





摘要
本研究旨在研究人类细支气管过氧化氢酶表达及其与吸烟和/或慢性阻塞性肺疾病(COPD)的关系,并确定小鼠对卷烟烟雾反应中细支气管过氧化氢酶表达的动态变化。
从36名受试者接受手术得到了用于外围肿瘤肺组织,包括终身非吸烟者和吸烟者具有或不COPD。至3个月,随后28天停止期间雄性C57BL / 6小鼠进行香烟烟雾暴露最多。我们量化使用激光捕获显微切割和定量逆转录 - 聚合酶链反应支气管过氧化氢酶基因。C22俱乐部细胞(克拉拉细胞)中培养暴露于香烟烟雾提取物,并监测存活当过氧化氢酶表达减少通过siRNA。
过氧化氢酶在mRNA和蛋白水平与COPD吸烟者减少在支气管上皮。在小鼠中,过氧化氢细支气管暂时在香烟烟雾暴露后第1天上调,而是通过反复烟雾暴露下调,而不是一次肺气肿开发撤出以后很久恢复。在C22细胞中降低过氧化氢酶表达导致更大的香烟烟雾提取物诱导的细胞死亡。
支气管过氧化氢酶减少与COPD有关。过氧化氢酶的调节取决于香烟烟雾暴露的持续时间,并播放针对吸烟诱导的细胞损伤的保护至关重要的作用。
介绍
肺部氧化应激被认为是慢性阻塞性肺疾病(COPD)发病机制的关键组成部分[1].动物研究还支持单独抵抗烟雾烟雾(CS)的能力,诱导肺抗氧化防御,所述肺抗氧化防御作为肺气肿发展的关键事件[2].
由于与环境的直接接触,呼吸道沿线的上皮细胞暴露于CS;因此,它们很可能参与了吸烟相关疾病的发病机制。特别是,晚期细支气管已知在各种吸烟相关肺部疾病中发挥关键作用,它们是COPD中气流限制的主要部位[3.].然而,吸烟者细支气管上皮抗氧化防御的信息有限,COPD患者的信息更少。我们最近研究了吸烟小鼠细支气管上皮细胞和分子的变化[4,5,以及它们与慢性阻塞性肺病的发展的关系[6- - - - - -8].在这项研究中,我们使用激光捕获显微切割(LCM)来分离终端支气管上皮和进行了cDNA阵列分析,着眼于应力和毒性的途径,用于筛选。这些数据表明,过氧化氢酶在正常(非吸烟者)成人肺的支气管上皮的大量表达基因的抗氧化剂,进而仅过氧化氢酶的mRNA在COPD患者的支气管上皮被显着降低。
过氧化氢酶是一种240 kDa的四聚体血红素蛋白,由于其将过氧化氢转化为氧气和水的能力,在肺的抗氧化筛选中起着核心作用[9].过氧化氢酶在肺部发育的后期表达,并在呼吸道和肺泡上皮细胞和成人巨噬细胞中构成思考型[10].到目前为止,一些研究集中在病理性肺状态中的过氧化氢酶[9- - - - - -11].然而,没有研究已经全面在过氧化氢酶的关联和吸烟或COPD进行。过氧化氢酶的肺防御的意义,特别是在细支气管水平,从而可能已被低估。与大多数的抗氧化剂,过氧化氢酶是不是在支气管上皮升高,健康人吸烟者[12].
因此,我们检测了CS暴露对小鼠远端气道上皮细胞中过氧化氢酶表达的影响。此外,我们还研究了过氧化氢酶是否可能在保护cs诱导的小鼠俱乐部细胞(Clara细胞)C22损伤中发挥作用。
材料和方法
本研究中使用的材料和方法的细节提供在线补充材料。
采集人体组织标本
肺组织标本从36例接受肺切除外周小的肿瘤获得。COPD患者是基于全球倡议的阻塞性肺疾病[准则选择13].研究获得了每位受试者的知情同意,日本札幌北海道大学医学院伦理委员会批准了该研究方案。一些患者是我们之前研究的对象[6- - - - - -8,14].
LCM-检索细支气管上皮细胞的cDNA阵列分析
LCM收集人细支气管上皮细胞,如前所述提取总RNA [6].含有96个基因的非放射性齿轮Q系列cDNA表达阵列过滤器,其表达水平指示应力和毒性途径(HS-012N; Superarray Inc.,MD,MD,USA)。
过氧化氢酶免疫组化及半定量评分
使用兔抗过氧化氢酶多克隆抗体(Calbiochem-Novabiochem, San Diego, CA, USA)对人肺中的过氧化氢酶进行免疫染色。过氧化氢酶免疫强度由两名独立观察员以盲法进行半量化,并报告平均评分。
老鼠吸烟模型
男性C57BL / 6J小鼠,9-10周(Charles River,Atsugi,Japan),每天暴露于全身主流CS 90分钟[7或每天60分钟只用鼻子的主流CS [15].使用任何一种暴露系统重复暴露3个月的CS会导致显著的空域扩大[7,15].处死小鼠,并在指示的几个时间点收集肺样品(每组n = 4-6),并且如前所述,通过LCM收获支气管上皮细胞[5].
原位杂交对过氧化氢酶
小鼠的肺膨胀固定,用10%中性缓冲的福尔马林,石蜡包埋,切成5微米的部分。脱蜡切片与对应于核苷酸鼠标过氧化氢酶基因的1215至1533年地高辛标记的RNA探针杂交。杂交后,使用碱性磷酸盐标记的抗洋地黄毒苷抗体(罗氏公司,巴塞尔,瑞士)进行地高辛检测。
实时rt - pcr
如前所述,进行RNA纯化、逆转录和定量PCR[6].Taqman基因表达检测探针Hs00156308_m1用于人过氧化氢酶,其水平对甘油醛-3-磷酸酶脱氢酶mRNA的影响正常,而Mm00437992_m1用于小鼠过氧化氢酶,其水平对β 2-微球蛋白mRNA的影响正常(应用生物系统,Foster City, CA, USA)。
CS提取物暴露于C22细胞,并通过siRNA抑制过氧化氢酶的
使用干扰素siRNA转染试剂(美国加利福尼亚州圣马科斯市Polyplus转染公司),用20 nM过氧化氢酶-siRNA双链(美国密苏里州圣路易斯市西格玛-奥尔德里奇)转染C22细胞,并如前所述暴露于无血清培养基中的稀释CS提取物中[4].为了评估细胞活力,如前所述,测定无细胞培养基的乳酸脱氢酶(LDH)活性[4].
数据展示和统计分析
所有数据均表示为平均值±扫描电镜或者中位数。在人类中,差异分析使用单因素方差分析,然后Fisher的保护最小显著差异检验作为一个后HOC.测试或Kruskall–Wallis测试和Mann–Whitney测试。在小鼠中,通过Dunnet多重比较分析确定统计显著性。
结果
人类个体的特性
我们收集了三组受试者:13名终身不吸烟者、13名非COPD吸烟者和10名COPD吸烟者。研究对象的临床特征总结在表1.研究对象均无哮喘病史,前一个月均无急性呼吸道感染。两组吸烟者的烟龄相似,但吸烟和戒烟的持续时间不同(表2.).所有COPD患者均在1秒内出现强迫呼气量(FEV)1)/强迫肺活量低于正常的下限[16].
用于应激和毒性途径基因的cDNA阵列揭示了具有COPD的吸烟者中的过氧化氢酶降低
为了解决这个问题在支气管上皮相关应力和毒性途径的基因表达的改变是否可能与在人体中的慢性吸烟史和/或COPD,完成cDNA阵列分析。因为在检索细胞LCM的限制,它在为了获得足够的RNA没有放大汇集从受试者的细胞各组(n = 10)中是必要的。中的组22的氧化和将cDNA阵列对代谢应激相关基因,过氧化氢酶是一种抗氧化剂基因在支气管上皮在正常成人肺部最大量表达,并且相比时仅过氧化氢酶显示出在患有COPD的吸烟者超过1.8倍的降低与不吸烟者(表S1)。这是符合N的调查结果惯性导航与制导等.[17,中度COPD患者与COPD高危患者相比,肺中过氧化氢酶表达下调。
定量分析证实了支气管上皮细胞酶表达的显着下调
为了确认CDNA阵列的数据,显示COPD患者的支气管过滤酶的下调,对从单个支气管上皮收获的RNA进行过氧化氢酶的定量RT-PCR(图1),与目前的cDNA阵列结果一致(表S1)。在吸烟者中,细支气管过氧化氢酶的个体表达水平与吸烟包年无关(图S1)。
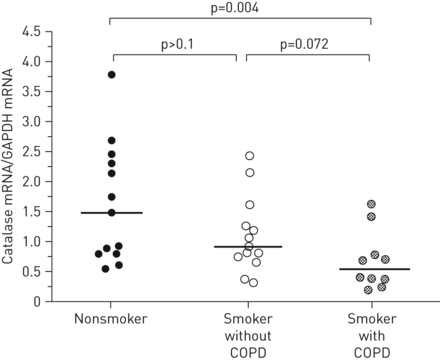
细支气管上皮中过氧化氢酶的表达。激光捕获微解剖获得人细支气管上皮细胞,用RT-PCR定量过氧化氢酶mRNA。与非吸烟者相比,慢性阻塞性肺疾病(COPD)吸烟者的细支气管上皮细胞中过氧化氢酶表达显著下调(中位数为0.6相对1.5; p<0.05),而吸烟者合并和不合并COPD之间的差异没有达到统计学意义(p=0.072)。中间带由水平线表示。GAPDH:甘油醛磷酸脱氢酶。
人COPD肺中细支气管过氧化氢酶蛋白减少
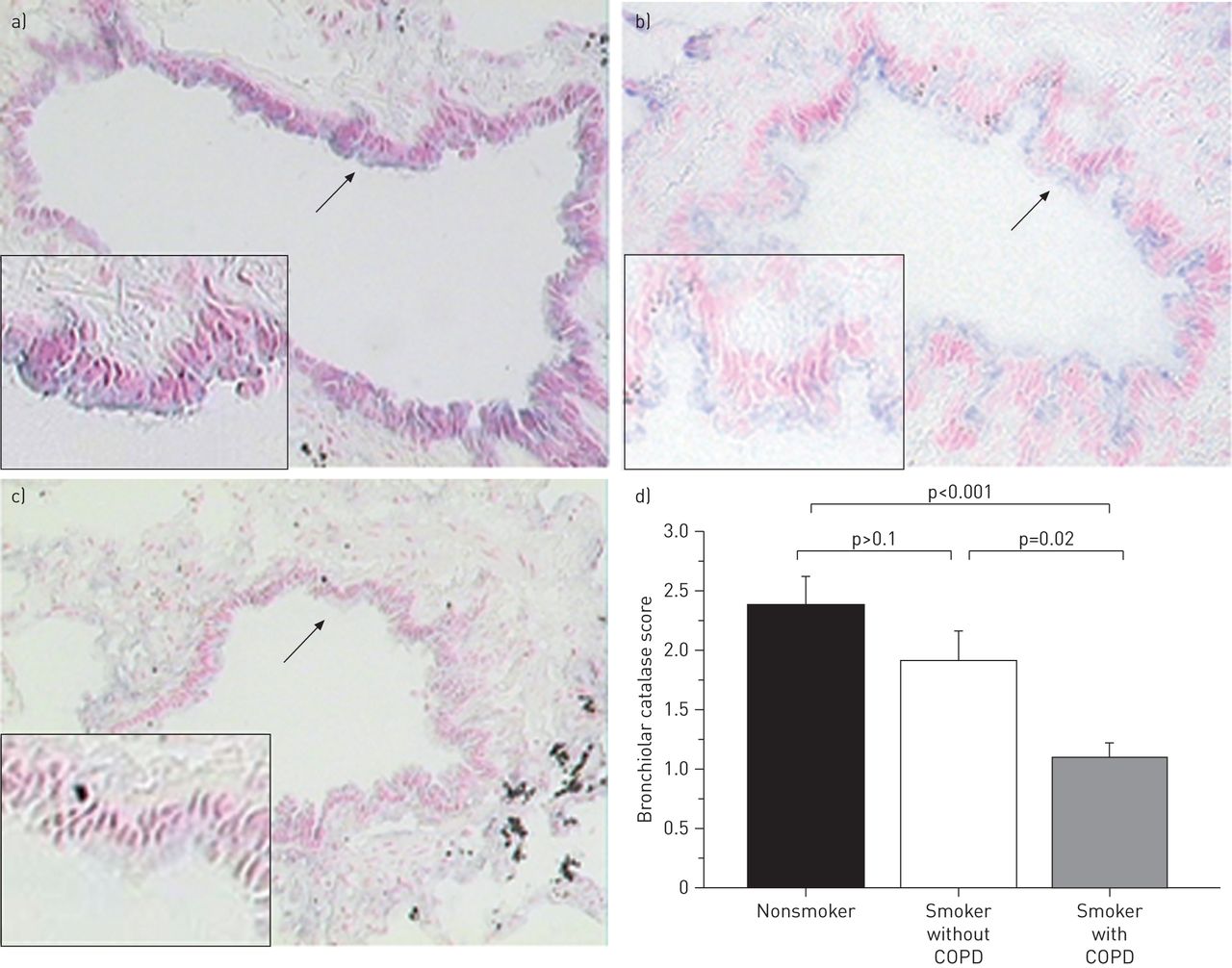
为了评估COPD患者支气管过氧化氢酶在蛋白水平上的降低,进行了免疫组织化学。过氧化氢酶主要定位于非吸烟者气道上皮细胞的顶端(图2a).即使是非吸烟者,肺泡组织中也未检测到过氧化氢酶(数据未显示)。这与K的发现一致aarteenaho -Wiik和K.妮乡[10,在人肺细支气管上皮中显示过氧化氢酶的免疫反应性。然而,尽管非COPD吸烟者的细支气管过氧化氢酶发生了微小变化,但在COPD吸烟者的晚期细支气管细胞中发现了过氧化氢酶染色的显著下降(图2b–d).免疫组化和FEV百分比之间的支气管硫醇过缩酶评分无显着相关性1或在COPD患者中计算断层扫描(CT)扫描的肺气肿程度(数据未显示)。
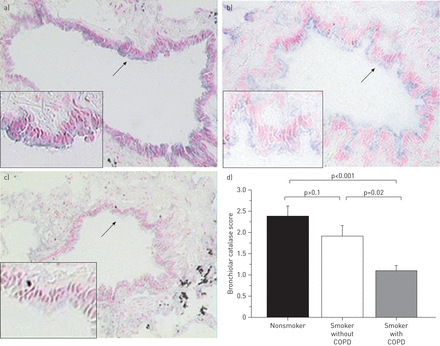
细支气管上皮组织中过氧化氢酶的免疫组化。a)不吸烟者,b)无慢性阻塞性肺病(COPD)吸烟者(年龄73岁,吸烟56包年),c) COPD吸烟者(年龄77岁,吸烟40包年)。免疫组化染色呈蓝紫色。箭头示细支气管上皮,抗人过氧化氢酶抗体免疫染色。d)通过评分,与不吸烟者和非吸烟者相比,COPD吸烟者的细支气管上皮中过氧化氢酶显著降低(平均值±)se1.1±0.1相对分别为2.4±0.2和1.9±0.2;P <0.05)。原始放大倍数(A-C)×200(插图×800)。
过氧化氢酶表达富含支气管上皮,在小鼠中Cs暴露后降低
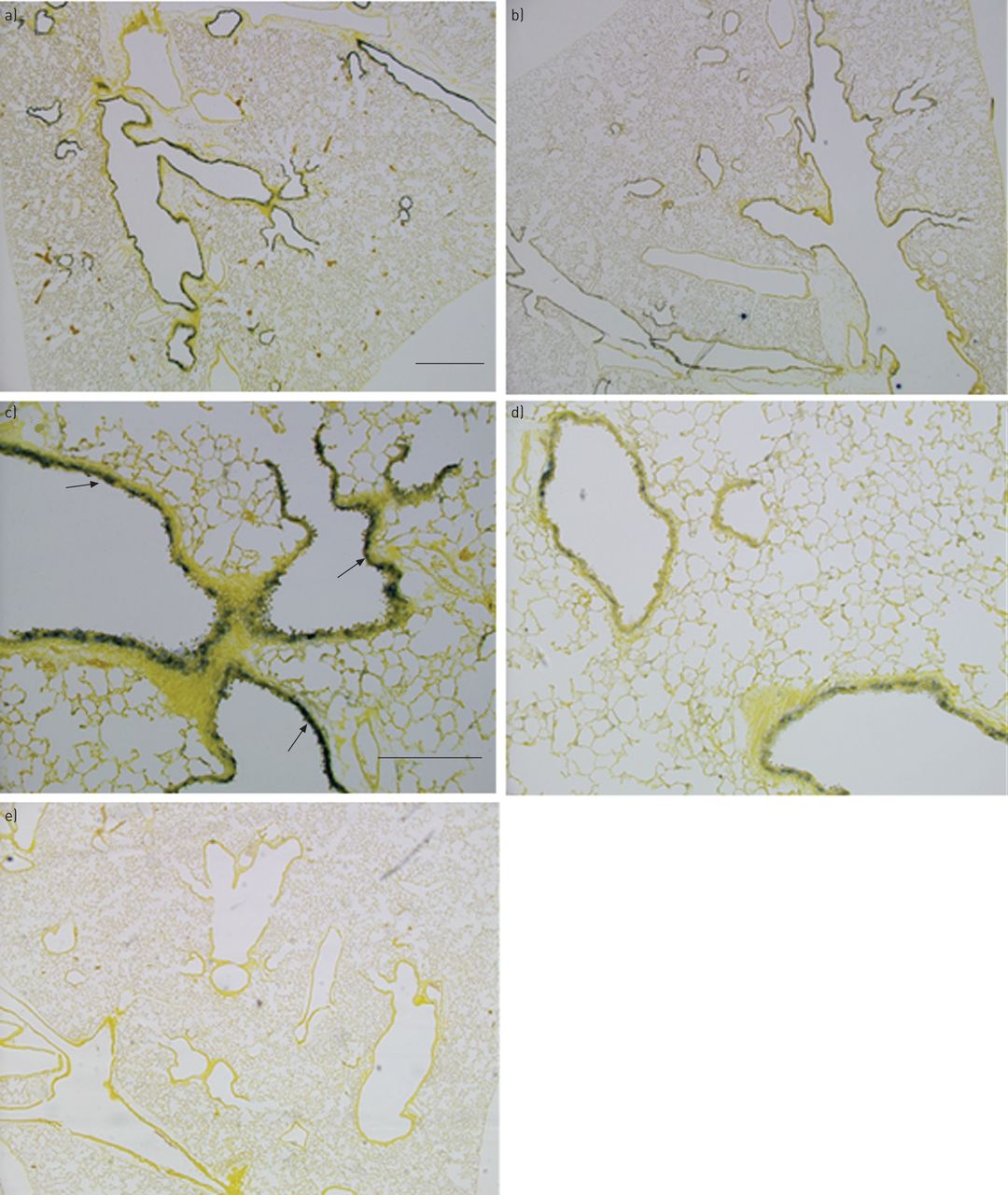
接下来,我们检测了小鼠肺中过氧化氢酶的表达,并检测了CS暴露对过氧化氢酶表达的影响。原位杂交在上皮细胞中揭示了衬里的上皮细胞中的高水平过氧化氢酶mRNA暴露的终端气道暴露C57BL / 6J对照小鼠(图3a和c),这与人类肺部的情况类似。在全体CS暴露10天的小鼠中,过氧化氢酶mRNA下降(图3b和d).即使在非cs暴露的对照组小鼠中,肺泡组织中也没有发现过氧化氢酶,这与在人类肺中的发现一致。
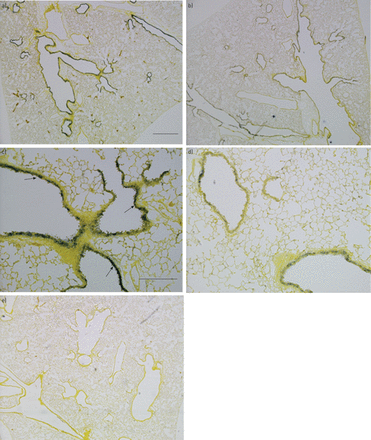
过氧化氢酶mRNA在小鼠肺中的定位:杂交肺切片的图像原位过氧化氢酶信使rna。在正常肺中,过氧化氢酶mRNA在细支气管上皮细胞中突出,从细支气管末端与肺泡管连接处开始,并沿细支气管近端至250 μm处(a和c)。细支气管上皮中过氧化氢酶mRNA降低(b和d)。e)使用感觉探针的对照组在正常肺中显示最小杂交。这些图像代表了每组的几只老鼠。箭头示细支气管上皮与与小鼠过氧化氢酶基因核苷酸相对应的地高黄素标记的RNA探针杂交。a, b, e)比例尺=600 μm。c, d)比例尺=20 μm。
在支气管上皮取决于CS暴露在小鼠中的持续时间过氧化氢酶表达的调控双峰
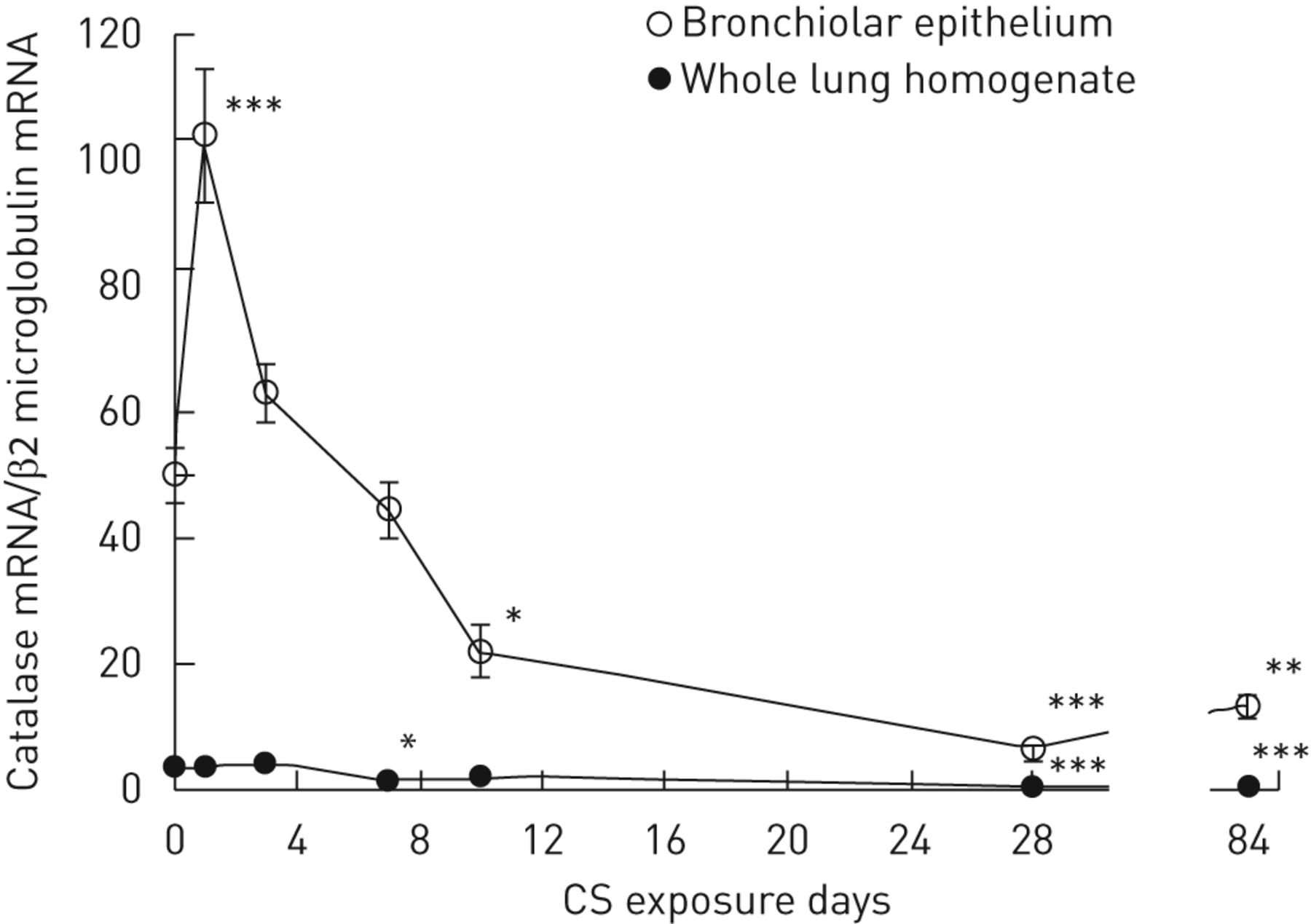
为了研究肺气肿的发展过程中支气管过氧化氢酶表达的动态变化,LCM-检索支气管上皮和全肺组织中的过氧化氢酶的表达在不同时间点以下,每天全身CS敞口长达3个月量化。值得注意的是,当与非暴露成年小鼠在整个肺组织中的水平(过氧化氢酶相比,mRNA的富集在支气管上皮14倍图4).
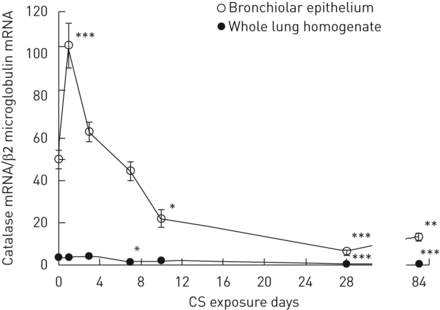
在香烟烟雾(CS)曝光期间小鼠的全肺和支气管过滤酶表达。显示的是在支气管上皮的重复全体CS暴露期间的过氧化氢酶mRNA表达的时间课程,并在全肺匀浆中进行84天。与非CS暴露的动物相比,在CS暴露后1天后,支气管过滤酶mRNA在时间上上调(104.1±12.5相对50.2±6.0,P <0.001);然而,经过10天(22.1±4.8下调相对50.2±6.0,P <0.05),28天(6.5±1.5相对(50.2±6.0,p<0.001), CS暴露84天(3个月)(13.4±2.2)相对50.2±6.0,p<0.01)。过氧化氢酶mRNA在整个肺组织中的表达随着时间的推移保持较低,但与未暴露CS的小鼠相比,在7天后,过氧化氢酶mRNA的表达较弱,但在统计学上有所下降(1.7±0.3)相对3.5±0.6,P <0.05),28天(0.5±0.1相对3.5±0.61,p<0.001)和84天(3个月)CS暴露(0.7±0.1相对3.5±0.6,P <0.001)。每个数据点代表平均值±扫描电镜六个动物。*: p<0.05, **: p<0.01, ***: p<0.001相对非公开的老鼠。
在CS暴露中使用鼻子模型也得到了类似的结果。与非CS暴露小鼠相比,CS暴露后10天和84天(3个月)细支气管上皮中过氧化氢酶表达降低,且CS诱导的细支气管上皮细胞凋亡增加(图5和图。S2)。这些数据表明,急性Cs暴露(1天)在支气管上皮诱导过氧化氢酶的上调,而慢性Cs暴露(最长3个月)降低了其表达,同时随着其他地方报告的空域扩大的发展恰逢[7,15].这一发现与我们之前在微阵列上分析的数据一致[4].
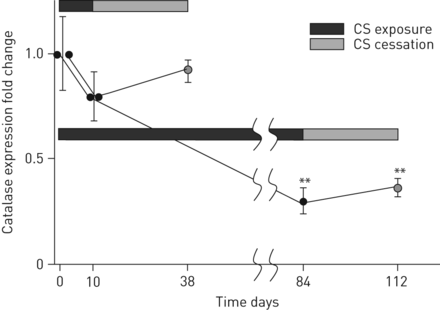
卷烟烟雾(CS)暴露后的支气管过缩酶表达的恢复。显示出在短期(10天)或长期(84天)CS暴露后,鼻子仅鼻子的CS暴露后折叠支气管过缩酶表达的变化。每个数据点代表平均值±扫描电镜为4 - 6的动物。**: p < 0.01相对非公开的老鼠。
在小鼠中停止慢性烟雾暴露后,未恢复下调的支气管过氧化氢酶表达
接下来我们讨论了CS暴露的停止是否可以恢复细支气管过氧化氢酶的表达。如上所述,与非CS暴露小鼠相比,CS暴露84天后(3个月)细支气管上皮中过氧化氢酶的表达降低了3倍以上(图5).停药28天后,过氧化氢酶表达略有升高;然而,细支气管上皮中过氧化氢酶的总体表达水平仍然受到抑制。这与一些COPD患者戒烟后过氧化氢酶表达受损长期存在的研究结果一致(表2.和图1).
相比之下,发生以下只有10天CS曝光的28天停止CS时期,支气管过氧化氢酶的表达恢复到接近基线水平(图5).此外,CS暴露的持续时间不仅影响炎性反应的到CS的类型(4:加1和巨噬细胞嗜中性粒细胞3个月后增加四倍淋巴细胞:巨噬细胞对嗜中性粒细胞1比后CS曝光的10天至2CS曝光),而且下面的CS停止恢复回弹性(约五倍的相对10天或3个月CS暴露后,在28天停止治疗后,总细胞数量分别减少了约3倍(表S2)。这些数据表明,从CS暴露的影响中恢复的能力(例如.降算酶表达和炎症分辨率的降低更依赖于CS暴露的持续时间而不是停止期的持续时间。
CS提取物诱导C22细胞过氧化氢酶基因表达
为了研究CS对小鼠终末细支气管(大于90%的Clara细胞)细胞中过氧化氢酶表达的直接影响,我们将C22细胞暴露于CS提取物中体外.与对照相比,Cs暴露在3,6和24小时的C22细胞中诱导在C22细胞中的过氧化氢酶表达的大致转换(P <0.05)(图6).这与研究结果一致,细支气管过氧化氢酶在1天CS暴露后暂时性上调体内在老鼠中。
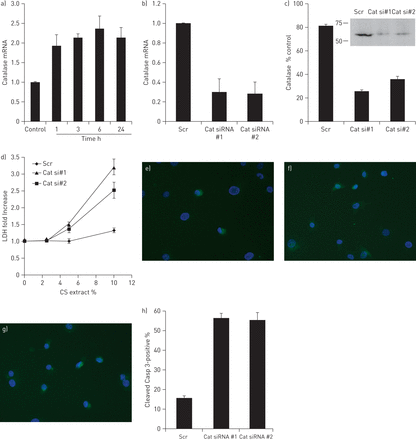
烟提取物对C22细胞过氧化氢酶mRNA表达的影响及过氧化氢酶对C22细胞死亡的影响a)从C22细胞中分离的rna暴露于含10% CS提取物的无血清培养基中,最长24小时,对过氧化氢酶进行实时RT-PCR。随后正常化到β2-微球蛋白,结果表现为未处理条件下的fold change。数据代表了三个独立的实验±扫描电镜一式三份。*:P <0.05相对于控制。用靶向过氧化氢酶表达(CAT)的两种不同的siRNA双链体或糖浆siRNA双链体(SCR)独立地转染C22细胞。转染后2天,分离RNA并进行实时RT-PCR以表达过氧化氢酶(B),分离出总蛋白质并进行Western印迹分析(C)。与用糖浆的siRNA处理的那些相比,用过氧化氢酶siRNA转染的C22细胞表达显着降低的过氧化氢酶mRNA水平(平均水平为70%),其通过Western印迹在蛋白质水平处确认。在单独的实验中,用炒或过氯酶siRNA双链体转染后2天,将C22细胞暴露于含有10%CS提取物的无血清培养基,测定培养基中的LDH的相对量。结果表示为相对于单独暴露于无血清培养基的SCR siRNA转染的C22细胞的倍数变化。另外,使用抗切割的细胞染色与SCR siRNA(e),猫siRNA#1(f)或猫siRNA#2(g)双链体接触并暴露于10%CS提取物的C22细胞进行染色24小时。用4',6-二氨基-2-苯基吲哚检测Caspase-3抗体和核。结果量化并表示为切割的Caspase-3阳性细胞的百分比±扫描电镜.
过氧化氢酶耗尽的C22细胞易受cs诱导的细胞死亡
为了确定过氧化氢酶的细支气管上皮细胞中的作用,C22细胞与siRNA双链的过氧化氢酶转(图6b和c).当过滤酶被击倒时,其他抗氧化剂和排毒基因没有显着变化(图S3)。在转染后2天后,通过单独或含有各种浓度的Cs提取物暴露于无血清培养基,测定培养培养基中的LDH活性的水平,细胞死亡的指标。当与暴露于0%CS提取物的细胞相比,用糖浆SiRNA转染的C22细胞在培养基中略微增加了培养基中的LDH活性。C22细胞暴露于≥20%的Cs提取物导致显着的细胞死亡,即使在用炒siRNA转染的C22细胞中也是显着的细胞死亡[4].相比之下,通过siRNA表达过氧化氢酶降低的细胞对CS暴露明显更敏感,这表明在条件培养基中LDH活性显著增加,即使在5% CS提取物浓度(图6 d).这些数据表明,过氧化氢酶在针对CS诱发支气管上皮细胞死亡的保护作用。
讨论
这项研究有关于过氧化氢酶在支气管上皮在人类和小鼠的重要发现。在人类中,过氧化氢酶是一种抗氧化剂基因在成年非吸烟者的支气管上皮表达最丰富的,而支气管上皮细胞过氧化氢酶在COPD患者的肺部明显下降。在小鼠中的实验表明,吸烟对支气管过氧化氢酶表达的作用是时间依赖性的,初始烟雾暴露后的早期增加,但与慢性暴露的下降和残留低甚至长烟雾暴露已终止后。我们还发现,过氧化氢酶的C22细胞耗竭增加易感性CS诱导的细胞死亡,这意味着过氧化氢酶的细支气管上皮细胞对CS诱导的细胞损伤中起关键作用。
在人类研究中,吸烟期的时期是吸烟者之间的变化。一方面,如果在受试者仍在吸烟的同时进行测量,则基因表达的变化将更加重要[18]. 另一方面,这项观察特别承认了一个事实,即并非所有的基因表达都因戒烟而恢复正常。基因表达的一些不可逆的变化,例如即使在戒烟和/或COPD患者患肺癌的高风险后,也可能与疾病的进展有关[19].在蛋白质水平上,患有慢性阻塞性肺病和吸烟对细支气管过氧化氢酶的抑制作用比不吸烟更大(图2),而COPD患者与非COPD患者在mRNA水平上差异无统计学意义(图1).这种差异表明,由于未知原因,COPD也与过度的营业酶蛋白蛋白质的速度或受损相关。
我们对小鼠的时间进程研究表明,CS暴露的急性影响不能自信地推断为吸烟的慢性影响。CS暴露对细支气管上皮细胞的影响可能是由几个具有不同时间框架的过程引起的。有趣的是,一旦空气扩大,停止CS暴露后,小鼠细支气管过氧化氢酶的下调仍然存在。这些特征可能模拟了人类中患有轻度COPD的前吸烟者的状态。在转录水平上,过氧化氢酶直接由FoxO3a和共激活因子、过氧化物酶体增殖物激活受体γ共激活因子1-α调控[20.].H王等.[21]最近报道,FoxO3主要局部地局部地局限于烟道/肺泡上皮,在吸烟者的肺部和COPD患者中减少,并且在暴露于Cs的小鼠的肺部下降。在该研究中,在FOXO3缺陷小鼠中,小鼠肺部的过氧化氢酶上调在小鼠肺部3天的肺部显着损害,尽管在野生型和稳定状态下肺部的过氧化氢酶表达水平没有差异FOXO3缺陷小鼠,表明FOXO3在过氧化氢酶转录调节中的关键作用。过氧化氢酶也可以受核因子(NF)-E2相关因子2(NRF2)的影响肺部至CS的反应[4].虽然Nrf2的表达水平在支气管上皮细胞没有下降[8],这些转录因子的基因或后生灭活可能参与这一机制的过氧化氢酶的下调坚持细支气管上皮细胞。这些研究都强调针对CS诱导肺上皮细胞损伤的保护抗氧化介导的细胞反应的重要性。慢性CS曝光后,将氧化应激变得比抗氧化潜力越大,过氧化氢酶的下调,并会在支气管上皮细胞发生CS诱导的凋亡。
因为过氧化氢酶处理过氧化氢及其有毒衍生物的细胞内负担,过氧化氢反向抑制过氧化氢酶活性并下调过氧化氢酶表达[22],过氧化氢酶缺乏的上皮细胞在细胞外环境中可能释放过多的过氧化氢。过氧化氢不是自由基;因此,它的活性较低,因此比其他活性氧更稳定[23].已经已知的是,COPD患者呼气更多过氧化氢,而不管当前吸烟的状态[24].据报道,丙酸癌小鼠更容易受到氧化剂组织伤害,导致肾纤维化[25]和腹膜纤维化[26].过氧化氢酶,过氧化氢也清除抑制气道烟雾引起的纤维化重塑[27].因此,吸烟小鼠细支气管上皮细胞过氧化氢酶表达降低也可能与细支气管周围纤维化有关[28]和慢性阻塞性肺病[3.].过氧化氢酶被证明可以通过快速调节生理ROS水平,防止氧化还原敏感的核转录因子核因子-κB激活导致肺部炎症的级联反应[29].通过B细胞淋巴瘤-2家族蛋白的调节在人巨噬细胞过氧化氢酶防止细胞凋亡,这意味着过氧化氢酶的不足上调和/或抑制可以在细胞凋亡参与[30.].
众所周知,CS旨在诱导肺泡巨噬细胞,肺内皮细胞和各种肺上皮细胞类型的细胞死亡。已经至少部分地示出了肺细胞对Cs的敏感性,以通过调节保护抗氧化系统来确定。在人类研究中,除了过氧化氢酶外,还有关于阵列上的其他基因是热休克70kDa蛋白1a(Hsp70-1a),加热休克70kDa蛋白1b(hspa1b),热休克70kda蛋白样1(hspa1l)和热量休克70 kDa蛋白6(HSP70B),所有这些都显示出> 1.5倍的吸烟者在与烟草的吸烟者中下调,与从未吸烟者相比。这是符合N的调查结果惯性导航与制导等.[17]与COPD高危患者(GOLD 0)相比,中度COPD(慢性阻塞性肺病全球倡议(GOLD)II期)患者肺中过氧化氢酶和HSP70表达下调。HSP70的减少不能抑制NF-κB的激活,而NF-κB的激活又会导致促炎症基因(如白细胞介素-8)的表达增强。这可能部分解释了过氧化氢酶和HSPs的减少仅与发展为COPD的易感吸烟者的小气道持续炎症相关。
我们的研究有几个局限性。首先,虽然我们的兴趣主要集中在轻度COPD的发病机制上,但未来对重度COPD患者的研究对于比较至关重要。然而,应该考虑到,即使是轻微的气流限制和存在轻微的实质破坏变化,都与细支气管上皮细胞中过氧化氢酶水平低有关,而与戒烟无关。其次,这三组人之间显著的性别差异提出了性别可能是造成肺细胞过氧化氢酶表达差异的原因之一。但COPD吸烟者的细支气管过氧化氢酶表达低于非COPD吸烟者。第三,本研究未检测过氧化氢酶活性。然而,测量全肺过氧化氢酶活性将完全忽略细支气管过氧化氢酶的变化,因为除细支气管外所有肺组织的过氧化氢酶水平都很低。
我们得出的结论是,烟雾暴露导致过氧化氢酶的诱导或失活受损,导致细支气管上皮细胞中不必要的氧化还原失衡,这可能导致各种吸烟诱导的肺疾病,包括慢性阻塞性肺病。我们的研究结果表明细支气管上皮是一种新的抗氧化治疗方法的细胞靶点,它可能保护肺免受氧化损伤。
致谢
笔者想感谢洋子铃木(医学北海道大学医学院,札幌,日本),晶科畑(帝人制药有限公司,日本东京),胜铃木(北海道大学医学院)提供援助,免疫组化和激光捕获显微切割,盖尔L.格里芬(医学华盛顿大学医学院和巴恩斯 - 犹太医院,圣路易斯,密苏里州,美国)与ISH,康幸Nasuhara(北海道大学医学院)援助得分CT扫描,Ichio滨村和高桥弘(帝人都制药有限公司),桂永井和Takyuki吉田(医学双方北海道大学医学院)的吸烟小鼠和Robert M.高级(医学华盛顿大学医学院和巴恩斯 - 犹太医院)的手稿的批判性阅读。
脚注
这篇文章有提供补充材料www.www.qdcxjkg.com
支持声明:本研究由日本厚生劳动省呼吸衰竭研究小组支持,日本文部科学省科学研究基金,NIH基金P50 HL084922和PO1 HL29594;艾伦和伊迪丝沃尔夫慈善信托基金/巴恩斯犹太医院基金会。
利益冲突:无声明。
- 已收到2012年4月9日。
- 接受2012年9月21日。
- ©2013年











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}