





摘要GydF4y2Ba
p38促分裂原活化蛋白激酶(MAPK)途径在慢性阻塞性肺疾病(COPD)被上调。迄今为止,双标记以识别磷酸化(磷酸)p38蛋白的细胞类型特异性存在还没有被执行。GydF4y2Ba
磷酸化p38 MAPK在20名COPD患者,12名吸烟者和使用免疫组织化学12名不吸烟者的肺组织中的多种细胞类型中进行定量。成对的血液和痰嗜中性粒细胞(来自七个受试者COPD),和CD8细胞和上皮细胞(来自三个受试者COPD)用一个p38蛋白抑制剂培养。上清液肿瘤坏死因子α和CXCL8水平通过ELISA进行分析。痰血中性粒细胞涂片进行了分析磷酸化p38 MAPK。GydF4y2Ba
COPD肺中支气管上皮细胞、巨噬细胞和CD20+、CD8+淋巴细胞中磷-p38 MAPK升高。所有患者组的痰液和肺组织中性粒细胞均无磷-p38。p38 MAPK抑制剂SB100对COPD肺CD8细胞和气道上皮的促炎介质释放作用减弱,但对COPD痰中性粒细胞无影响。GydF4y2Ba
我们的数据显示了p38 MAPK抑制在肺中的细胞特异性抗炎作用。GydF4y2Ba
介绍GydF4y2Ba
慢性阻塞性肺疾病(COPD)以可逆性差的气流阻塞为特征。也有进行性气道炎症的证据[GydF4y2Ba1GydF4y2Ba]。糖皮质激素是COPD最常用的抗炎药物,但对气道炎症和疾病进展的影响有限[GydF4y2Ba2GydF4y2Ba,GydF4y2Ba3GydF4y2Ba]。需要针对慢性阻塞性肺病炎症的新的治疗方法。GydF4y2Ba
p38促分裂原活化蛋白激酶(MAPK)的细胞内信号传导途径是由多种细胞外刺激,包括促炎细胞因子和Toll样受体(TLR)激动剂激活GydF4y2Ba4GydF4y2Ba,GydF4y2Ba五GydF4y2Ba]。p38蛋白激活引起的基因的一个子集的启动子区域内的组蛋白修饰;这个增加的辅助功能转录因子如核因子κB到这些区域增强炎性基因表达GydF4y2Ba6GydF4y2Ba]. 另外,p38 MAPK通过稳定mRNAs在转录后发挥作用,并促进蛋白质翻译[GydF4y2Ba7GydF4y2Ba]。p38蛋白抑制剂减少由肺泡巨噬细胞的细胞因子产生[GydF4y2Ba8GydF4y2Ba-GydF4y2Ba11GydF4y2Ba],并正在进行治疗慢性阻塞性肺病的临床研究[GydF4y2Ba12GydF4y2Ba,GydF4y2Ba13GydF4y2Ba]。GydF4y2Ba
COPD患者肺部炎性细胞数量增多,包括淋巴细胞、巨噬细胞和中性粒细胞[GydF4y2Ba1GydF4y2Ba]. COPD患者的肺淋巴滤泡数量也增加了[GydF4y2Ba1GydF4y2Ba],可能作为抗原提呈位点,促进自身免疫过程[GydF4y2Ba14GydF4y2Ba]. 右GydF4y2Ba恩达GydF4y2Ba等。GydF4y2Ba[GydF4y2Ba15GydF4y2Ba]使用单标签免疫组织化学证实在COPD患者的肺泡巨噬细胞与对照组相比激活p38蛋白的表达增加。在其他相关的肺细胞,例如上皮细胞,淋巴细胞和嗜中性粒细胞活化的p38蛋白的特异性表达,还没有描述过。此外,尽管p38蛋白激酶抑制剂对细胞因子产生的抗炎作用从COPD肺泡巨噬细胞是有据可查的[GydF4y2Ba8GydF4y2Ba-GydF4y2Ba11GydF4y2Ba],关于该类药物对COPD患者肺内其他相关免疫细胞类型的影响的数据很少。GydF4y2Ba
p38 MAPK有四种亚型,分别由不同的基因编码;p38α、p38βp38δp38γ。这些亚型的表达因组织和细胞类型而异[GydF4y2Ba16GydF4y2Ba]。p38α和p38β亚型扮演主要角色在免疫细胞的激活,所以大多数p38 MAPK抑制剂用于治疗炎症都是针对这些亚型,以避免不必要的生理效应通过p38γ和p38δ抑制。它已被证明在肾小球肾炎,p38α是最高度表示对肾脏浸润白细胞(同种型GydF4y2Ba17GydF4y2Ba),但也有证据表明p38β和p38γ同种型表达在细胞结构类型。p38 MAPK亚型在COPD患者肺组织中的表达水平尚未得到定量研究;这将确定与COPD病理生理相关的亚型。GydF4y2Ba
本研究的目的如下:1)采用双标记免疫荧光法,比较活化的p38 MAPK在COPD肺中的细胞特异性表达;2)探讨p38 MAPK抑制对慢性阻塞性肺疾病(COPD)分离的中性粒细胞、上皮细胞和淋巴细胞产生细胞因子的抗炎作用;3)与对照组比较,探讨COPD患者肺组织中p38 MAPK亚型的表达情况。GydF4y2Ba
方法GydF4y2Ba
研究对象GydF4y2Ba
我们招募了接受手术切除的疑似或确诊肺癌患者。根据全球慢性阻塞性肺疾病行动指南,既往诊断为慢性阻塞性肺疾病的患者[GydF4y2Ba18GydF4y2Ba]具有至少一个10香烟包装年的历史和在1秒(FEV定义为用力呼气量气流阻塞GydF4y2Ba1GydF4y2Ba)<80%,FEVGydF4y2Ba1GydF4y2Ba/用力肺活量(FVC)<70%被招募。对照由> 10包年的吸烟史和肺功能正常和终生不吸烟者。有些控件有FEV的证据GydF4y2Ba1GydF4y2Ba/ FVC <由于70%至肿瘤阻塞。在另一项研究中,COPD患者,吸烟和不吸烟的诱导痰和COPD患者的隔离中性粒细胞的工作,被招募。此外,三名COPD患者招募支气管镜气道上皮细胞的分离。人口统计数据示于GydF4y2Ba表1GydF4y2Ba。所有患者签署知情同意书。该研究经当地伦理委员会。GydF4y2Ba
P38的mRNA水平分析GydF4y2Ba
p38α水平、p38βp38δ和p38γ衡量qPCR在慢性阻塞性肺病,吸烟和不吸烟的病人。表达水平被归一化到GAPDH进行分析(在线补充材料中有描述)。GydF4y2Ba
免疫组织化学GydF4y2Ba
从肺尽可能远离肿瘤尽可能的面积得到的组织块,并如前所述进行处理[GydF4y2Ba19GydF4y2Ba]。块使用抗磷酸化p38 MAPK初级抗体标记。嗜中性弹性蛋白酶,CD20,CD8或CD4:用磷酸化p38双标签免疫荧光用下列一抗中的一个执行。的方法和抗体的进一步细节在网上补充材料说明。GydF4y2Ba
图像分析GydF4y2Ba
定量分析炎性卵泡和上皮下的磷酸化p38+CD20+、CD8+和CD4+细胞百分率。同时还测定了磷酸化p38+中性粒细胞、小气道上皮细胞和巨噬细胞(通过形态学鉴定)的总数。对于双标记图像,采集同一区域的荧光图像并进行数字合并以确定磷酸化p38阳性细胞。数码显微照片是通过使用尼康Eclipse 80i显微镜(尼康英国有限公司,英国泰晤士河上的金斯敦)获得的,该显微镜配备了一个可改变图像的数码相机(媒体控制论,英国马洛)和ImagePro Plus 5.1软件(媒体控制论)。使用ImagePro Plus 5.1软件对细胞计数、卵泡面积、上皮和上皮下长度进行定量分析。细胞计数标准化为每毫米阳性细胞数GydF4y2Ba2GydF4y2Ba感兴趣的区域。GydF4y2Ba
细胞培养GydF4y2Ba
中性粒细胞GydF4y2Ba
分离血液和痰嗜中性粒细胞(在在线补充材料中提供的细胞分离细节;痰细胞计数以示出GydF4y2Ba表2GydF4y2Ba)使用p38α和p38βMAPK选择性抑制剂SB100(葛兰素史克公司,史蒂芬奇,英国)预处理,在添加脂多糖(LPS)(100 ng·mL)之前使用最终浓度10–1000 nMGydF4y2Ba-1GydF4y2Ba) (Sigma Aldrich, Poole, UK) 24小时,LPS 100 ng·mLGydF4y2Ba-1GydF4y2Ba常用于嗜中性粒细胞刺激的GydF4y2Ba20GydF4y2Ba],因此我们确认这是对细胞因子的产生(图S1A)次优浓度。除去上清液,并储存于-80℃下用于细胞因子分析。剩余的细胞被去除并离心(涓GR4i离心机;赛默飞世尔科技),在400×GydF4y2BaGGydF4y2Ba在4℃10分钟。细胞生存力和计数细胞离心涂片制备和免疫细胞化学分析之前4%多聚甲醛固定之前进行测定(参见方法在线补充材料)。为了测定细胞活力,台盼蓝排除之前和SB100(1000 nm)的治疗后评价。为了通过末端脱氧核苷酸转移酶dUTP缺口末端标记(TUNEL)和分别核形态(在线补充材料所述)进行了评估之前和SB100(1000 nm)的治疗,DNA片段化和冷凝的核的存在之后确定细胞凋亡的程度。细胞凋亡是通过检查核叶(早期凋亡)和收缩或核(晚期凋亡)的碎片之间的桥梁染色质的消失确定。出现正常,以及早期中性粒细胞和晚期凋亡中性粒细胞百分比,由总共300个嗜中性粒细胞计数评估。GydF4y2Ba
CD8细胞GydF4y2Ba
分离肺CD8细胞(隔离协议在网上补充材料详述)以5×10接种GydF4y2Ba4GydF4y2Ba每孔细胞用SB100(0.1-1000 nM)的前处理之前。然后将细胞刺激24小时,用白细胞介素(IL)-12(10毫微克·毫升GydF4y2Ba-1GydF4y2Ba;派普泰克公司,伦敦,英国)和IL-18(10毫微克·毫升GydF4y2Ba-1GydF4y2Ba;派普泰克公司制)收获上清液用于测定前24小时,干扰素(IFN)-γ通过ELISA(eBioscience公司,哈特菲尔德,UK)。这些细胞因子的浓度通过在血液中分离CD8细胞初步浓度 - 反应实验来确定;次优加上p38磷酸IFN-γ生产中观察到(图S1b中和c)。GydF4y2Ba
上皮细胞GydF4y2Ba
上皮细胞的分离在在线补充材料进行说明。上皮细胞在96孔板中培养,直到80%汇合。将细胞用SB100(1μM)1个小时进行预处理与聚肌苷培养之前:胞苷酸(聚I:C)(10微克·毫升GydF4y2Ba-1GydF4y2Ba;Invivogen,San Diego,CA,USA)培养24小时。收集上清液,用ELISA(英国阿宾顿R&D Systems)检测趋化因子(C-C motif)配体5(CCL5)、CXCL8和IL-6。GydF4y2Ba
细胞因子测量GydF4y2Ba
肿瘤坏死因子(TNF)-α的上清液的水平,CXCL8,IFN-γ,IL-6和CCL5是根据制造商的说明书,使用ELISA确定。检测的对TNF-α,CXCL8的下限,IL-6和CCL5分别为15.6皮克·毫升GydF4y2Ba-1GydF4y2Ba32.5 pg·毫升GydF4y2Ba-1GydF4y2Ba9.375微克·毫升GydF4y2Ba-1GydF4y2Ba和15.6皮克·毫升GydF4y2Ba-1GydF4y2Ba,分别为(R&d系统)。检测的IFN-γ的下限为7.8皮克·毫升GydF4y2Ba-1GydF4y2Ba(eBioscience公司,哈特菲尔德,英国)。GydF4y2Ba
统计分析GydF4y2Ba
使用Kolmogorov-Smirnov检验评估正常值。COPD患者、吸烟者和非吸烟者之间的比较采用单因素方差分析(one-way ANOVA),然后采用Bonferroni’s后测法对参数化分布的免疫组化数据进行比较。PCR数据采用组间比较的非参数方差分析(Kruskal-Wallis检验)和组内分析的重复测量方差分析(Friedman检验),然后是Dunn的后检验。采用配对t检验比较SB100对细胞培养的影响。使用GraphPad InStat软件3.06版(GraphPad software, Inc., San Diego, CA, USA)进行分析。GydF4y2Ba
结果GydF4y2Ba
在肺组织中的p38同种型的表达GydF4y2Ba
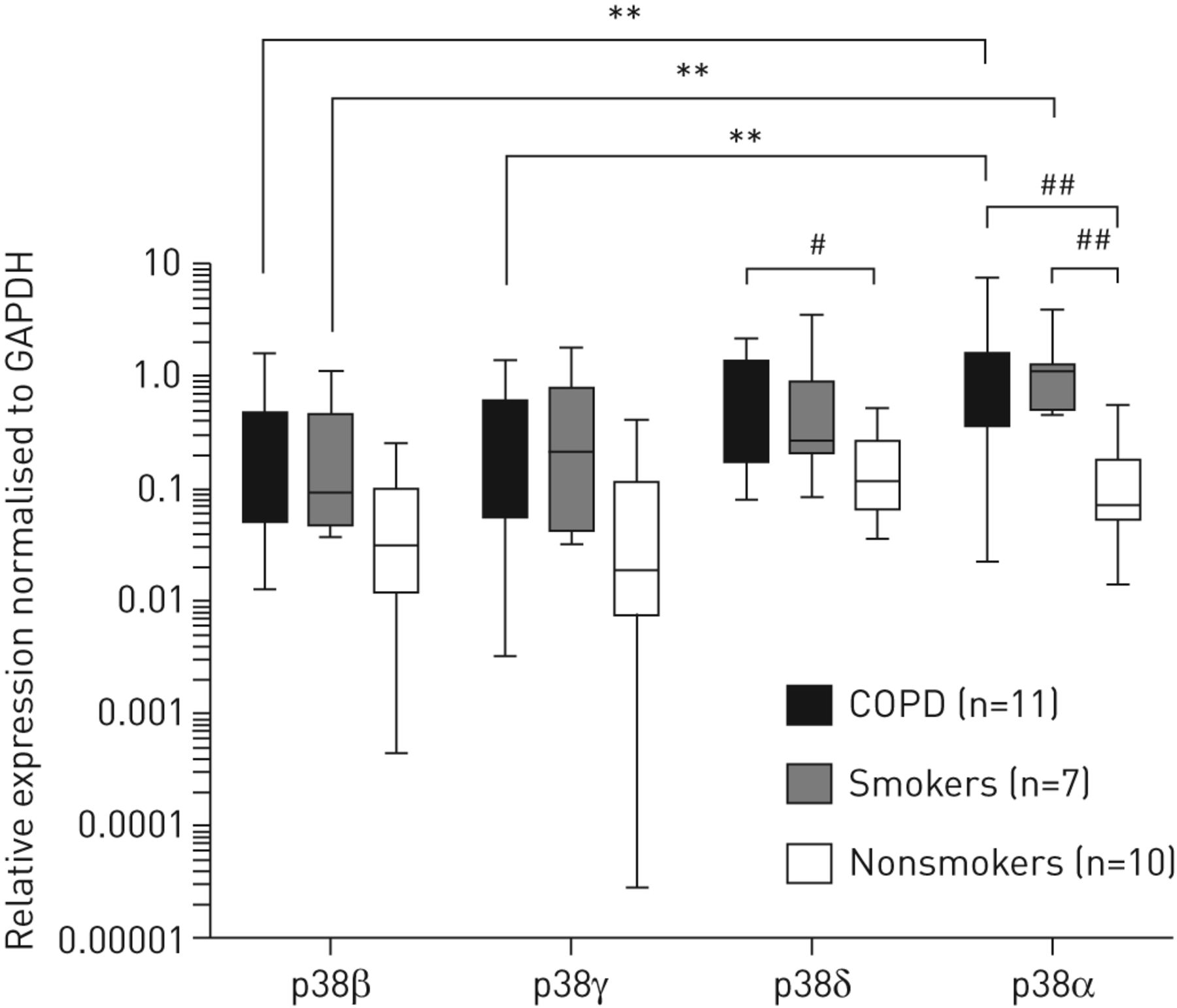
p38的α,β,γ的基因表达水平和δ分别通过qPCR在RNA分析了来自COPD萃取(N = 11),吸烟(N = 7)和不吸烟的(N = 10)的肺组织(GydF4y2Ba图。1GydF4y2Ba). COPD患者肺组织中α和δ亚型与不吸烟组相比显著升高(p<0.01和p<0.05),而p38α在吸烟组与不吸烟组相比也显著升高(p<0.01)。GydF4y2Ba

慢性阻塞性肺病(COPD)患者肺溶出物中p38亚型的表达,以及吸烟和非吸烟对照组中p38亚型的表达。p38β、γ、δ和α亚型分析qPCR和正常3 -磷酸甘油醛脱氢酶(GAPDH)表达不吸烟(n = 10),吸烟(n = 7)和慢性阻塞性肺病(n = 11)患者的肺溶解产物。数据以中位数(范围)表示。GydF4y2Ba#GydF4y2Ba:P <0.05;GydF4y2Ba# #GydF4y2Ba:P <0.01,组之间的差异达到统计学意义(不成对数据)。**:P <0.01,组内差达到统计学意义(成对数据)。GydF4y2Ba
p38α表达增加与p38β和p38γ相比在慢性阻塞性肺病患者(p < 0.01两种亚型),和p38α表达也与之相比,吸烟者p38β增加(p < 0.01)。在非吸烟者中,亚型的表达水平没有差异。GydF4y2Ba
淋巴细胞磷酸化p38 MAPKGydF4y2Ba
毛囊GydF4y2Ba
双标签免疫荧光显示,磷酸化p38 MAPK存在于所有的毛囊进行分析。GydF4y2Ba
b细胞GydF4y2Ba
Bonferroni多重比较试验表明,CD20 +磷酸化p38 +细胞的百分比是显著更高COPD患者与吸烟者和非吸烟者相比(P <0.001对于两个比较;平均86.6%,分别为44.1%和30.9%)。CD20 +磷酸化p38 +的数目也与不吸烟者(P <0.05)(比较吸烟者显著更大GydF4y2Ba无花果2GydF4y2Ba和GydF4y2Ba3GydF4y2Ba)。GydF4y2Ba
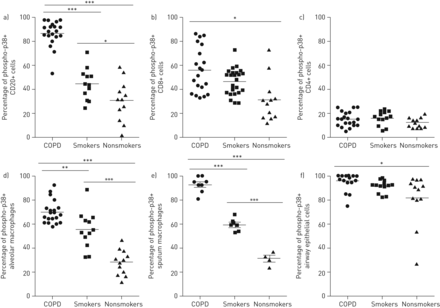
磷酸化(磷酸)的p38促分裂原活化蛋白激酶(MAPK)+ A)卵泡CD20 + B细胞,B)的平均百分比卵泡CD8 +细胞,c)中的卵泡的CD4 +细胞,d)肺组织巨噬细胞,E)痰巨噬细胞和F)小气道上皮细胞。慢性阻塞性肺病:慢性阻塞性肺疾病。患者组之间的差异对于每个小区类型:*:P <0.05;**:P <0.01;***:P <0.001。GydF4y2Ba
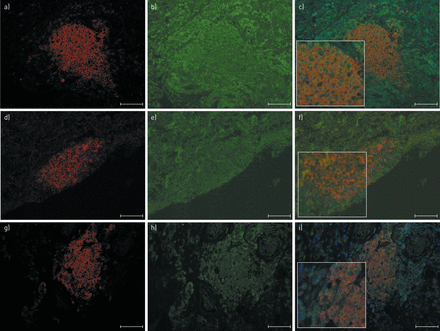
双标免疫荧光法检测人肺组织内炎性滤泡CD20+ b细胞中磷酸化(phospho-)p38丝裂原活化蛋白激酶(MAPK)的代表性图像。代表性图像来自a-c) 20例慢性阻塞性肺疾病患者,d-f) 12例吸烟者和g-i) 12例非吸烟者。用4′,6-二氨基-2-苯基吲哚(蓝色)对细胞核进行反染。利用Alexa-488标记的山羊抗小鼠二级抗体(red;a、d、g),利用Alexa 468标记的山羊抗兔二抗(绿色;(b, e, h).复合图像显示(c, f, i)。绿色/黄色荧光是由固有的荧光组织成分引起的,如弹性纤维和红细胞。通过形成红、绿、蓝通道的合成图像,可以将自发荧光与阳性荧光区分开来。在所有三个通道中都能看到自发荧光,因此呈现为三种颜色的融合。阳性荧光仅在一个通道可见,因此呈现为纯色。放大×200。 Scale bars=75 μm.
CD8细胞GydF4y2Ba
Bonferroni多重比较试验表明,CD8 +磷酸化p38 +细胞的百分比(分别平均56.2%和31.5%,)为显著更高COPD患者与非吸烟者(P <0.001)。还有向CD8 +磷酸化p38 +细胞在COPD与吸烟者(平均43.6%),并与不吸烟者(平均31.5%)相比,吸烟者相比数量增加的数值的趋势,但这种差异不是统计学显著(P> 0.05两者比较)(GydF4y2Ba无花果2 bGydF4y2Ba和GydF4y2Ba4GydF4y2Ba)。GydF4y2Ba
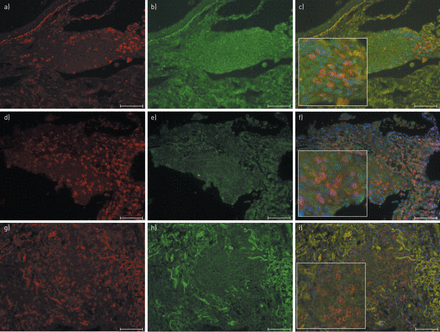
肺组织炎性滤泡内CD8+细胞磷酸化p38丝裂原活化蛋白激酶(MAPK)双重免疫荧光检测的代表性图像。显示了来自a–c)20名慢性阻塞性肺疾病患者、d–f)12名吸烟者和g–i)12名不吸烟者的代表性图像。细胞核用4′,6-二氨基-2-苯基吲哚(蓝色)复染。CD8+细胞用Alexa-488结合羊抗鼠二级抗体(红色;a,d和g)鉴定,磷酸p38-MAPK用Alexa 468结合羊抗兔二级抗体(绿色;b,e和h)检测。还显示了合成图像(c、f和i)。放大倍数×200。比例尺=75μm。GydF4y2Ba
CD4细胞GydF4y2Ba
所有患者组炎性滤泡内的磷酸化p38+ CD4+细胞数量均明显减少;典型地<20%的CD4+细胞对phospho-p38 MAPK呈阳性。从统计学上看,两组之间没有任何差异(GydF4y2Ba图。2CGydF4y2Ba)GydF4y2Ba
上皮下GydF4y2Ba
上皮下淋巴细胞的数量和上皮下淋巴细胞阳性磷酸化p38 MAPK的数量在所有三个病人组中相似(GydF4y2Ba表3GydF4y2Ba)。磷酸化p38 MAPK在皮下CD4 +淋巴细胞缺失。CD20 +和CD8 +磷酸化p38 +细胞数量分别显著降低与毛囊内磷酸化p38 +淋巴细胞(ANOVA P <0.0001两种细胞类型的比较)进行比较。GydF4y2Ba
巨噬细胞磷酸化p38 MAPKGydF4y2Ba
肺泡巨噬细胞GydF4y2Ba
磷酸化p38 +肺泡巨噬细胞的百分比在与吸烟者和非吸烟者(平均70.0%相比,COPD患者显著较大,分别为56.4%和28.5%,Bonferroni多重比较试验COPDGydF4y2Ba与GydF4y2Ba吸烟者,P <0.01;慢性阻塞性肺病GydF4y2Ba与GydF4y2Ba非吸烟者,P <0.001),而在吸烟者不吸烟者(Bonferroni多重比较检验P <0.001相比)(GydF4y2Ba无花果2 dGydF4y2Ba和GydF4y2Ba5A-CGydF4y2Ba)。GydF4y2Ba
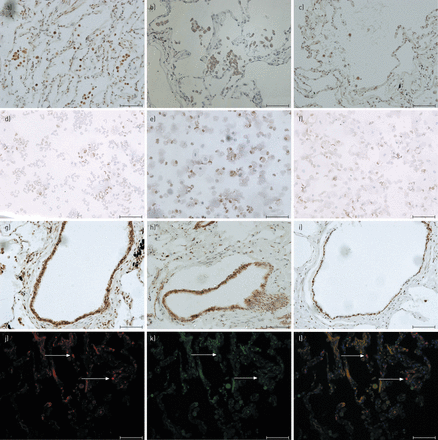
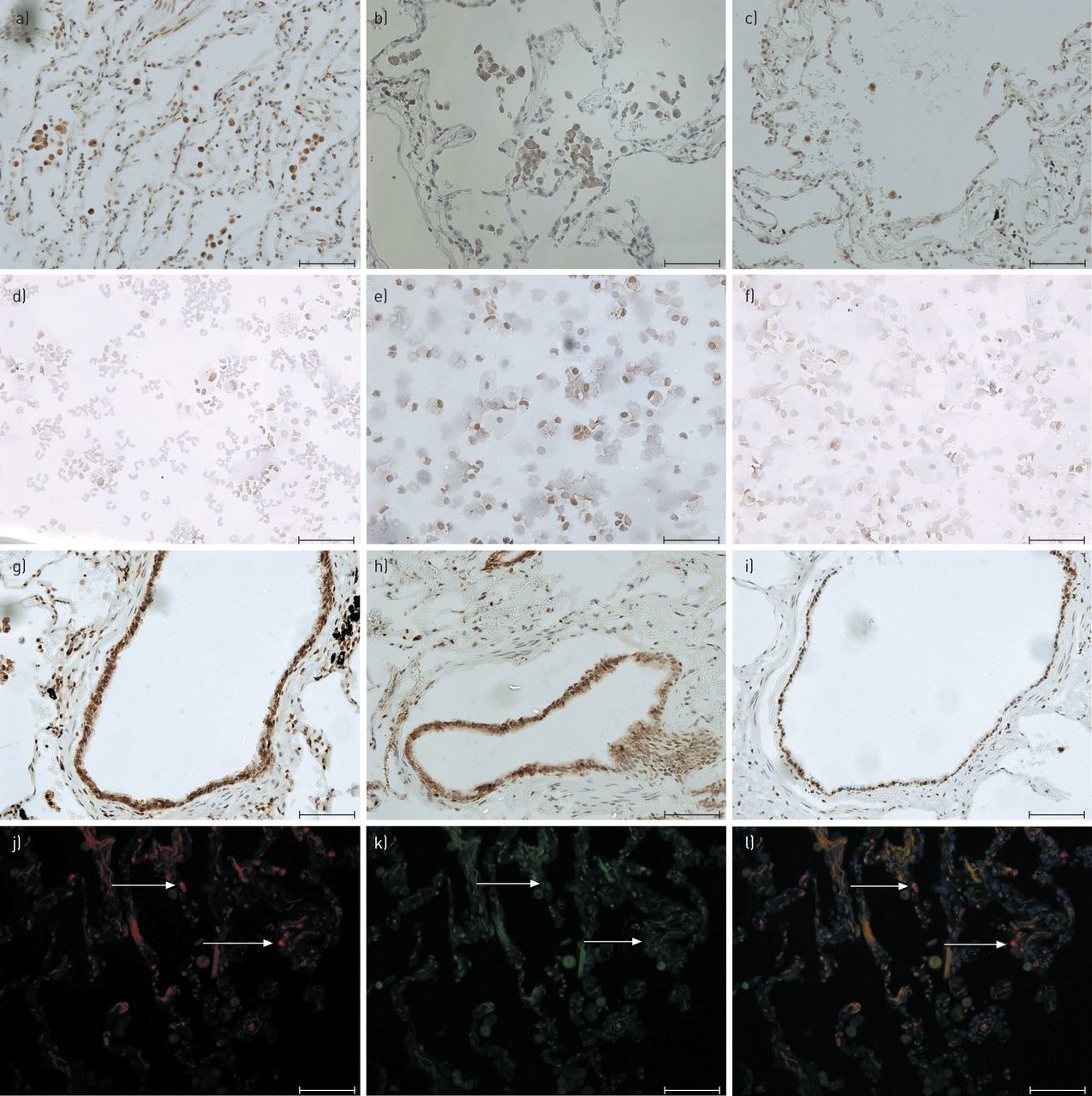
代表性图像的免疫组织化学和双标签免疫荧光检测在人肺组织和细胞离心涂片痰磷酸(磷酸)p38促分裂原活化蛋白激酶(MAPK)的。为(A-I)的免疫细胞化学和从(N = 20; d:A,G和J N = 8)(J-L)免疫荧光组织分析显示代表图像慢性阻塞性肺疾病的患者,(B,H和K:N = 12; E:N = 6)吸烟者和(c,i和L:N = 12; F:N = 4)不吸烟者被示出。细胞核用任一Mayer氏苏木精复染(蓝色; A-I)或4',6-二脒基-2-苯基吲哚(蓝色; J-1)。对于免疫组织化学分析磷酸化p38 MAPK的表达是使用3,3'- diaminobenzadine检测(棕色; A-I)。对于双标签免疫荧光(J-L)被使用的Alexa-488缀合的山羊抗小鼠第二抗体鉴定肺组织嗜中性粒细胞(红色; j)的和磷酸化p38 MAPK使用的Alexa 468缀合的山羊抗兔第二抗体检测(绿色; K)。双标签免疫荧光合成图像还示出(1)。在肺泡巨噬细胞磷酸化p38 MAPK表达(褐色; A-C),痰巨噬细胞(褐色; d-f)和小气道上皮细胞(褐色; G-I)。肺组织嗜中性粒细胞(红色)的表达磷酸化p38 MAPK(绿色; J-1)。放大×200。 Scale bars=75 μm.
痰巨噬细胞GydF4y2Ba
COPD患者(n=8)痰中磷酸p38+巨噬细胞百分比明显高于吸烟者(n=6)和非吸烟者(n=4)(平均值分别为92.8%、59.3%和31.8%;所有比较p<0.001)(GydF4y2Ba无花果2 eGydF4y2Ba和GydF4y2Ba5D-FGydF4y2Ba)。GydF4y2Ba
p38 MAPK在上皮细胞中的表达GydF4y2Ba
在所有三组患者中分析的大多数小气道上皮细胞中均存在Phospho-p38 MAPK。Bonferroni的多重比较试验表明,COPD与非吸烟者之间的磷-p38+上皮细胞比例显著增加(分别为96%和81%;p < 0.05)。与吸烟者相比,COPD患者中磷-p38+上皮细胞百分比也有上升趋势(平均91%),尽管这在统计学上没有意义(GydF4y2Ba无花果2 fGydF4y2Ba和GydF4y2Ba5G-我GydF4y2Ba)。GydF4y2Ba
中性粒细胞磷酸化p38 MAPKGydF4y2Ba
所有患者组的肺组织中性粒细胞均无磷酸化-p38 MAPK免疫反应性(GydF4y2Ba图5 j-lGydF4y2Ba). 痰中性粒细胞也缺乏磷酸化p38 MAPK(GydF4y2Ba图。5D-FGydF4y2Ba)。我们从COPD患者的血液中分离嗜中性粒细胞检测磷酸化p38 MAPK。嗜中性粒细胞从血液中分离后立即检查没有磷酸化p38 MAPK,但磷酸化p38 MAPK诱导下用LPS(文化GydF4y2Ba图。图6a和bGydF4y2Ba)。尽管在嗜中性粒细胞的痰,观察到p38蛋白表达(图至S1d),磷酸化p38 MAPK在用LPS培养24小时痰嗜中性粒细胞不存在(GydF4y2Ba图6c和dGydF4y2Ba),这表明p38蛋白激酶途径不是在肺嗜中性粒细胞的活性。GydF4y2Ba
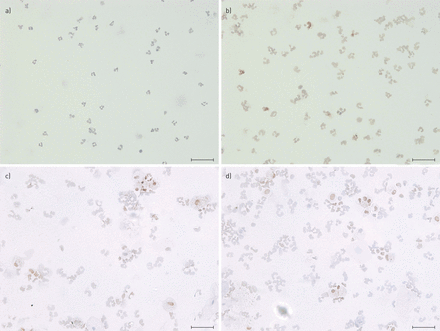
慢性阻塞性肺疾病(COPD)患者血(a、b)和痰(c、d)中性粒细胞磷酸化p38丝裂原活化蛋白激酶(MAPK)免疫组化检测的代表性图像。用梅耶苏木精(蓝色)复染细胞核。直接免疫组化(brown)后,用3,3′-二氨基苯扎定检测磷酸化p38 MAPK的表达。a) 基础血中性粒细胞中缺乏磷酸化p38 MAPK的表达。b) 1μg·mL刺激后中性粒细胞产生磷酸化p38 MAPK(brown)GydF4y2Ba-1GydF4y2Ba脂多糖(LPS)。磷酸化p38 MAPK是不存在于c)中基本的和d)LPS刺激的嗜中性粒细胞的痰。酒吧= 75μm规模。GydF4y2Ba
p38 MAPK抑制的细胞特异性效应GydF4y2Ba
CD8细胞GydF4y2Ba
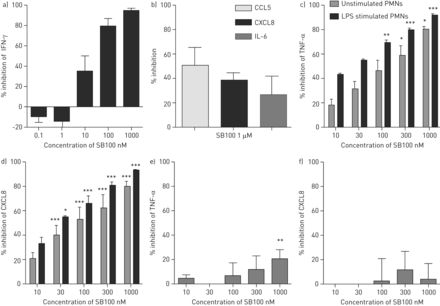
从三个COPD患者的肺组织中分离CD8细胞用SB100结合IL-12和IL-18刺激前进行预处理。IFN-γ的生产增加,从5.3皮克·毫升平均基础水平GydF4y2Ba-1GydF4y2Ba为是指1780皮克·毫升GydF4y2Ba-1GydF4y2Ba。SB100抑制以剂量依赖性方式产生IFN-γ,与最大抑制的94.8%观察到(GydF4y2Ba图7aGydF4y2Ba)。GydF4y2Ba
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
p38丝裂原活化蛋白激酶(MAPK)抑制对慢性阻塞性肺疾病(COPD)细胞的影响。a) 白细胞介素(IL)-12(10ng·mL)刺激COPD肺CD8细胞(n=3)24小时后,SB100(0.1-1000nm)抑制IFN-γ释放GydF4y2Ba-1GydF4y2Ba)和IL-18(10毫微克·毫升GydF4y2Ba-1GydF4y2Ba)。b)抑制趋化因子(碳碳主题)配体5 (CCL5) CXCL8和上皮细胞il - 6与COPD肺组织(n = 3) SB100(1000海里)后24小时刺激polyinosinic: polycytidylic酸(多聚肌苷酸)(10μg·毫升GydF4y2Ba-1GydF4y2Ba)。的c)中肿瘤坏死因子抑制(TNF)-α和d)在CXCL8在用SB100(0.1-1000 nM)的24小时温育刺激和未刺激分离的COPD血嗜中性粒细胞(N = 7)。电子商务抑制)TNF-α和f)从痰嗜中性粒细胞(N = 7)以下用SB100(0.1-1000 nM)的24小时温育CXCL8释放。数据被呈现为平均值±GydF4y2Ba扫描电镜GydF4y2Ba. SB100治疗后细胞因子释放的显著减少用*:p<0.05;*:p<0.01;和*:p<0.001表示。GydF4y2Ba
上皮细胞GydF4y2Ba
下24小时:将细胞用SB100(1000 nm)的1个小时用聚I刺激之前预处理。IL-6和CXCL8的平均基础水平85.5皮克·毫升GydF4y2Ba-1GydF4y2Ba和15 258 PG·毫升GydF4y2Ba-1GydF4y2Ba分别。CCL5的基础水平低于检测限。聚I:C的刺激增加IL-6,CXCL8和CCL5释放到1580.1 PG·毫升GydF4y2Ba-1GydF4y2Ba,56 335 PG·毫升GydF4y2Ba-1GydF4y2Ba2643微克·毫升GydF4y2Ba-1GydF4y2Ba, 分别。SB100分别引起50.7%,38.7%和IL-6,CXCL8和CCL5,26.7%抑制(GydF4y2Ba图7bGydF4y2Ba)。GydF4y2Ba
中性粒细胞GydF4y2Ba
分离血嗜中性粒细胞(来自七个COPD患者),用于具有和不具有LPS 24小时进行培养。未刺激的TNF-α和CXCL8释放的平均水平是527皮克·毫升GydF4y2Ba-1GydF4y2Ba和2857 PG·毫升GydF4y2Ba-1GydF4y2Ba,分别上升至2706 pg·mLGydF4y2Ba-1GydF4y2Ba和7161 PG·毫升GydF4y2Ba-1GydF4y2Ba,分别为LPS刺激后。SB100对受刺激和未受刺激的中性粒细胞(GydF4y2Ba图。7c和dGydF4y2Ba)。有在刺激或未刺激的嗜中性粒细胞的TNF-α的抑制百分比和CXCL8之间没有差异。GydF4y2Ba
同时采集了7例COPD患者的痰标本。以前的研究表明,LPS对痰中性粒细胞产生细胞因子没有影响[GydF4y2Ba21GydF4y2Ba,我们也观察到(数据未显示)。的平均水平如果TNF-αCXCL8释放681 pg·毫升GydF4y2Ba-1GydF4y2Ba和4532 pg·毫升GydF4y2Ba-1GydF4y2Ba在痰中性粒细胞。在如果中性粒细胞分离出的痰,SB100只抑制TNF-α最高浓度(1000 nM)和没有影响CXCL8生产(GydF4y2Ba图。7E和fGydF4y2Ba)。SB100有LPS刺激和-unstimulated血嗜中性粒细胞在所有浓度都为TNF-α和CXCL8(P <0.05对所有浓度的比较使用一个尾配对t检验)相比于嗜中性粒细胞的痰一个显著降低效果。GydF4y2Ba
细胞活力之前和SB100(1000 nm)的治疗(N = 4)后进行评估。有SB100的痰嗜中性粒细胞或嗜中性粒细胞的血液活力没有效果显著,通过台盼蓝排除,形态学分析凋亡和TUNEL测定(图S2和S3)进行测定。GydF4y2Ba
讨论GydF4y2Ba
我们已经证明,与对照组相比,COPD患者肺内特定细胞类型中磷-p38表达增加;与对照组相比,COPD患者肺标本中表达磷酸化p38免疫反应活性的滤泡B细胞和CD8淋巴细胞、小气道支气管上皮细胞和巨噬细胞的比例增加。在这些特定的细胞类型中,我们观察到的总体模式是吸烟增加了phospho-p38 MAPK,而COPD的发展导致其进一步增加。之前有报道称COPD肺泡巨噬细胞中表达磷酸化-p38 MAPK的巨噬细胞比例增加[GydF4y2Ba15GydF4y2Ba];我们现在已经确定了其他肺细胞类型,这些类型在COPD患者中也显示出这种磷酸p38 MAPK表达增加的模式。p38 MAPK在COPD肺CD8细胞和上皮细胞中的抑制降低了细胞因子的产生,表明这些细胞中磷酸化p38的表达与促炎症功能有关。GydF4y2Ba
无论是COPD患者还是对照组,肺中性粒细胞都缺乏磷-p38 MAPK免疫反应活性。此外,p38 MAPK抑制对COPD肺中性粒细胞产生的细胞因子没有影响;这提示p38 MAPK信号在COPD肺中性粒细胞促炎活性中不发挥作用。这与COPD血液中性粒细胞形成对比,在中性粒细胞中检测到磷酸-p38并在功能上参与细胞因子的产生。目前正在开发p38 MAPK抑制剂作为COPD的抗炎药;这些药物对某些类型的肺细胞如b细胞、CD8细胞、巨噬细胞和上皮细胞有抗炎作用,但对肺中性粒细胞无抗炎作用。GydF4y2Ba
我们观察到,p38αMAPK同种型的基因表达水平在肺组织中COPD患者和吸烟增加与非吸烟者相比,具有COPD患者和吸烟之间没有差异;这表明吸烟上调的p38α基因表达。p38α也是最高度表达亚型;这是多数的p38蛋白激酶抑制剂的主要同种型靶。p38δ表达是在与非吸烟者相比,COPD患者中增加,虽然有COPD患者和吸烟之间没有差别。p38δ先前已经显示在巨噬细胞[待表达GydF4y2Ba9GydF4y2Ba]我们的研究结果提示COPD患者肺组织中该亚型表达上调。我们还证明了p38γ在COPD肺组织中的表达水平与对照组相似;这种亚型在糖皮质激素抵抗性炎症中起作用[GydF4y2Ba22GydF4y2Ba]。GydF4y2Ba
在COPD患者小气道淋巴滤泡的数量增加与更严重的疾病有关[GydF4y2Ba1GydF4y2Ba]。我们观察到,与对照组相比,这些滤泡中的b细胞核心和CD8细胞增加了COPD患者的磷-p38 MAPK。有趣的是,与对照组相比,COPD患者上皮下区域内的磷-p38+ b细胞和CD8细胞数量没有增加。这表明滤泡内的淋巴细胞与其他肺淋巴细胞具有不同的生理功能。这可能并不奇怪,因为滤泡淋巴细胞位于类似于淋巴结而不是正常肺组织的环境中,并且是抗原呈递的重要场所[GydF4y2Ba23GydF4y2Ba,GydF4y2Ba24GydF4y2Ba]。GydF4y2Ba
所有患者组的卵泡磷-p38+ CD4+细胞数量相似。卵泡内的磷-p38+ CD4+细胞数量明显低于CD8+细胞和CD20+细胞。此外,在上皮下的CD4细胞中,磷-p38免疫反应性缺失。不同淋巴细胞亚型间磷酸化-p38表达的这种显著差异表明,p38 MAPK信号可能在肺CD4细胞的生理活动中起不了中心作用。然而,对这些从快照中获得的免疫组化数据的解释应该谨慎,因为在COPD CD4细胞中,p38 MAPK信号可能在其他时间被激活,例如在病情加重期间。GydF4y2Ba
先前的研究表明,药物抑制p38 MAPK可以减少COPD巨噬细胞产生促炎性细胞因子[GydF4y2Ba9GydF4y2Ba-GydF4y2Ba11GydF4y2Ba]。我们现在表明,p38蛋白激酶抑制也减少了从肺CD8细胞和COPD患者的上皮细胞促炎细胞因子的产生。p38蛋白的抑制从COPD肺显著降低从分离的CD8细胞的IFN-γ释放。已知的是,p38蛋白信号传导参与从血液CD8细胞[细胞因子的产生GydF4y2Ba25GydF4y2Ba],我们现在确认这个途径在肺CD8细胞类似的作用。最近已经证明,从支气管肺泡灌洗淋巴细胞的IFN-γ释放糖皮质激素的作用COPD患者减少与对照组[对比GydF4y2Ba26GydF4y2Ba]。此外,IFN-γ的原因glucocortiocoid不敏感的细胞因子产生中COPD肺泡通过信号转导转录的巨噬细胞和活化剂(STAT)1个活化GydF4y2Ba27GydF4y2Ba]. 因此,抑制肺CD8细胞p38-MAPK信号传导可以靶向糖皮质激素不敏感机制,如CD8细胞产生IFN-γ,以及随后巨噬细胞产生STAT1介导的细胞因子。GydF4y2Ba
香烟烟雾诱导支气管上皮细胞释放多种促炎介质GydF4y2Ba体外GydF4y2Ba[GydF4y2Ba28GydF4y2Ba-GydF4y2Ba31GydF4y2Ba],暗示在COPD的发病机制中的上皮。在本研究中,积极为磷酸化p38 MAPK小气道上皮细胞的百分比在COPD肺显著较高。在气道上皮细胞中增加的磷酸化p38 MAPK的表达也已在重度哮喘实测值[GydF4y2Ba32GydF4y2Ba]。COPD和重度哮喘的特征在于在气道中促炎介质的持续增加的水平[GydF4y2Ba33GydF4y2Ba,GydF4y2Ba34GydF4y2Ba]。的p38蛋白激酶途径由一系列炎性刺激的激活,并且似乎支气管上皮细胞中对这些刺激受p38 MAPK的活化既严重哮喘和COPD响应。GydF4y2Ba
肺中性粒细胞缺乏在我们所有的患者群体,包括吸烟和不吸烟的对照磷酸化p38 MAPK免疫反应。嗜中性粒细胞肺用LPS刺激不诱导磷酸化p38 MAPK活化,这表明肺嗜中性粒细胞的促炎活性是不依赖于p38蛋白信令。这是由缺乏SB100对促炎性细胞因子的产生从肺嗜中性粒细胞的效果证实。相反,在血液嗜中性粒细胞中观察到磷酸化p38 MAPK活化,并从这些细胞中细胞因子的产生是由SB100抑制。我们的研究结果表明,SB100同时抑制预成型和GydF4y2Ba新创GydF4y2Ba在嗜中性粒细胞的血细胞因子合成的,如在两个未刺激和LPS刺激的细胞中观察到类似的抑制。我们的研究结果表明,嗜中性粒细胞离开血液和进入肺的表型发生改变,改变的重要细胞功能所需的细胞内信号通路的活性。同样,我们先前观察到正常的糖皮质激素受体的表达在嗜中性粒细胞的血液,但在嗜中性粒细胞呼吸道[耗尽的糖皮质激素受体的表达GydF4y2Ba35GydF4y2Ba]。这些数据突出使用血液嗜中性粒细胞为嗜中性粒细胞肺模型的潜在的缺陷,因为似乎是负责细胞因子产生的信令机制差异。GydF4y2Ba
先前的研究已经证明p38 MAPK抑制可以减弱趋化性[GydF4y2Ba36GydF4y2Ba-GydF4y2Ba39GydF4y2Ba]和超氧化物生成[GydF4y2Ba40GydF4y2Ba]中,除了促炎介体代[GydF4y2Ba41GydF4y2Ba-GydF4y2Ba43GydF4y2Ba]在血液嗜中性粒细胞。缺乏肺嗜中性粒细胞活化的观察到p38蛋白的使得它不太可能p38蛋白抑制会对趋化和在肺嗜中性粒细胞的任何作用产生超氧化物。肺嗜中性粒细胞的改变的表型,与p38蛋白的减少的活化和表达糖皮质激素受体的[GydF4y2Ba35GydF4y2Ba],提示在COPD中需要针对这种细胞类型的特异性抗中性粒细胞疗法,而不是广泛的抗炎药。GydF4y2Ba
在肺泡巨噬细胞中,我们之前已经发现p38 MAPK活化对糖皮质激素具有抵抗性[GydF4y2Ba11GydF4y2Ba]。因此,p38 MAPK抑制剂针对的是COPD患者肺内激活的糖皮质激素耐药通路,我们在这里表明,这些药物可以抑制COPD肺淋巴细胞和上皮细胞产生的细胞因子。p38 MAPK与糖皮质激素存在协同作用;这两种药物联合使用对慢性阻塞性肺病肺泡巨噬细胞产生的细胞因子的抑制作用大于相加作用[GydF4y2Ba11GydF4y2Ba]. 有一些分子机制可以解释这种现象,如糖皮质激素诱导MAPK磷酸酶上调[GydF4y2Ba44GydF4y2Ba],使p38 MAPK去磷酸化。糖皮质激素和p38 MAPK抑制剂联合治疗也可能对淋巴细胞和上皮细胞产生的细胞因子产生协同作用。GydF4y2Ba
我们和其他人报告了p38的抑制对炎症介质的肺泡巨噬细胞释放的影响COPD患者和对照之间没有差异[GydF4y2Ba9GydF4y2Ba,GydF4y2Ba11GydF4y2Ba]。我们现在报告P38抑制肺癌CD8细胞和上皮细胞的作用COPD患者,这将有兴趣知道,如果这些影响与对照不同。GydF4y2Ba
综上所述,我们发现COPD患者肺中p38 MAPK通路的细胞特异性激活。p38 MAPK抑制剂抑制COPD肺淋巴细胞和上皮细胞产生细胞因子,但对肺中性粒细胞无影响。因此,这些新药物针对的是慢性阻塞性肺病的部分(但不是全部)炎症过程。GydF4y2Ba
脚注GydF4y2Ba
这篇文章有补充资料GydF4y2Bawww.www.qdcxjkg.comGydF4y2Ba
利益冲突:可以在本文的在线版本中找到披露GydF4y2Bawww.www.qdcxjkg.comGydF4y2Ba
- 收到GydF4y2Ba2011年10月3日。GydF4y2Ba
- 公认GydF4y2Ba2012年9月27日。GydF4y2Ba
- ©ERS 2013GydF4y2Ba
工具书类GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba
- ↵GydF4y2Ba










