亡率(72年,73年),如下所示:多环芳烃(组1);PH值由于肺部疾病如慢性阻塞性肺病和肺气肿和/或缺氧(组3);慢性血栓栓塞肺动脉高压(CTEPH) (4);PH值与不清楚多因子的机制,如血液学的障碍(5)组(74年]。在所有这些PH值子组,肺内皮功能障碍导致肺血管重塑(75年,76年]。特别是,主要在肺血管内皮功能改变在PAH,包括等:1)从静止状态没有粘附能力和粘附能力激活状态(77年];2)一个异常的前导和apoptosis-resistant表型(78年];3)炎性表型的特点是过度释放等关键细胞因子和趋化因子白介素(IL) 1α,IL - 6,引发、IL - 12和趋化因子(碳碳主题)配体2 (CCL2) /单核细胞趋化蛋白1 (MCP-1) [77年];和4)过度生产和分泌各种关键生长因子包括纤维母细胞生长因子2 (FGF-2) [78年,79年),血管紧张素II (Ang) [80年和瘦素81年- - - - - -83年]。额外的洞察改变肺EC表型和血管细胞(内皮沟通与居民即。PA-SMCs myofibroblasts)和免疫细胞更好地理解多环芳烃发病机制的先决条件,可能导致新的治疗策略。
最近,内皮沟通与周报告,一个关键类型的祖细胞,是周围发现pre-capillary动脉,毛细血管和post-capillary小静脉。他们还占据了战略地位在血液循环和间隙空间之间的接口,和接近ECs PA-SMCs。Ricardet al。(84年)展示了过度的周皮细胞覆盖率(增加2 - 3倍)在人类的多环芳烃,远端肺动脉的现象,导致肺血管重塑的smooth-muscle-like细胞。周血管发展的中央监管机构,稳定和成熟,以及调制改造的影响:1)电子商务增长,增殖,分化和迁移;2)PA-SMC收缩;3)免疫细胞功能;4)作为祖细胞和可能在SMC-like细胞和树突状细胞分化。
破译调节肺血管重塑的细胞内途径为PH值的精确分子靶向治疗的新策略(85年]。例如,一个现在可以选择特定的和非特异性ET-1受体,或合成的刺激或抑制降解的关键细胞内第二信使,环鸟苷酸(cGMP),改变肺血管病变的州的语气。虽然预后仍然贫困,这些额外的途径可能代表创新工具在未来治疗PH值。
PH值由于肺部疾病,如慢性阻塞性肺病、肺血管损伤是非常普遍和大约40%的全球倡议对慢性阻塞性肺疾病患者(黄金)阶段4 COPD患者和27%的黄金阶段3显示增加慢性阻塞性肺病肺动脉平均(86年]。患者没有PH值在休息时,很大一部分开发它在运动(87年,88年]。这些发现同意频繁的观察组织学变化,主要包括内膜的增生,在小肺动脉终末期疾病患者接受肺移植(5),即使在轻度到中度疾病患者(6,89年]。除了血管重塑、血管特异表达的语气是一个主要因素在慢性阻塞性肺病PH值:以挪士的表达(90年),国网公司(16)和PGI2合酶(36)在慢性阻塞性肺病患者的肺动脉减少,从而减少他们的血管舒张药和抗增殖能力(图3)。
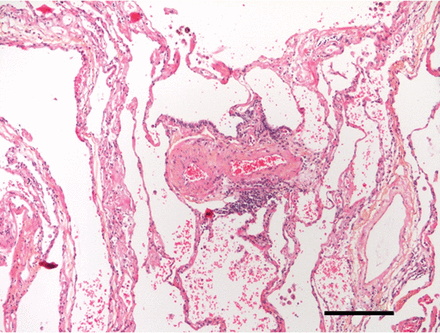
肺动脉在慢性阻塞性肺疾病患者。船舶在两个分支点(如图所示)提供了重要的内层的纤维化。注意轻微血管周的渗透(主下动脉)。周围的肺实质显示气性改造。200μm比例尺表示。
与这一背景下,有趣的是,一些广泛的肺气肿患者出现严重的PH值(91年肺气肿的破坏之间的],显示一个链接肺实质和肺血管损伤的严重程度。事实上,在移植肺的慢性阻塞性肺病病人肺移植,肺气肿的严重程度明显高于在这些患者PH值(92年]。因此,血管重塑的结合,内皮功能障碍和实质错乱可能解释的发展严重的PH值在COPD患者的一个子集93年]。
在慢性阻塞性肺病肺动脉内皮代表了一个有趣的治疗目标。在实验模型研究表明,针对NO-cGMP信号通路与国网公司刺激器(16]或phosphodiesterase-5 (PDE5抑制剂)(94年,95年)防止PH值的发展,肺血管重塑和右心室肥大。有趣的是,国网公司刺激器和PDE5抑制剂也减毒肺气肿的发展(16,95年),这种效果与中性粒细胞的数量的减少和肺组织肺泡巨噬细胞,以及减少白细胞粘附[16]。总的来说,这些发现表明,治疗干预NO-cGMP信号通路产生双重影响:1)放松和最终衰减通过依赖蛋白激酶G细胞增殖的平滑肌细胞机制;2)减少白细胞粘附在ECs,可能通过下调P-selectin [96年]。
这些结果取得了使用“预防性实验设计”,但它仍然显示为治疗干预在多大程度上产生同样的效果时肺损伤已经建立(即。使用“治疗设计”)。事实上,治疗慢性阻塞性肺病患者PH值使用PDE5抑制剂或国网公司刺激器一直无力改善运动耐量(86年]。然而,宽容也许不是最适当的代理运动治疗对血管内皮细胞的影响。进一步的实验和临床研究解决干扰内皮的治疗干预措施的效果才能阐明这种方法在慢性阻塞性肺病的重要性。
这样的PH值由于血液疾病,镰状细胞病(SCD) [97年- - - - - -101年],溶血性贫血和血管病变之间的机械连接已被广泛研究的主题在临床前动物模型和血管研究的病人,以及在大型人类群组研究[99年,102年,103年]。血管内溶血释放出游离血红蛋白(Hb)不能清除的等离子体,产生活性氧,影响氧化还原平衡,导致增殖系统和肺血管病变(103年,104年]。它最近指出,产品发布在红细胞溶血可视为危险分子模式(抑制)或红细胞抑制(eDAMPs) [105年]。SCD支持这些研究更一般的病理作用的血管内溶血和Hb游离在各种人类疾病和输血医学(106年]。
生物因素评估肺动脉内皮功能障碍
根据所发挥的关键作用肺动脉内皮细胞在急性和慢性肺部疾病,识别生物因素评估(来)函数的字段是必需的;然而,这是一个真正的挑战为临床医师和研究人员。事实上,广阔的升值的内皮细胞的许多功能需要评估大型面板的分子在血液循环内皮来源(表1前)和全面的评估和验证在大型和特征明显群病人在常规临床使用。这些电子商务产品可能包括等:措施没有生物学,microrna循环,细胞因子,趋化因子,循环粘附分子、生长因子和监管者的血栓形成,以及内皮损伤的标记(即。超然的成熟ECs或派生内皮微粒(emp))和内皮修复(即。循环内皮祖细胞的数量和功能特征)。
除了血管活性的介质的变化,慢性阻塞性肺病患者存在肺内皮结构损伤。地区之间细胞剥蚀和超然的ECs已被证明在慢性阻塞性肺病肺动脉科目(107年]。内皮损伤的慢性阻塞性肺病的存在也证明了增加员工的数量(108年,109年]。循环是员工可能源自EC凋亡或激活,可以识别的表达CD31 CD62E标记,分别。循环是员工的数量成反比的用力呼气量1 s [109年]。有趣的是,尽管CD31 EMPs(凋亡)已经轻微的慢性阻塞性肺病患者显著升高,CD62E EMPs(激活)只有在严重疾病患者增加。循环是员工的数量进一步增加在恶化事件(110年)也与肺气肿的严重程度(109年),指出实质破坏和内皮损伤之间的连接。此外,在慢性阻塞性肺病,循环EMPs也与系统性动脉的血管功能。增加循环是员工数量与内皮功能受损和刚度增加系统性动脉(111年]。慢性阻塞性肺病患者也显示骨髓衍生的数量减少循环内皮祖细胞(epc) [112年- - - - - -115年],它扮演着一个关键角色,修复受损的内皮细胞和内皮功能密切相关116年]。研究在过去年作出了巨大努力,试图识别、定义和特征与表型细胞群,内皮祖细胞的生化、分子和功能特性117年,118年]。内皮祖细胞被认为是一个珍贵的源主管ECs生成功能,适合各种临床应用。然而,这些祖细胞的缺乏和所涉及的技术难题在体外增长代表一个重要限制其使用。有趣的是,最近的研究导致造血的识别组织(血液、骨髓和脐带血)非常罕见的内皮祖细胞不是源自骨髓。这些内皮细胞克隆形成(ECFCs)或“晚ECs产物”能产生大量的子代的表型和功能能力成熟的ECs在体外并维持在活的有机体内angiogenetic过程(119年,120年]。这样的内皮祖细胞也存在在血管壁的水平,特别是在血管内皮内膜(117年,118年]。这种方法可以很容易地扩展到循环内皮祖细胞的研究,可以促进和改善内皮祖细胞的功能描述在不同病理条件下,尤其是在各种肺部疾病。
结论
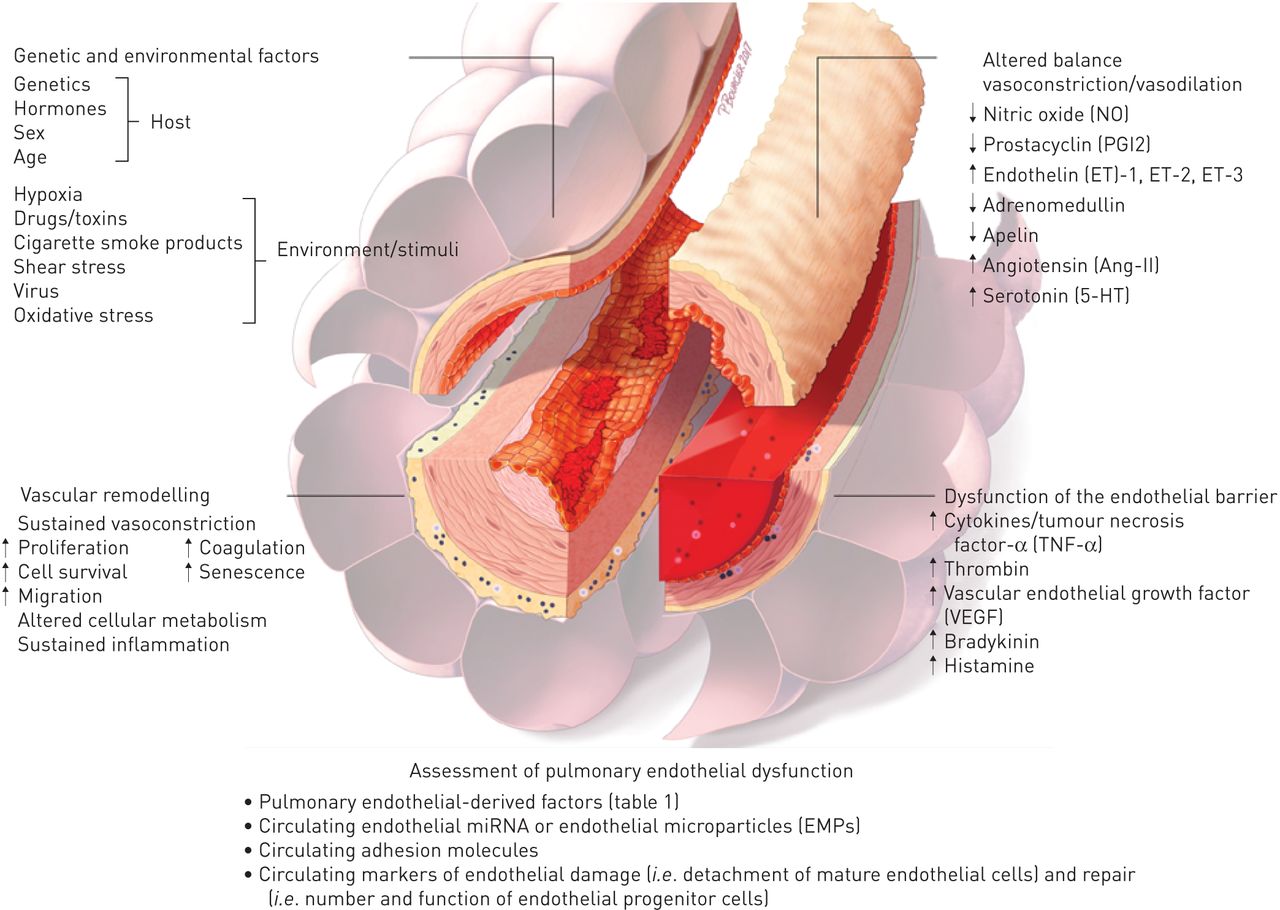
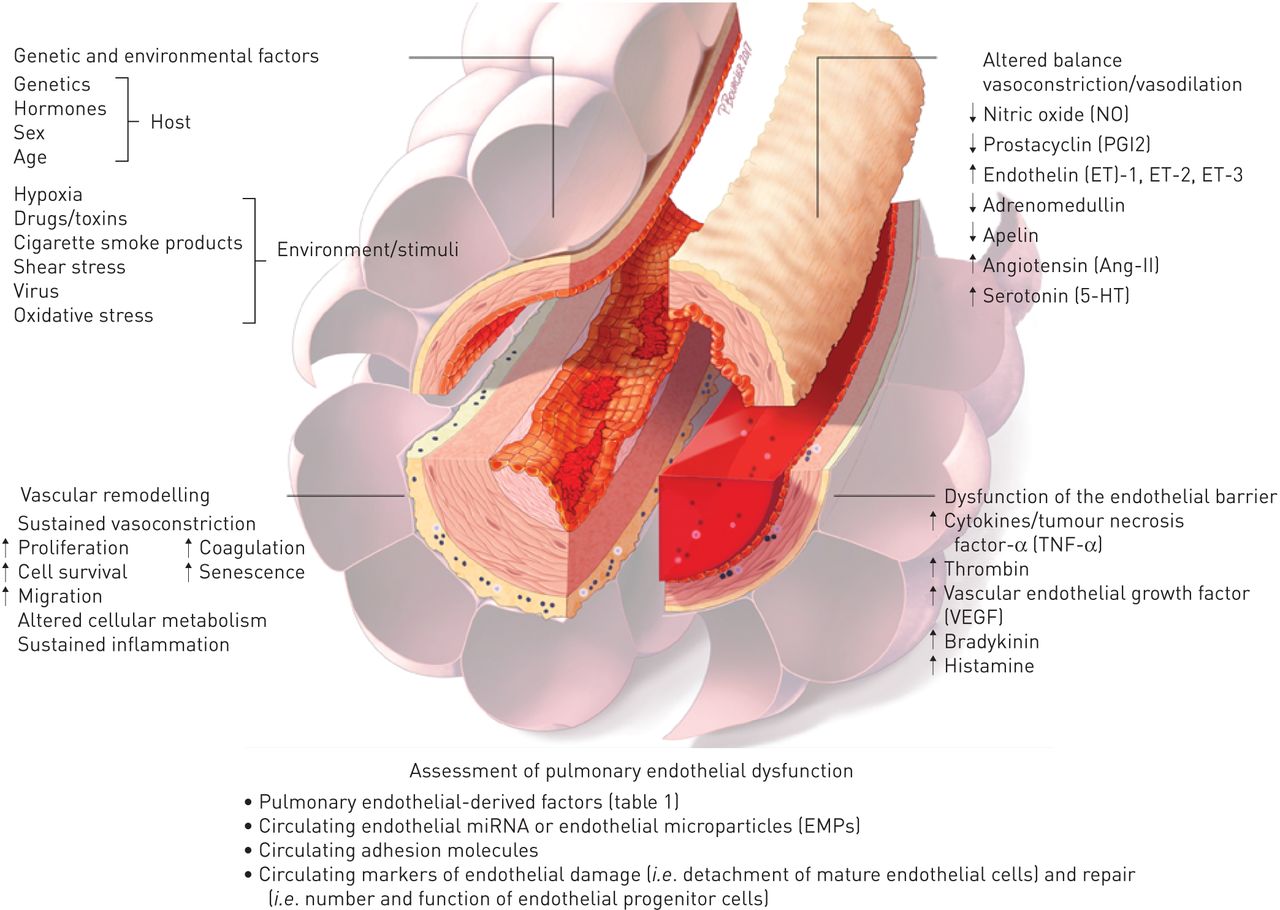
总之,现在好了,肺血管内皮细胞参与大多数(如果不是全部的话)急性和慢性肺部疾病,这些疾病过程的主要决定因素或附带损害的受害者(图4)。尽管已经取得了相当大的进展的理解肺EC(来)功能,触发,机制和后果内皮功能紊乱的肺部疾病,急性和慢性,仍然没有被完全了解。此外,还需要进一步努力,阐明肺内皮的显著的异质性在空间和时间,在结构和功能,在健康和疾病。这些关键方面的分子基础的了解将有助于发现新的疾病生物标记物和/或新药保持体内平衡的反应伤害和疾病,减少内皮生物医学中的“实验到临床”的差距。
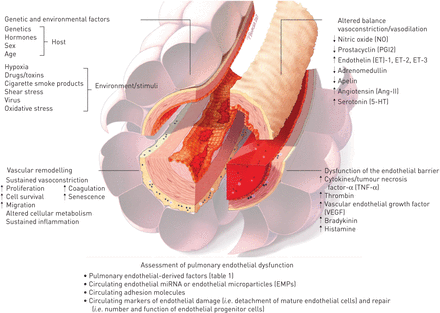
原理概述肺内皮功能障碍的主要特征。
确认
欧洲呼吸学会的组织者(ERS)研究研讨会谢谢188bet官网地址教员,椅子,人的科学活动助理和所有的参与者:a . Abd Al-Aziz(埃及开罗),e·巴雷罗(西班牙巴塞罗那),h . Bendjenana(法国巴黎),即Benhamou-Tarallo(法国巴黎),o·伯纳德(法国巴黎),即Bonovolias(希腊塞萨洛尼基),j . Bordenave(法国巴黎),即Campean(媒体、罗马尼亚),p . Dybantsa(法国巴黎),美国Fairhall(英国索尔兹伯里),m·法尔顿(德国柏林),p·费雷拉(波尔图,葡萄牙),n Frossard (Illkirch、法国),c·吉伯特(法国波尔多),b . Jugg(英国索尔兹伯里),即Khachatryan(亚美尼亚埃里温),m . Kontic(贝尔格莱德,塞尔维亚),K.B. Kurakula(阿姆斯特丹,荷兰),A.K. Larsson-Callerfelt(瑞典Lund),大肠Letsiou(德国柏林),n Mesropyan(亚美尼亚埃里温)诉米歇尔(法国波尔多),r·帕尔马(巴西圣保罗),c .表象(法国巴黎),e·罗西(法国巴黎),k .翅果(希腊雅典),d·桑托斯里贝罗(布鲁塞尔,比利时),美国Schmitt-Grohe(德国波恩),a关根身上(日本千叶)诉闷烧(西班牙巴塞罗那),r . Szulcek(阿姆斯特丹,荷兰),y谷口(神户,日本),r . Thuillet(法国巴黎),l .涂(法国巴黎),o .发信息给鲍思高堂另Ceide(西班牙巴塞罗那),j . Weatherald(卡尔加里,加拿大)。
脚注
利益冲突:正当巴贝拉已收到个人费用(按照顾问委员会,咨询和发言)从拜耳Actelion股价和葛兰素史克公司,从拜耳和机构资助,Actelion股价,葛兰素史克和辉瑞公司在提交工作。p·巴奇收到了诺华的个人费用讲座,Permamed AG) MSD Mundi制药公司,咨询公司和个人费用从拜耳公司,在提交工作。p . Dorfmuller已收到个人费用从默沙东公司Actelion股价,在提交工作。M.T. Gladwin有nitrite-related专利广泛和版税支付相关肺动脉高压。m·亨伯特与制药公司的关系包括Actelion股价,拜耳,葛兰素史克、诺华制药和辉瑞。除了试验涉及到这些公司的调查员,关系包括咨询服务和科学顾问委员会的成员。o·桑切斯已经收到BMS的个人费用,资助,个人费用和非金融支持默沙东,Actelion股价和拜耳,赠款从波尔图和第一三共制药和个人费用和非金融基耶西的支持,在提交工作。l . Savale已收到拨款,个人费用和非金融Actelion股价的支持,默沙东公司,拜耳和赠款和非金融葛兰素史克公司的支持,在提交工作。核磁共振威尔金斯专利ZIP12作为治疗目标待定。
支持声明:研究研讨会是由欧洲呼吸协会(人)与金融支持无限制的拨款从协会de矫揉造作的en Physiopathologie188bet官网地址 Respiratoire, Elivie,葛兰素史克,Miltenyi研究,Oxyvie Teva, Vivisol。资金信息,本文已沉积的Crossref资助者注册表。
- 收到了2017年4月10日。
- 接受2018年2月3日。
- 版权©2018人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}