


摘要
皮质类固醇不敏感(CI)是治疗严重哮喘的主要障碍。尽管研究深入,但CI的分子机制仍不确定。本研究的目的是确定严重哮喘中皮质类固醇作用的异常,并确定长效β的分子机制2肾上腺素能激动剂(LABAs)福莫特罗和沙美特罗对体外恢复重度哮喘患者皮质类固醇敏感性的作用。从16例重度皮质激素不敏感哮喘患者、6例轻度皮质激素敏感哮喘患者和11名健康志愿者中采集外周血单个核细胞(PBMCs)。以肿瘤坏死因子-α (TNF-α)诱导的白细胞介素(IL)-8的产生来测定皮质类固醇(地塞米松)的敏感性。采用免疫沉淀- western blotting分析和激酶磷酸化阵列技术,对IL-2/ il -4处理的PBMCs糖皮质激素不敏感模型中糖皮质激素受体(GR)磷酸化和激酶磷酸化进行评价。重度哮喘患者体外皮质类固醇对TNF-α-诱导的IL-8产生的敏感性明显低于健康志愿者和轻度哮喘患者。在严重哮喘中观察到的CI与GR核易位减少和GR过度磷酸化有关,这可被LABAs逆转。在IL-2/ il -4处理的pbmc中,LABAs抑制Jun-NH的磷酸化2-末端激酶和p38丝裂原活化蛋白激酶-γ (p38MAPK-γ)以及GR。此外,通过RNA干扰敲低p38MAPK-γ的细胞在IL-2/IL-4存在下不发生CI。此外,p38MAPK-γ蛋白在一些严重哮喘患者的PBMCs中表达上调。综上所述,p38 MAPK-γ激活会损害皮质类固醇的作用,而LABAs抑制p38 MAPK-γ具有治疗严重哮喘的潜力。
介绍
大多数哮喘患者的症状现在通过吸入皮质类固醇得到有效控制。然而,大约5%的哮喘患者对皮质类固醇反应不佳,或需要大剂量吸入或口服皮质类固醇来控制哮喘症状,尽管副作用仍然是一个问题。因此,皮质类固醇不敏感(CI)带来了相当大的管理问题,占哮喘医疗保健支出的不成比例(Leung and Szefler, 1998;Adcock and Ito, 2004)。
糖皮质激素的生物学作用是由糖皮质激素受体(gr)介导的,这些受体通常位于细胞质中。皮质类固醇穿过细胞膜与GR结合,然后GR易位进入细胞核,其同型二聚体与皮质类固醇反应的抗炎基因(如分泌性白细胞蛋白酶抑制剂)启动子区的糖皮质激素反应元件DNA结合(SLPI)、丝裂原活化激酶磷酸酶-1 (MKP-1)和糖皮质激素诱导亮氨酸拉链(GILZ),增加基因转录。除了这种GR-糖皮质激素反应元件结合外,GR还可以通过与促炎dna结合转录因子如激活蛋白-1和核因子-κB (NF-κB)形成抑制性相互作用,或通过募集组蛋白去乙酰化酶2 (伊藤等人,2006,b)。因此,GR核易位是皮质类固醇作用的必要和关键步骤。然而,正如我们之前所报道的(Matthews et al., 2004),一些严重哮喘患者出现GR核易位缺陷。
大量研究证明了皮质类固醇不敏感的可能机制,如转录因子的过度表达(Adcock et al., 1995),组蛋白去乙酰化酶还原(Cosío et al., 2004;Hew et al., 2006)和增加的诱饵受体(Leung et al., 1998)。GR的翻译后修饰,如磷酸化、乙酰化和泛素化,也是皮质类固醇抗性机制的重要组成部分(伊藤等人,2006,b)。例如,Rogatsky et al. (1998)研究表明,一旦大鼠GR的Ser467(相当于人GR的Ser226)被c-Jun n末端激酶(JNK)磷酸化,GR的转录激活能力就会降低。Irusen等人(2002)结果表明,p38丝裂原活化蛋白激酶(MAPK)-α和-β同工型抑制剂可抑制白细胞介素(IL)-2/IL-4诱导的全细胞提取物中GR磷酸化Rogatsky et al. (1998)结果表明GR at Ser-246未被p38 MAPK磷酸化。因此,据报道GR磷酸化与CI相关,但尚未在临床样本中检测到GR磷酸化。
p38mapk -γ是p38MAPKs的四种同工异构体之一(Mertens et al., 1996;昆达等人,1997)。这种激酶也被称为应激活化蛋白激酶-3、细胞外信号调节激酶6或MAPK12,能够磷酸化突触后密度95/大圆盘/闭塞带基元的蛋白,如应激活化蛋白(SAP) 90和SAP97。p38MAPK-γ被环境应激激活,如氧化应激和渗透应激,或促炎细胞因子,并磷酸化几个下游靶点。p38 MAPK-γ在T淋巴细胞、巨噬细胞和骨骼肌细胞中均有表达,但其功能尚不明确。
长效β的组合2-激动剂(LABA)与低剂量吸入皮质类固醇相比,比单独使用该药物或单独使用高剂量吸入皮质类固醇(Reynolds et al., 2005;Miller-Larsson和Selroos, 2006)。单独的LABAs已被证明可诱导平滑肌细胞和成纤维细胞的GR核易位(Eickelberg等人,1999),并增强体内及体外的皮质类固醇作用(Pang和Knox, 2000;Roth et al., 2002;Usmani et al., 2005)。因此,LABAs可能会增强皮质类固醇的抗炎作用,但其分子机制尚未完全阐明。本研究表明,p38 MAPK-γ通过GR的过度磷酸化导致严重哮喘患者对皮质类固醇不敏感。此外,我们发现类固醇敏感性和缺陷机制被LABAs逆转。
材料与方法
材料。
Formoterol [rac- (R,R)-N-[2-羟基-5-[1-羟基-2-[1-(4-甲氧基苯基)丙烷-2-氨基]乙基]苯基]甲酰胺]和沙美特罗[(R,年代) 2-(羟甲基)-4-{1-羟基-2-[6-(4-苯基丁氧基)己胺]乙基}苯酚]由阿斯利康(瑞典隆德)和葛兰素史克(英国格林福德)分别提供。地塞米松((8年代,9R, 10年代, 11年代, 13年代, 14年代、16R, 17R) 9-fluoro-11 17-dihydroxy-17 - (2-hydroxyacetyl) -10年,13日,16 - trimethyl-6, 7, 8, 9, 10, 11, 12日,13日,14日,15日,16日17-dodecahydro-3H-cyclopenta [一个]菲-3- 1]和SB203580[4-[4-(4-氟苯基)-2-(4-甲基亚砜苯基)-1]H-咪唑-5-基吡啶]购自Sigma-Aldrich (Poole, UK)。
科目。
11例健康非吸烟受试者(平均年龄±s.e.m., 35.8±1.6岁;三个女人;1 s内平均用力呼气量(FEV)1±s.e.m.,预测的98.2±2.2%),轻度哮喘6例(41.5±3.3年;三个女人;FEV1,为预测的84.5±4.6%),重度哮喘16例(年龄,35.1±2.6岁;11个女人;FEV1,预期55.0±3.4%)(表1)。本研究经皇家布朗普顿和哈里菲尔德医院国家卫生服务信托伦理委员会批准,所有受试者均给予书面知情同意。取50毫升血液,用Ficoll-Paque (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK)梯度分离PBMCs。
Corticosteroid-Insensitive模型。
健康志愿者pbmc与人重组IL-2 (2 ng/ml)和IL-4 (10 ng/ml)孵育48 h。
FITC-Dexamethasone合并。
pbmc与fitc偶联地塞米松(FITC-Dex;10−6M)在37°C下放置30分钟。在10的存在下测定非特异性FITC扩散−5M非共轭Dex,从总FITC荧光值中减去。核部分用低渗缓冲液(Active Motif, Rixensart, Belgium)制备,孵育10分钟,然后用含有PBS的0.1% NP-40脉冲涡流。用0.5% NP-40(含PBS)在冰上提取细胞核中的fitc -地塞米松20 min,用标准曲线测定不同浓度的FITC-Dex的浓度。在荧光板读取器中检测488 nm处的FITC。
GR和磷酸化GR的检测。
全细胞提取液采用改良放射免疫沉淀缓冲液(伊藤等人,2000)。用tris -甘氨酸SDS/聚丙烯酰胺凝胶电泳分离全细胞提取物,并将其转移到硝化纤维素膜上检测GR。GR水平归一化为β-actin表达。为了检测磷酸化的GR,使用抗GR抗体偶联琼脂糖A/G (Santa Cruz Biotechnology Inc., Santa Cruz, CA)对GR进行免疫纯化,并使用sds -聚丙烯酰胺凝胶电泳/Western blotting对GR进行分离。用抗泛磷酸丝氨酸抗体(Santa Cruz Biotechnology Inc.)检测磷酸化水平,并归一化为GR表达。此外,使用抗磷酸化(S226) GR抗体(New England Biolabs UK Ltd., Hitchin, Hertfordshire, UK)检测Ser226位点磷酸化的GR。使用Labworks软件(Ultra-Violet Products, Cambridge, UK),通过密度测定法(UVP Bioimaging Systems, Cambridge, UK)计算波段密度。
一。
收集细胞进行总RNA分离。市售试剂盒用于提取细胞总RNA (RNeasy;QIAGEN, Crawley, UK)并进行逆转录(Omniscript RT;试剂盒)。p38 MAPK-α, -γ, -δ和管家基因的基因转录水平GNB2L1或GAPDH采用TaqMan PCR试剂盒(Applied Biosystems, Warrington, UK),在Rotor-Gene 3000 PCR仪(Corbett Research, Mortlake, NSW, Australia)上进行实时PCR定量。
ELISA。
细胞用地塞米松(10−12-10年−6M)在存在或不存在LABA的情况下刺激30分钟,然后用TNF-α (1 ng/ml)或抗人CD3 (10 μg/ml)和CD28抗体(8 μg/ml)的组合刺激过夜(BD Biosciences, Oxford, UK)。采用夹心ELISA法测定上清液中IL-8和IL-2水平(人IL-8 Duoset ELISA;R&D Systems Europe, Abingdon, UK)根据制造商的说明。
激酶剖析。
根据制造商的说明,使用Human Phospho-MAPK Array Kit Proteome Profiler (R&D Systems Europe)评估19种不同激酶的磷酸化。Western blotting检测HSP27(磷酸化和总)和p38MAPK-γ(磷酸化p38MAPK/应激激活蛋白激酶和总)。所有抗体均购自欧洲研发系统公司。
细胞中磷酸化和总p38MAPK-γ的测定。
采用p38MAPK-γ (Thr183/Tyr185)磷酸化和基于细胞的总酶联免疫吸附试验(Duoset胞内酶联免疫吸附试验)检测健康受试者外周血中p38MAPK-γ磷酸化和总p38MAPK-γ。简单地说,用人重组IL-2 (2 ng/ml)和IL-4 (10 ng/ml)刺激细胞48小时,然后用福莫特罗、沙美特罗或沙丁胺醇处理20分钟。根据制造商的说明,收集细胞并使用裂解缓冲液裂解。
RNA干扰。
p38 MAPK-δ (MAPK13)和p38 MAPK-γ (MAPK 12)的短干扰RNA (siRNA)购自Dharmacon Inc. (Colorado Springs, CO, USA),使用AMAXANucreofector (Lonza GmbH, Cologne, Germany)按照制造商的说明书进行转染(各100 nM)。细胞孵育24小时,然后再用IL-2/IL-4刺激48小时。非特异性对照双工(混乱的寡核苷酸,47% GC含量)也从Dharmacon RNA Technologies (Lafayette, CO)购买。
统计分析。
结果用均数±标准差表示,方差分析采用Kruskal-Wallis分析;当有意义时,由曼·惠特尼进行比较U使用PC分析软件包SPSS 10.0 (SPSS Inc., Chicago, IL)或Prism 4 (GraphPad Software, San Diego, CA)进行测试。采用Welch’s分析各处理组体外数据的差异t测试。用Spearman方法确定两个参数之间的相关性。一个p值< 0.05认为有统计学意义。
结果
重度哮喘患者因GR核易位缺陷而对皮质激素不敏感。
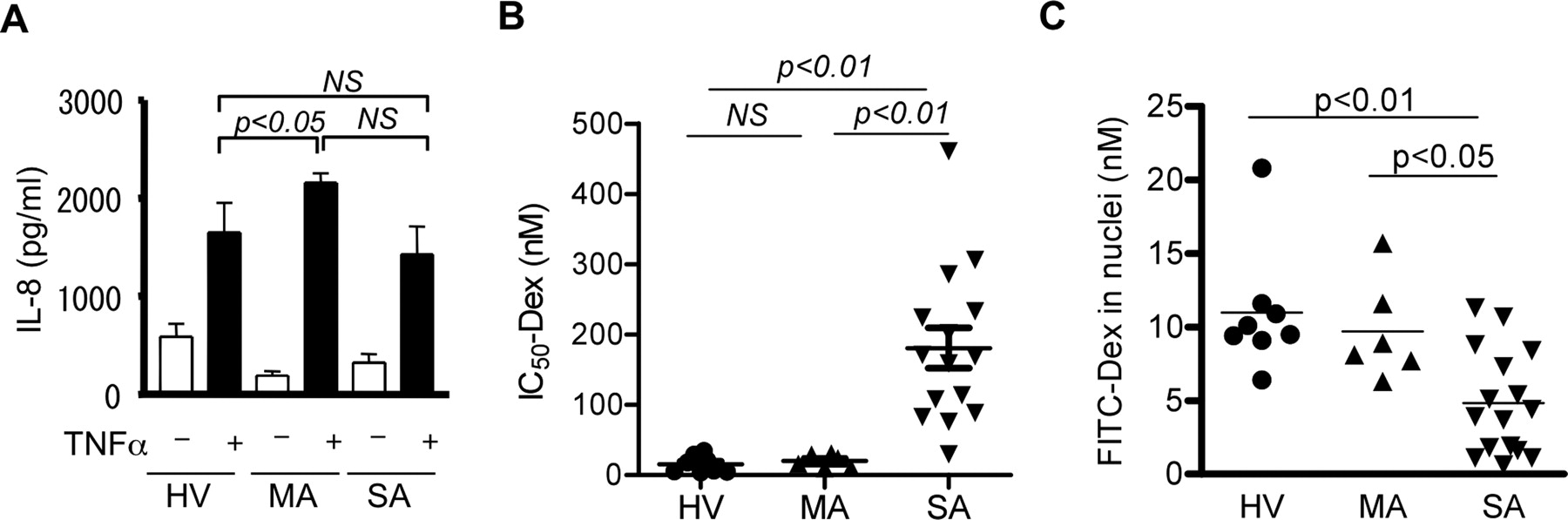
如图所示图1A,严重哮喘患者在TNF-α刺激下PBMCs产生IL-8 (SA;1430±286 pg/ml),与健康志愿者相似(HV;1650±304 pg/ml),但轻度哮喘患者IL-8的产生明显更高(MA;(2160±94.9 pg/ml)高于HV组。相反,当地塞米松(Dex-IC)的抑制活性达到50%时50)对TNF-α-诱导的IL-8释放的影响,作为皮质类固醇敏感性指数Dex-IC50SA患者的PBMCs值(181±28.7 nM)显著高于HV患者(15.5±4.2 nM);pMA患者(20.0±3.8 nM;p< 0.01) (图1B)。

TNF-α-诱导IL-8产生(A),地塞米松敏感性抑制这一反应(IC)50敏捷;B),并通过FITC-Dex掺入试验(C)测定从HV和MA或SA患者获得的pbmc的GR核易位。NS,不显著。
在PBMCs中,GRα mRNA的表达差异无统计学意义(GRα/GNB2L1: SA, 70.4±17.8;Hv, 102.4±34.4;MA, 89.3±24.3),GRβ mRNA表达量(GRβ/GNB2L1: SA, 0.00042±0.00015;Hv, 0.000002±0.0000005;MA, 0.00043±0.00029)或GRα蛋白表达(GRα/β-actin: SA, 1681±205;Hv, 4914±763;Ma, 3050±670)。
作为配体结合后GR核易位的标志,我们测定了细胞核中FITC-Dex的含量。因为FITC-Dex的抗炎效果比未标记的Dex (IC)低10倍50值,FITC-Dex, 5.6 × 10−8M与未标记的Dex, 4.3 × 10−9M对il -1β诱导的A549细胞IL-8产生的影响),我们使用了相对高浓度(10−6M)的FITC-Dex的分析。如图所示图1C、SA细胞GR核易位明显受损[细胞核FITC-Dex, 4.8±0.9 nM;p< 0.01 vs HV(11.0±1.5 nM);p< 0.05 vs MA(9.7±1.4 nM)]。此外,IC之间存在显著的负相关50-Dex值对TNF-α-诱导的IL-8释放及细胞核中FITC-Dex含量的影响(Spearmanr=−0.55;p= 0.0035),表明右美托咪定疗效较差与GR核易位缺陷有关。
福莫特罗通过增强GR核易位逆转SA患者PBMCs的皮质类固醇不敏感。
福莫特罗(formoterol, 1 nM)治疗可降低Dex-IC50TNF-α诱导SA患者外周血中IL-8释放的价值(Dex-IC)50加与不加FOR分别为42.8±21.0 nM和182.5±28.2 nM;n= 6,p< 0.05;图2A)和沙美特罗(SAL, 100 nM) (Dex-IC)也有类似的变化,但效果较低50有无SAL,分别为83.7±16.7 nM和180.7±28.7 nM;n= 15,p< 0.05) (表2)。在SA患者中,1 nM FOR治疗的改善指数优于SAL (SA: FOR 4.3 vs SAL 2.2;表2),但两组疗效差异无统计学意义。对于HV (Dex-IC)患者,FOR和SAL均未显著改变皮质类固醇敏感性50,带FOR, 21.5±4.8 nM;SAL为31.0±8.3 nM;未处理,15.5±4.2 nM;表2)或MA患者(Dex-IC)50SAL组为14.7±3.5 nM,未处理组为20.0±3.8 nM;未在MA患者中检测FOR)。在SA (Dex-IC)中,皮质类固醇对tnf α-诱导的IL-8释放的敏感性降低的同时,皮质类固醇对CD3/ cd28诱导的PBMC中IL-2释放的敏感性也降低50SA为77.6±25.4 nM;n= 8,而HV为11.5±3.3 nM;n= 7, MA为40.1±16.4 nM;n= 6),经SAL (100 nM)逆转(Dex-IC)50SAL为30.4±14.4 nM;p= 0.0065;未检测FOR),提示SAL增加了Dex的敏感性。此外,在SA中,通过FITC-Dex评估,FOR (1 nM)增强了GR核易位(核FITC-Dex与未FOR相比,9.5±1.5 vs 4.8±0.9 nM;p< 0.05;图2B)。SAL (100 nM)的结果相似(核FITC-Dex,有与没有SAL, 14.2±2.7 vs 4.8±0.9 nM;p< 0.05;n= 16)。Dex-IC的改进50增加Dex-IC的比例50没有FOR和Dex-IC50与FOR相比,没有FOR的细胞核中FITC-Dex比有FOR的细胞核中FITC-Dex的比例降低;斯皮尔曼r=−0.77,p= 0.042)。此外,Dex-IC的改进50与细胞核中的FITC-Dex值也呈负相关(Spearmanr=−0.73,p= 0.0013;图2C),提示有GR核易位缺陷的pbmc对FOR治疗更敏感。

1 nM FOR对地塞米松敏感性抑制TNF-α-诱导的IL-8产生的影响50敏捷;A)和FITC-Dex掺入法测定的GR核易位(B)在SA患者的pbmc中进行评估。C,细胞核中FITC-Dex与Dex-IC比值的相关性50作为皮质类固醇敏感性改善的指标。
GRs在严重哮喘的PBMCs中高度磷酸化,福莫特罗使其去磷酸化。
如图所示图3, A和B, ppbmc细胞质中的GR在SA的丝氨酸残基处被高度磷酸化(磷酸化-GR/GR比值为0.48±0.065;图3B)与HV组比较(0.22±0.083;图3B)和MA(0.25±0.050;没有显示)。孵育20 min后,FOR显著抑制GR磷酸化(磷酸化-GR/GR比值,SA为0.58±0.093,10 nM FOR为0.22±0.037,1 M FOR为0.25±0.050;p与未处理SA相比< 0.05;图3C)。

GR磷酸化在重度哮喘中的作用及福莫特罗的恢复作用。SA和HV患者外周血中免疫沉淀GR的丝氨酸磷酸化(A和B)。FOR对SA患者外周血中GR磷酸化的影响(C)和IL-2/ il -4处理的HV外周血中GR磷酸化的影响(D和E)。p经Wilcoxon秩对检验,与严重哮喘PBMCs (C)或与IL-2/ il -4治疗无FOR的PBMCs (E)相比< 0.05。F、GR 226丝氨酸磷酸化也在HV (n= 4), SA患者(n= 6);#,pSA与HV的比值< 0.05。
IL-2/IL-4对HV诱导的皮质类固醇不敏感PBMCs GR核易位受损的治疗(细胞核中FITC-Dex,有与没有IL-2/IL-4, 2.3±1.1 vs 10.7±1.3 nM;p< 0.05)。IL-2/IL-4处理的PBMCs也可诱导GR磷酸化(有IL-2/IL-4与无IL-2/IL-4处理的磷酸化GR/GR之比分别为0.42±0.047和0.11±0.024;p< 0.05;图3(D和E),而FOR显著抑制了这种效应(10 nM FOR时,phospho-GR/GR的比值为0.17±0.044;0.17±0.048,1 nM FOR;为,p< 0.05,对照组为0.42±0.047;图3(D和E)。此外,在相同的样品中测定了Ser 226位点磷酸化的GR。如图所示图3F, GR中Ser 226位点的磷酸化也显著(p< 0.05), SA患者PBMCs升高。
p38 MAPK-γ激活引起皮质类固醇不敏感并被福莫特罗抑制。
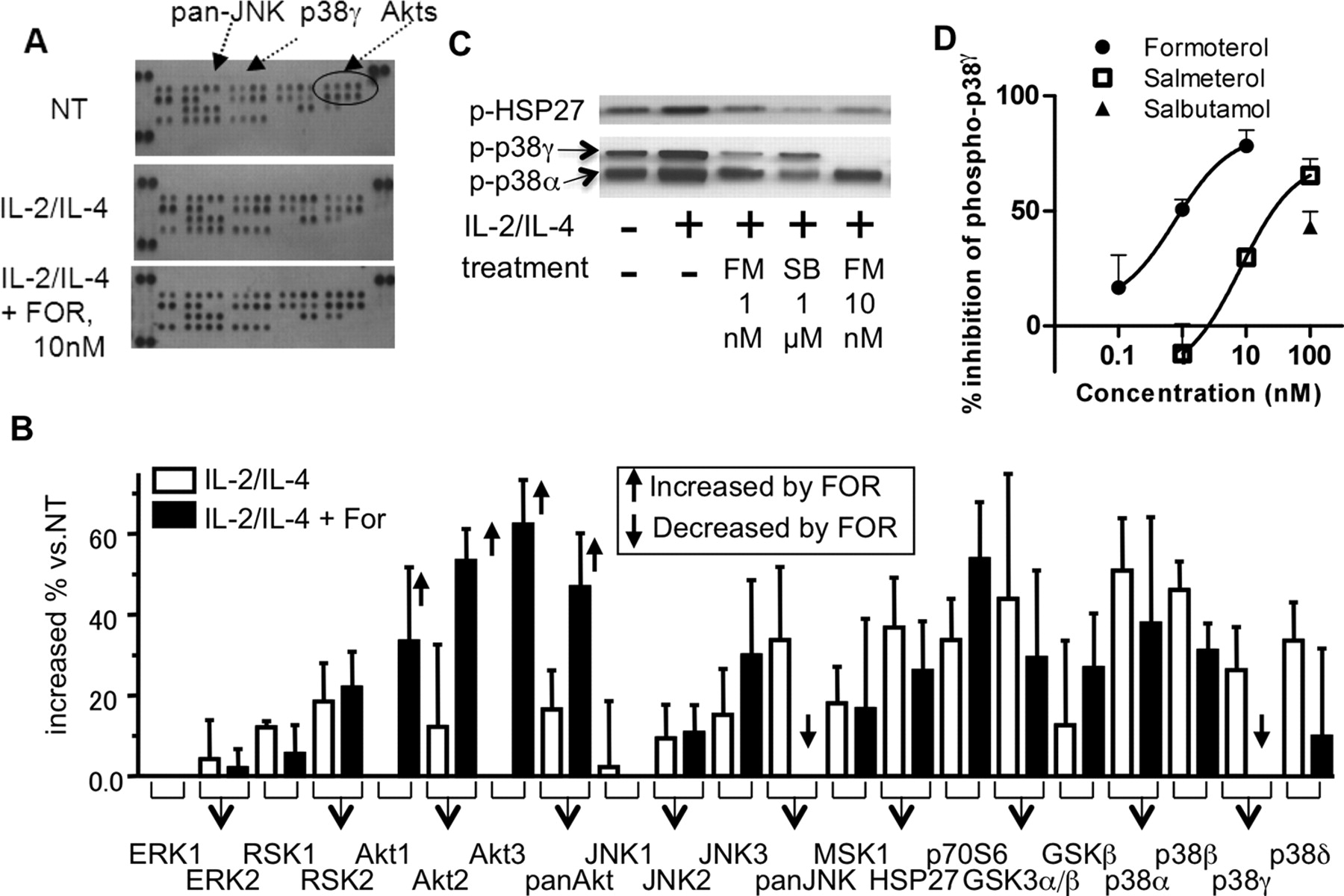
为了确定IL-2和IL-4在细胞孵育48小时后激活的激酶(直接或间接)可能参与GR磷酸化,我们对健康志愿者的PBMCs进行了激酶磷酸化阵列分析。暴露于IL-2/IL-4 48小时后,p38 MAPK (α、β、γ和δ)、RSK1和-2、Akt2(和pan-Akt)、JNK2、JNK3(和pan-JNK)、MSK1、HSP27、糖原合成酶激酶3α和-β、p70S6 (图4(A、B)。本实验采用较高浓度的FOR (10 nM)达到最大效果。FOR仅显著抑制pan-JNK和p38 MAPK-γ的磷酸化(图4IL-2/IL-4上调p38 MAPK-α和-β的磷酸化,而FOR不抑制p38 MAPK-α和-β的磷酸化。Western blot分析还显示,FM (1 nM和10 nM)降低了p38MAPK-γ的磷酸化,但没有降低p38α的磷酸化(图4相比之下,尽管p38MAPK α/β抑制剂SB203580抑制p38MAPK-α下游分子HSP27的磷酸化,但它不抑制p38MAPK-γ的磷酸化(图4C)。这些结果也被量化并显示在补充图1中。此外,通过基于细胞的酶联免疫吸附试验(ELISA)评估β-肾上腺素能受体激动剂对p38MAPK-γ磷酸化的抑制作用。如图所示图4D、FOR和SAL浓度依赖性地抑制p38MAPK-γ的磷酸化和IC50值分别为0.97和26 nM,但沙丁胺醇在100 nM处有部分抑制作用。

IL-2/ il -4处理的健康志愿者pbmc的激酶谱分析。激酶磷酸化阵列的代表性图像(A)和密度分析(B)。将pbmc与IL-2/IL-4孵育48 h,再与10 nM的for孵育20 min,计算每个点的密度,并测定与未处理相比增加的百分比。开放栏表示单独使用IL-2/IL-4治疗,封闭栏表示IL-2/IL-4联合FOR治疗。用粗体箭头表示FOR的显著诱导或减少。数据以均数±标准差绘制n= 3个独立实验。C, p38MAPK-α和-γ磷酸化和HSP27的Western blot。细胞处理如A. SB, SB203580所示。D,基于细胞的p38MAPK-γ磷酸化ELISA。福莫特罗、沙美特罗和沙丁胺醇分别在处理IL-2/IL-4 48 h后处理20 min,计算并绘制与IL-2/IL-4对照的抑制效果。
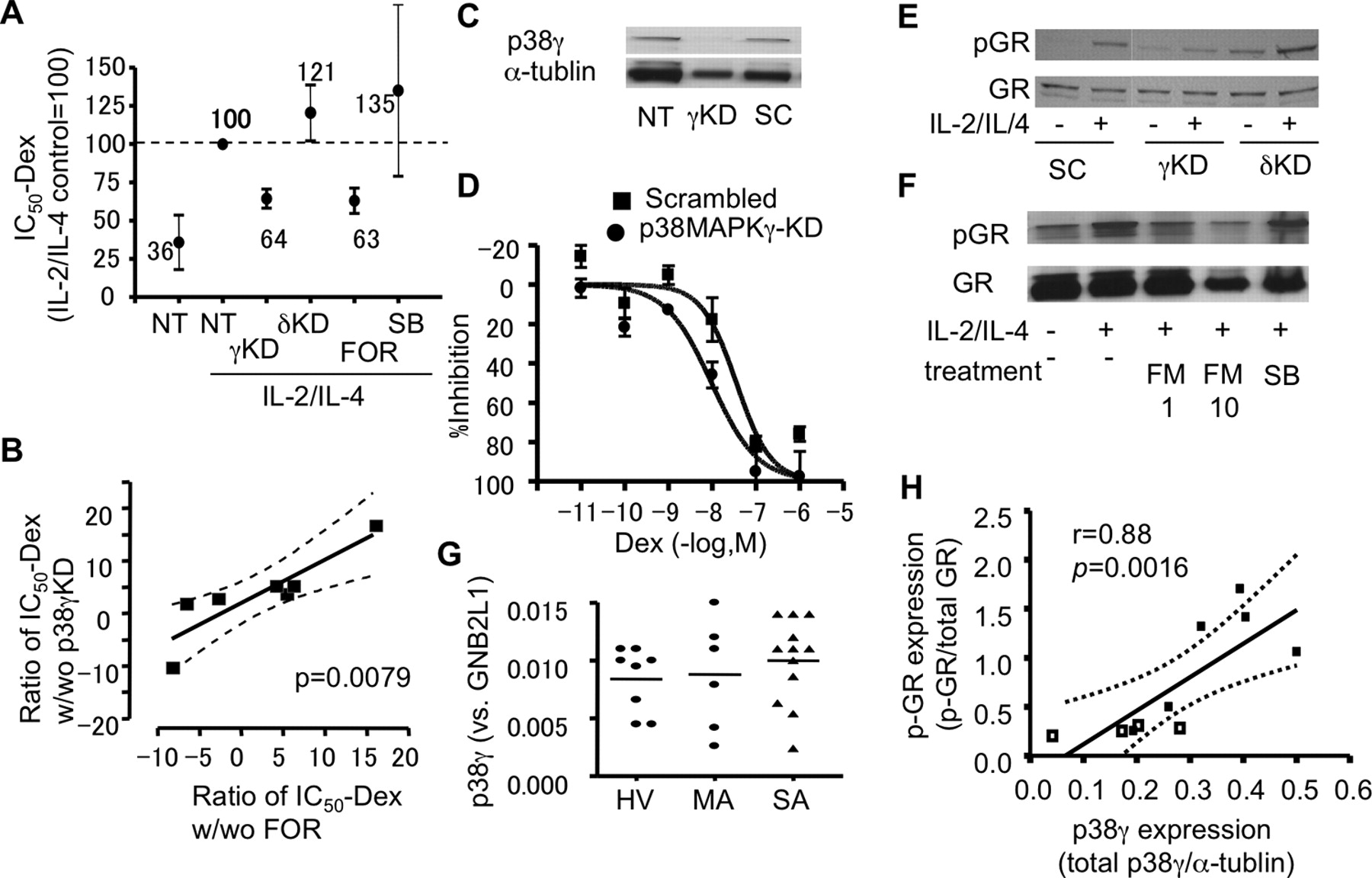
将针对p38 MAPK-γ和-δ的sirna转染到健康志愿者的pbmc中以获得敲低(KD)。24 h后,p38MAPK-γ和-δ mRNA水平降低75%以上。用IL-2和IL-4处理KD细胞(和正常细胞)48小时,在Dex存在或不存在的情况下用CD3/CD28刺激以测定Dex- ic50IL-2的产生在RNA干扰p38 MAPK-γ KD的细胞中,未观察到预期的IL-2 / il -4诱导的皮质类固醇抵抗CD3/ cd28诱导的IL-2释放(图5相比之下,p38 MAPK-δ KD并不能阻止皮质类固醇不敏感(图5A)。尽管FOR (1 nM)逆转了IL-2/ il -4依赖性皮质类固醇的不敏感性,但选择性p38 MAPK-α和-β抑制剂SB203580并没有恢复IL-2/ il -4处理的PBMCs的皮质类固醇敏感性(图5A) Dex-IC的改进50对于CD3/ cd28诱导IL-2释放的影响与p38 MAPK-γ KD的改善密切相关(r= 0.53,p= 0.0079;图5B)相同的科目。

p38 MAPK-γ导致皮质类固醇不敏感。A, RNA干扰p38 MAPK-γ或-δ KD对健康志愿者PBMCs中IL2/ il -4诱导地塞米松抵抗CD3/ cd28诱导的IL-2释放敏感性的影响。在CD3/CD28处理前,FOR (1 nM)或SB-203580 (SB, 1 μM)也预孵育20 min。B、健康志愿者各样本福莫特罗疗效与p38 MAPK-γ KD疗效的Spearman相关性分析。C, U937细胞中p38MAPK-γ敲低的代表性图像。SC,混乱的寡核苷酸。E, p38 MAPK-γ对U937细胞地塞米松抗炎作用对TNF-α-诱导的IL-8 (D)和IL-2/IL-4对GR磷酸化的影响。F, FOR(1或10 nM)或SB-203580 (SB;1 μM)对U937细胞GR磷酸化的影响。G、p38 MAP-γ mRNA在SA、HV和MA患者外周血中的表达。 H. The relationship between phosphorylated GR (p-GR) corrected to total GR expression and p38MAPK-γ protein expression corrected to α-tublin, evaluated by Western blot analysis in PBMCs from three healthy volunteers and six patients with SA (Supplemental Fig. 2).
U937细胞(一种单核细胞系)转染针对p38 MAPK-γ或重组寡核苷酸的sirna作为对照24小时图5C, p38MAPK-γ在这种情况下明显被敲低。然后用IL-2/IL-4处理细胞48 h,用TNF-α (1 ng/ml)刺激细胞,在Dex存在或不存在的情况下测定Dex- ic50值。Dex通过IC抑制tnf α诱导的IL-8生成50但在IL-2/IL-4 (Dex- ic)存在下,Dex效价降低50, 36 nM)。然而,p38 MAPK-γ KD使Dex的剂量响应曲线向左移动(Dex- ic)50, 9.6 nM与36 nM,用打乱寡核苷酸处理作为对照;所有细胞均用IL-2/IL-4处理;p< 0.05),提示Dex可抵消IL-2/ il -4诱导的皮质类固醇不敏感(图5相反,p38 MAPK-δ的KD不影响IL-2/ il -4诱导的皮质类固醇不敏感(Dex-IC)50, 21 nM与36 nM混合寡核苷酸处理;不显著的;数据未显示)。此外,p38 MAPK-γ KD,而不是p38 MAPK-δ KD,也抑制了IL-2/IL-4对GR的磷酸化(图5E),其中FOR而不是SB203580抑制了U937体系中GR的磷酸化(图5当p38MAPKs mRNA在PBMCs中被检测时,HV和SA患者在编码p38MAPK -α, -β和-δ(数据未显示)或p38MAPK-γ (图5G)。然而,p38 MAPK-γ的mRNA表达与GR核易位(核FITC-Dex)之间存在良好的相关性(p< 0.05,数据未显示),提示p38MAPK-γ升高导致GR功能缺陷。更重要的是,当分析3例HV和6例SA患者外周血中p38MAPK-γ蛋白的表达并将其归一化为α-微管蛋白(一种管家基因)表达时(补充图2),GR磷酸化与p38MAPK-γ蛋白表达之间存在显著相关性(图5为了进一步证实p38MAPK-γ在皮质类固醇不敏感中的作用,p38MAPK-γ在U937细胞和Dex-IC中也过表达50是确定。p38MAPK-γ过表达的U937细胞Dex-IC明显升高50IL-2/IL-4处理后tnf α诱导的IL-8释放(补充图3)。
讨论
严重哮喘以皮质类固醇不敏感炎症为特征。我们在这里展示了IC50在SA患者的PBMCs中,Dex对TNF-α刺激的IL-8释放的值比HV或MA患者高约10倍,表明SA患者的PBMCs在体外也对类固醇不敏感。
在分子水平上,在SA患者的细胞中观察到的皮质类固醇反应性降低归因于GR数量减少,配体对GR的亲和力改变,GR与DNA结合能力降低,竞争DNA结合的炎症转录因子(如激活蛋白-1)表达增加,或组蛋白去乙酰酶-2减少(Adcock et al., 2006)。在本研究中,GRα mRNA和蛋白的表达无显著差异。SA患者的GRβ mRNA表达可能升高,但差异不显著,可能是由于缺乏动力所致;然而,一些报告显示,GRβ过表达对SA患者的皮质类固醇不敏感并不重要(Irusen et al., 2002;Torrego et al., 2004)。Irusen等人(2002)也证实了SA患者细胞核中GR的亲和力降低,尽管我们在本研究中没有分析细胞核中的GR功能(Irusen et al., 2002)。相反,我们在细胞质中发现了GR的缺陷。
细胞核内活性GR的增加对GR的作用至关重要。对于细胞核中配体结合GR的检测,我们通过测定细胞核中fitc共轭Dex的量,而不是使用经典的免疫细胞化学或Western blotting方法,这些方法不定量,耗时且需要大量细胞。由于fitc偶联的Dex对il -1β诱导的A549细胞IL-8产生的抗炎作用比未标记的Dex弱10倍,因此我们使用了相对高浓度(10−6M)的FITC-Dex的分析。免疫细胞化学检测中GR阳性细胞核的百分比与健康志愿者PBMC细胞核中FITC-Dex的绝对值有很好的相关性(数据未显示,r= 0.65,p< 0.05),提示FITC-Dex方法可用于定量少量细胞GR核易位。我们发现,在SA PBMCs中,GR核易位明显受损(图1C),这是由先前的报告(Matthews et al., 2004)。此外,IC之间存在显著的负相关50-Dex值对TNF-α-诱导的IL-8释放和细胞核中FITC-Dex的量的影响,表明GR核易位越少的患者对皮质类固醇不敏感(Spearmanr=−0.55p= 0.0035)。据报道,在一些体外系统和临床试验中,甚至在我们的PBMC系统中,LABAs能够增强皮质类固醇的敏感性;我们还证实,加用FOR (1 nM)和SAL (100 nM)可降低Dex-IC50TNF-α-诱导的白介素8在SA患者外周血中释放的价值(图2A),尽管SAL对皮质类固醇敏感性的恢复效果弱于FOR。无论FOR还是SAL都不能改变HV和MA患者的皮质类固醇敏感性(表2)。此外,用FITC-Dex评估FOR (1 nM)增强SA患者GR核易位(图2B).皮质类固醇敏感性水平和FOR或SAL的恢复不受当前药物(口服类固醇或吸入类固醇治疗;数据未显示)。Dex-IC的改进50与FOR (Dex-IC比)50有或没有FOR)与FOR对细胞核中FITC-Dex积累的改善密切相关(Spearman)r=−0.77,p= 0.042),提示FOR通过增强GR核易位逆转皮质类固醇不敏感。Dex-IC的改进50与细胞核的FITC-Dex值呈负相关(Spearmanr=−0.73;p= 0.0013;图2C),提示GR核易位较少的患者对皮质类固醇耐药的for依赖性逆转更敏感。
据报道,GR是一种磷酸化蛋白,非活性GR的磷酸化可能阻断随后的激素结合,影响GR亚细胞定位和GR核胞质通过核孔复合物的运输(Ismaili and Garabedian, 2004)。我们证明,与HV和MA患者相比,SA患者细胞质中的GR在丝氨酸残基处高度磷酸化(图3(A和B), GR磷酸化与FITC-Dex核易位(作为GR核易位能力的指标)之间存在良好的相关性。据报道,Ser226的磷酸化会导致gr介导的转录激活缺陷(Rogatsky et al., 1998)或加强核出口(Itoh et al., 2002)。非常有趣的是,与HV患者相比,SA患者Ser226的GR磷酸化水平更高(图3更重要的是,FOR显著抑制GR的磷酸化(图3C). IL-2/IL-4治疗,已知与SA中出现的皮质类固醇不敏感相似(Kam et al., 1993;Larsson et al., 1997;Irusen et al., 2002),也诱导了GR (图3D和E),并被FOR (图3D和E)。
几种激酶,如MAPK、周期蛋白依赖性激酶、糖原合成酶激酶-3和JNK被报道可使GR磷酸化,每种激酶对潜在的磷酸化位点都有不同的特异性(伊藤等人,2006)。激酶磷酸化阵列分析表明,IL-2/IL-4处理上调了几种应激激酶的磷酸化,包括p38 MAPK (α, β, γ, δ)和JNK (图4非常有趣的是(10)−8M)仅显著抑制pan-JNK和p38 MAPK-γ的磷酸化(图4, A和B)。据报道,JNK催化GR Ser226磷酸化并使GR失活(Rogatsky et al., 1998)。富含亮氨酸的序列位于Ser246在大鼠GR中(在人GR中Ser226)也被报道参与核输出(Itoh et al., 2002),该位点的磷酸化可能增加GR核输出,从而使GR转录增强失活,最终导致磷酸化的GR在细胞质中积累。因此,在这一发现中,JNK将是GR磷酸化的关键激酶,但p38 MAPK-γ在GR磷酸化和皮质类固醇效应中的作用之前尚未报道。
在U937细胞和pbmc中,p38 MAPK-γ被RNA干扰敲低,IL2/IL-4暴露不诱导皮质类固醇不敏感(图5此外,p38 MAPK-γ KD,而不是p38 MAPK-δ KD,抑制了IL-2/IL-4对GR的磷酸化(图5因此,p38 MAPK-γ似乎是调节皮质类固醇敏感性的关键激酶,可能是通过细胞质GR的磷酸化来实现的。事实上,Ser226处GR磷酸化水平较高的pbmc显示出更高水平的p38 MAPK-γ mRNA表达(图5SA患者的p38MAPK-γ蛋白表达也高于健康受试者(补充图2)。对U937细胞中p38MAPK-γ过表达的进一步分析(补充图3)支持了SA中p38MAPK-γ过表达可能是类固醇不敏感的分子机制之一的发现。
在我们的系统中,在IL-2/IL-4处理48小时后,FOR在20分钟内将磷酸化的GR和p38 MAPK-γ转化为非磷酸化形式,这表明FOR可能通过p38 MAPK-γ的去磷酸化而不是直接抑制p38 MAPK-γ来实现GR的去磷酸化。事实上,FOR(或SAL) (100,1000 nM)并没有直接抑制p38 MAPK-γ激酶活性[激酶谱仪分析(Millipore, Billerica, MA;数据未显示]。也就是说,FOR可能会增强一种特定的磷酸酶,使p38 MAPK-γ去磷酸化。特异性地去磷酸化p38 MAPK-γ的磷酸酶尚未被确定。然而,据报道,cAMP-PKA信号可增强蛋白磷酸酶(PP)2A (Feschenko et al., 2002)。PP2C也被称为camp偶联磷酸酶(横山等人,1995),据报道PP1和PP5参与了GR定位(DeFranco et al., 1991;Dean et al., 2001;Hinds and Sánchez, 2008)。LABA对p38MAPK-γ(和GR)去磷酸化的分子机制有待进一步研究。
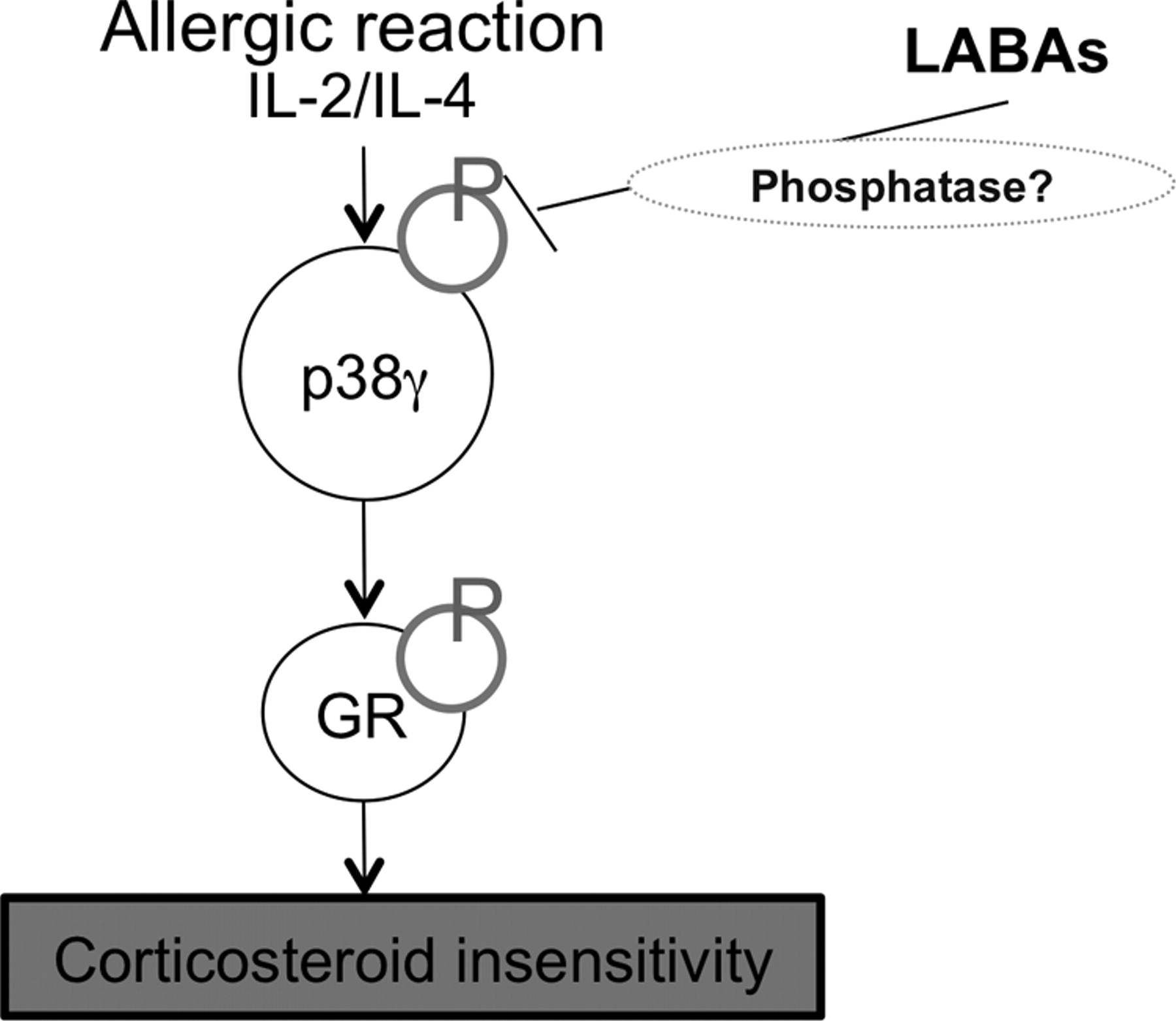
因此,LABAs恢复了磷酸化缺陷的皮质类固醇敏感性(图6)。据报道,皮质类固醇本身可增加β-肾上腺素能受体的表达(Aksoy et al., 2002)和LABAs也被报道能促进GR核易位(Eickelberg等人,1999;Usmani et al., 2005)。这应该是一个由LABA和吸入皮质类固醇联合治疗诱导的自我强化循环。皮质类固醇不敏感的严重疾病是异质性的,但我们的研究表明,至少有一个SA患者亚群的特征是GR核易位缺陷和GR过度磷酸化,这可以通过p38 MAPK-γ依赖机制被LABA逆转(图6)。我们的研究为炎症的调节提供了新的见解,并提出了治疗SA和其他炎症性疾病的新型化合物的前景。

福莫特罗恢复重度哮喘患者皮质类固醇敏感性的确切机制。通过p38MAPK-γ激活GR磷酸化,与IL-2和IL-4的产生相关的严重过敏性炎症诱导皮质类固醇不敏感。福莫特罗抑制p38MAPK-γ磷酸化,可能通过磷酸酶激活,抑制GR磷酸化。
作者的贡献
参与研究设计:梅尔卡多,托,阿德考克和伊藤。
进行实验:梅尔卡多,托,小林和伊藤。
进行数据分析:梅尔卡多,托,小林和伊藤。
手稿的:撰写或参与撰写手稿的:巴恩斯和伊藤。
致谢
我们感谢Sergei A. Kharitonov博士、Debby Campbell博士和Sally Meah博士协助提供临床样本,Misako Ito博士和Masashi Deguchi博士协助进行体外分析。
脚注
↵
 本文的在线版本(可在http://molpharm.aspetjournals.org)包含补充材料。
本文的在线版本(可在http://molpharm.aspetjournals.org)包含补充材料。本研究得到Asthma UK的支持[Grant 04-56];英国医学研究理事会[Grant G0401662];葛兰素史克公司;阿斯利康(AstraZeneca)。
文章,出版日期和引用信息可在http://molpharm.aspetjournals.org。
doi: 10.1124 / mol.111.071993。
缩写:
- CI
- 皮质类固醇不敏感
- GR
- 糖皮质激素受体
- NF -κB
- 核因子-κB
- 物
- c-Jun-NH2终端激酶
- MAPK
- 丝裂原活化蛋白激酶
- 伊尔
- 白介素
- 腊八粥
- 长效β2受体激动剂
- SB203580
- 4 - [4 - (4-fluorophenyl) 2 (4-methylsulfinylphenyl) 1H-imidazol-5-yl]吡啶
- FEV1
- 1秒内用力呼气量
- PBMC
- 外周血单核细胞
- FITC
- 异硫氰酸荧光素
- 敏捷
- 地塞米松
- 聚合酶链反应
- 聚合酶链反应
- ELISA
- 酶联免疫吸附试验
- HSP
- 热休克蛋白
- KD
- 可拆卸的
- 为
- formoterol
- 萨尔
- 氟替卡松加沙美特罗
- 高压
- 健康志愿者(年代)
- SA
- 严重的哮喘
- 妈
- 轻度糖皮质激素敏感性哮喘
- 页
- 蛋白磷酸酶。

- 收到了2011年2月26日。
- 接受2011年9月6日。
- 版权所有©2011美国药理学和实验治疗学会









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}