





文摘
健康的肺曾被认为是一个无菌器官因为标准微生物培养技术持续产生负面的结果。然而,文化无关的技术报告,大量的微生物共存的肺。有许多未知的方面,但可用的报告显示,下呼吸道微生物群:1)类似的健康受试者口咽壁厚菌门的微生物群,由成员,拟杆菌和变形菌门的门;2)显示改变吸烟者和明确的慢性呼吸道疾病的差异,尽管这些变化的时空动力学只有部分已知;和3)显示相对丰富non-cultivable细菌在慢性阻塞性肺疾病、特发性肺纤维化、囊肿性纤维化和支气管扩张,每个疾病与特定的模式。在所有这些疾病,失去多样性,平行的代表比例变形菌门(失调),一直与疾病严重程度和发作有关。然而,未知是否失调的原因或结果损害支气管肺泡表面。
最后,对细菌之间的交互功能和病毒、真菌和细菌。预计未来的研究在细菌基因表达式中,宏基因组的纵向分析和host-microbiome动物模型将有助于朝着目标微生物对呼吸道疾病的干预。
文摘
呼吸系统细菌社区是由特定类群变化在慢性呼吸道疾病http://ow.ly/j68Z30967DB
介绍
健康的肺是传统上被认为是无菌器官因为标准微生物培养技术不断产生负面的结果(1]。然而,在过去的十年里,使用文化无关的分子技术已经证明这种教条是错误的,大量的微生物有机体,包括细菌、真菌和病毒,统称为微生物,共存的肺健康受试者和呼吸道疾病患者2,3),挑战我们的理解在呼吸医学微生物学(3]。事实上,解决肺微生物群之间的关系的性质和呼吸道上皮表面似乎是最有前途的研究领域之一,呼吸医学(1]。例如,大量的证据现在支持这一概念,异常的监管host-microbiota相声在身体不同器官和不同表面可能发挥重要的致病作用几个慢性炎性疾病(4- - - - - -7]。因此,越来越多的兴趣决定的潜在价值的描述呼吸道微生物成分作为预后标记或一个元素能够指导治疗在一些呼吸道疾病(3]。这手稿反映了当前的知识水平在呼吸道微生物(见箱1为当前的术语),其unspecificities可以本质上的异质性有关的呼吸系统疾病临床分层目前正在使用。考虑到这些因素,巴塞罗那呼吸网络组织了一个国际多学科研讨会于6月3日,2016年,讨论并确定研究的挑战,重点和差距,研究未来的发展方向以及对病人和医疗保健系统的影响。相关讨论,以及车间的主要结论,总结如下。完整的演示视频,在巴塞罗那呼吸免费在线网络网站(www.brn.cat microbiome2016)。
箱1通用术语
微生物群:微生物群落成员与定义的栖息地,如人体。
微生物:遗传信息(基因)和推断基因产物的理化性能的微生物群。
人类微生物组:微生物共同发现了人体的内部和外部的栖息地。
宏基因组:猎枪随机序列的DNA样本,包括DNA的宿主和微生物来源、分析、组织和标识使用序列数据库和计算工具。
16 s rRNA (16 s rRNA)基因:30年代的小亚基的原核生物的核糖体。用于重建的发展史由于极慢的这个基因的进化速度和变量和常量的存在区域允许放大。
16 s rRNA基因高变区:一个DNA序列,表明在不同种类的细菌多样性。
16 s rRNA基因分析(或基因测序):一个常见的扩增子测序方法用于识别和比较细菌在一个给定的样本。16 s rRNA基因测序是一种行之有效的方法,研究样本的发展史和分类复杂的微生物或学习环境是困难的或不可能的。
扩增子:DNA DNA扩增的产物通过PCR。
鸟枪测序:DNA测序方法,DNA测序的分散成段。
失调:改变微生物群的组成与当地生态环境的扰动,通常与受损宿主的相互作用有关。
挑战不同的科学学科
生物信息学的观点
16 s rRNA基因有几个变量区域可用于细菌和古菌的分类(即。分类法)[8- - - - - -10]。进一步,因为其测序是快速和相对廉价的11),它常被用来确定组成、丰度和多样性的细菌和古细菌隐藏在不同的生态系统,如人类的呼吸道(12- - - - - -14]。然而,这种方法有一些重要的局限性。首先,在任何研究活动,研究人员必须确定正确的问题和选择适当的工作流从一系列可用的生物信息学工具正确地解决问题(15),因为太多的分析可以产生混乱,从而导致损失的研究焦点。其次,适当控制潜在的变异来源的研究,包括病人的多样性、抽样方法、DNA提取程序,扩增和测序批次,在微生物的研究至关重要,因为他们可以很容易引入不必要的可变性和意想不到的偏见16)(表1)。如下面所讨论的,试图保持尽可能低的这些变异的来源是最好的策略来克服这些障碍。第三,16 s rRNA基因测序不提供信息,病毒和真菌,或对他们的交互与细菌微生物群,需要调查使用替代方法(如宏基因组测序和/或内部转录间隔区。最后,从纯生物信息学的角度来看,很多问题不完全相关的数据库和方法论的约束(见以下讨论框2目前的术语)也需要考虑。
箱2生物术语
OTU(操作分类单位):集群的微生物,按特定的DNA序列相似性分类分组标记基因,如。16 s rRNA。辣子鸡是用作微生物“物种”代理在不同分类等级:门,类,秩序、家庭、属和物种。序列相似性的定义是基于相似性的标准;如。测序读有97%的相似度可以聚集在一起,代表的是单一的OTU,和对一些细菌可以达到等效的物种水平。
多样性:的数量和分布不同辣子鸡样本或原始人口。因此,所谓alpha-diversity估计描述物种的数量(或相似度量)在一个示例中,虽然beta-diversity估计描述样本之间的物种多样性的差异。一种广泛使用的多样性指数是Shannon-Wiener多样性指数。
相对丰度:如何相对于其他常见或罕见OTU辣子鸡社区,测量的百分比辣子鸡的人口总数。因此,OTU富足是当作一个代孕细菌物种丰度的测量。
均匀度:相似性的测量的相对丰度不同的辣子鸡。
分类:的一个或多个的生物体或生物种群组成一个单元。
数据库的约束
目前现有的16 s rRNA基因数据库提供(部分或全部)基因序列为超过170万个细菌和古菌(17),并足够详细分类细菌在不同分类水平,从门(高分类水平)属(分类水平低)(图1)。然而,这些数据库包含一些未解决的信息序列,所以了解识别不是实现对某些微生物(18]。也有可能,由于高水平的16 s序列之间的同源性物种,一个序列给多个了相同的分数在两个或两个以上不同的记录在数据库中,表示无法区分它们。为了解决这种情况,通常使用“最低的共同祖先概念”(19]。这种方法后,分类单元的分配不是给定的物种,一些细菌,达到只属水平。例如,是这样的链球菌属,这是普遍的呼吸系统,包括致病细菌物种等肺炎链球菌和同桌的如草绿色链球菌。这种限制物种分配明显抑制识别微生物的范围归因于这些属。所有这些原因,生物信息学工具用于微生物研究集群通常使用一个共同的方法测序读在某种程度上一般术语操作下的相似性分类单元(辣子鸡)。因此,序列相似性至少97%的16 s rRNA序列的参考数据库通常可接受的考虑认为辣子鸡是相当于物种水平,或属当相似性水平仅达到94%。
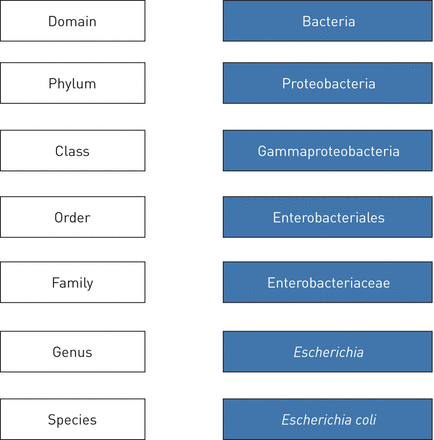
分类的分类大肠杆菌。
方法论的问题
如上所示,16 s rRNA基因有几个变量地区(V1-V3或V3-V5)可用于细菌的分类(3,8,9]。然而,目前尚不清楚谁提供了最好的评估呼吸道微生物。此外,它已经表明,不同的测序平台,包括454年Illumina公司HiSeq MiSeq,可以产生不同的结果(16]。这部分是因为16 s基因的特定变量地区使用,使用的引物,扩增子的长度由不同的平台。降低测序错误,再读取是首选16]。另外一个方法论的问题就是,使用不同的算法,假设和参数会导致不同的结果(13,19,20.]。因此,重要的是要注意这些限制,如果可能的话,使用不同的测序和生物信息学工具(如。标记基因,猎枪基因组或转录组测序)与不同的方法比较结果。最后,值得说的是,16 s测序提供了定性而不是定量微生物组的信息,和补充定量PCR或数字PCR等方法推荐完成信息获得通过分析16 s rRNA的基因。
其他生物信息学挑战
其他生物信息学挑战考虑包括以下。首先,大部分的研究直到现在估计每执行分类单元的相对含量,根据16 s rRNA基因的拷贝数恢复序列库(21]。然而,基因丰度的变化会导致不同的实际细菌负荷或从一个特定的细菌类群的基因组拷贝数可以达到在分析过程。这两个因素的相对权重的估计微生物群落结构是未知,但可以系统性偏差的来源研究中使用16 s rRNA测序。有方法,正确的拷贝数16 s rRNA基因,但这个调整是对< 5%的已知细菌物种(22]。同样值得注意的是,其他基因cpn06也可以用来推断细菌群落多样性(23];因此,使用多个基因的可能性也应该被考虑。第二个,研究之间的比较结果在不同的实验室进行建议使用模拟社区,创建在体外与一个预定义的内容肺部微生物(细菌操纵子的特定的24),但它可能是更方便建立联盟,将执行所有分析在一个单一的中心。第三,不同的DNA提取(25)和PCR扩增的方法也可以引入methodology-related可变性(26]。
视图从呼吸医学
微生物的健康的肺
正常的人类肺微生物的研究仍处于起步阶段,但现在很清楚的是,健康的肺港口系统不同的微生物群落(2,3,27- - - - - -31日]。发表的研究结果有些有限体积小和缺乏纵向采样,而是表明,在健康受试者,厚壁菌门,拟杆菌和变形菌门是最经常发现细菌在门级(32]。在属级,普氏菌、韦永氏球菌属和链球菌主要的微生物,从常见的致病性变形菌门包括最小的贡献嗜血杆菌(32]。健康的航空公司是具有挑战性的样本,因为健康受试者不产生自发的痰,所以抽样需要支气管镜检查,和重复健康个体的内窥镜手术是繁琐,限制有纵向数据的可能性。然而,最近的研究,包括支气管镜的抽样的近端和远端支气管树报道,口咽的微生物群,支气管和肺泡表面有类似的成分在健康个体29日]。这种相似性归因于愿望口咽分泌物在睡眠的33- - - - - -35]。在呼吸道疾病,这种情况可能会改变生长条件的扰动支气管和肺实质促进微生物群落组成的变化,与潜在的致病细菌能够持续的时间更长3,29日- - - - - -31日)(图2)。
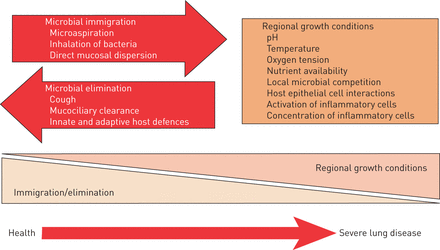
关键因素决定呼吸道微生物:微生物移民、微生物消除和其成员的相对生殖率。在健康受试者中,微生物主要取决于移民和消除。然而,在严重的肺部疾病,地区经济增长条件微生物组成的主要决定因素。从[102]复制与许可。
在任何微生物污染是一个问题,研究和降低气道的潜在污染样品的口咽微生物群是一个主要问题是明确呼吸道疾病(3]。大量的微生物呼吸系统分会低于其他人体表面,和低生物样品如通过一个受保护的标本刷(公安局)或支气管肺泡灌洗(BAL)可能无法提供足够的DNA,而背景信号从试剂可能会被误解为一个真正的信号(36]。因此,为了区分噪声和信号,适当的技术控制急需在样本序列分析,特别是呼吸道样本,可能遭受稀释效应。
信息的长期影响吸烟对健康受试者的呼吸道微生物是稀缺的,显然需要研究。初步研究修改报告的吸烟者的口咽微生物群的微生物组成、影响主要是壁厚菌门的门奈瑟氏菌属足以被视为很重要的物种,生态失调(37,变形菌门的相对丰度的下降;这些修改不会戒烟后恢复(38]。相比之下,支气管分泌物呼吸道微生物群的研究没有发现吸烟者和不吸烟者之间的显著差异(37),戒烟后也没有相关的细菌多样性的变化(39),这表明暴露于吸烟导致近端微生物变化不反映在相应的下游支气管树中的改变,至少在缺席的情况下呼吸道疾病。口腔微生物的差异与前吸烟者没有呼吸道疾病没有正确评估,然而,目前还不可能正确地辨别暂时失调引起的失调的接触刺激物和急性损伤与慢性疾病有关。
慢性阻塞性肺疾病
支气管殖民化的潜在的病原微生物一直建立在慢性阻塞性肺疾病(COPD)由几个以前的研究(40,41),但这个殖民之间的因果关系的方向和气道炎症,气流限制,和支气管和肺实质破坏仍悬而未决。有证据表明之间的关系恶化的症状的出现和新菌株的收购40),但是这个菌群的变化只是部分证明急性加重的外观。
患者的临床稳定的慢性阻塞性肺病,几项研究已经报道了丰富的肺部微生物,显然是不同于健康对照(2,27,30.,31日,42- - - - - -46]。常见的门在这些患者中有变形菌门、拟杆菌、放线菌和壁厚菌门假单胞菌,链球菌,普氏菌和嗜血杆菌在这些患者常见的属(2,27]。
大多数数据可以从慢性阻塞性肺病来自从活检获得的样本(46),肺组织外植体(30.),BAL或公安局2,27,31日,46),痰液(43- - - - - -47]。不同地区不同的抽样程序目标的呼吸系统,然而,结果表明,痰港口在支气管肺泡样品中微生物群落,不同于那些[46),已经证实,事实上,支气管和肺泡的COPD患者包含一个独特的微生物(3)(见图3在迪克森et al。(3])。
发作期间,有些属增加相对丰富而不显著改变(42,44,48,49]。此外,加重似乎不仅代表的孤立属有关,而且还与抵押品微生物组成的变化作为一个整体,进而出现与增加炎症相关的标记在落下帷幕41,50]。此外,似乎有病毒感染和细菌群落组成,之间的相互作用与增加的相对丰度变形菌门实验后鼻病毒感染(48]。类似的真菌和细菌之间的相互作用提出了(51]。此外,治疗期间发作时影响呼吸道微生物不同抗生素的基础上,减少细菌丰度,主要变形菌门,与口服类固醇,当管理系统不影响细菌丰富但支持特定的代表类群(40,52]。
最后,一些挑战需要解决之前受益于微生物研究慢性阻塞性肺病可以有效地纳入临床实践:1)关于报道呼吸道微生物的差异远端和近端支气管,分别落下帷幕的目标和痰(46),有意义的阈值需要根据确定的临床重要的细菌代表对所有样本类型;2)non-cultivable但潜在的致病微生物的作用被微生物研究尚不清楚,需要调查;和3)之间的相互作用细菌、病毒和真菌与宿主需要有针对性。
总之,尽管这些重要障碍,肺微生物研究有可能解开新的和相关见解COPD发病机制,可能导致更好的慢性阻塞性肺病的临床管理。具体来说,有一个明确的需要了解当前标准的慢性阻塞性肺病治疗的影响,特别的吸入型皮质类固醇激素,COPD气道微生物,因为这些药物被证实能够减少发作的频率,但与此同时,增加肺炎的风险,可能通过直接调制的呼吸道微生物。最终,微生物的变化可能成为重要机制(即。endotypes)潜在的不同的临床表现(即。慢性阻塞性肺病的表型)。
囊肿性纤维化和支气管扩张
呼吸道细菌感染是我们理解核心囊性纤维化的病理生理学(CF)和(non-CF)支气管扩张。传统的微生物培养技术揭示了著名的病原体等的重要性流感嗜血杆菌,铜绿假单胞菌和莫拉克斯氏菌属复活在支气管扩张53),另外金黄色葡萄球菌和伯克不过在CF (54]。微生物的研究正在向前我们对这两种疾病的理解。例如,一些病人先前未被发现的生物丰富,无论是在CF (55,56)在支气管扩张(57,58]。此外,研究描述呼吸道微生物抗生素治疗后显示的抵抗细菌社区随时间变化在这些病人57,59,60];抗生素治疗主要导致细菌多样性的减少,但这种效果在几周后就会消失,恢复以前的微生物组成(57]。总细菌的多样性,用复合Shannon-Wiener多样性指数等指标,与气流限制目前的水平和其他疾病严重程度的标记在CF和支气管扩张。此外,澳大利亚non-CF支气管扩张患者的随机临床试验表明,潜在的病原微生物的相对丰度假单胞菌增加患者接受长期治疗属大环内酯类(60),但在多大程度上微生物的变化归因于抗生素机制尚不清楚。真菌、病毒的作用分枝杆菌(不确定标准细菌16 s rRNA测序)尚不清楚在CF和支气管扩张,并且需要未来的研究(61年]。同样,其他重要问题需要检查在临床包括16 s rRNA基因测序的程度提供有用的临床信息以外的文化,与宿主的相互作用,可能基于微生物组概要文件,选择抗生素治疗的有用性微生物结果来评估治疗反应,微生物组的预后影响的影响分析和抗生素对新出现的病原体。的痰可以获得在这些患者群体促进在未来几年大规模研究。
间质性肺疾病
传统上,间质性肺病(ILD)一直被视为非感染性肺实质疾病。然而,近期呼吸道微生物组的描述特发性肺纤维化(IPF)已显示出代表特定的生物等链球菌,普氏菌和葡萄球菌在这些患者与健康对照组相比62年,63年)(图3)。他们是否可以推动疾病进展是一个假设的优点未来的研究(63年]。
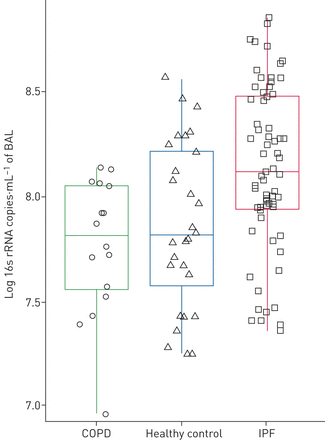
细菌负荷(16 s拷贝数毫升−1患者的支气管肺泡灌洗(BAL)特发性肺纤维化(IPF),慢性阻塞性肺疾病(COPD)和健康对照组。IPF患者(红色;n = 64)细菌负担明显高于慢性阻塞性肺病患者绿色;n = 17)和健康对照组(蓝色;n = 27)(分别为p = 0.006, p = 0.0007)。盒子表明第25和第75百分位数,中位数是由短线内框表示。从[53]复制与许可。
IPF急性加重的存在已经日益被视为这些患者的死亡率的主要原因(64年]。这些事件的确切发病机制仍不清楚,目前的诊断标准具体要求排除任何感染引发的65年]。尽管如此,有证据支持IPF发作的传染性的假设:1)随机对照试验显示死亡率降低病人预防性复方磺胺甲恶唑(66年),2)免疫抑制与急性发作率增加(67年),3)更高比例的急性加重发生在冬季,和4)传染性事件赋予一个相同的死亡率non-infective加重(63年]。因此极大的兴趣在使用文化无关的分子技术探索IPF急性加重感染的作用,虽然这些事件的不可预测性和取样困难限制因素在解决这个话题。
微生物的研究在整个范围的不同ILDs应当建立1)如果有任何角色的肺部微生物组成发生和演化;和2)最优抽样方式在这些患者中,鉴于这些实质疾病可能不是适当由支气管痰等样品。
肺移植
由于长期使用预防和/或治疗免疫抑制药物和抗生素,降低航空公司的肺移植受者为居民提供一个特殊的利基微生物群(68年,69年]。事实上,改变当地条件在第一个月移植后促进下呼吸道感染由于机会性致病菌。迟钝的炎症状态普遍盛行移植后6 - 12个月,与一个强大的优势细菌通常发现在口咽微生物群(70年]。修改在呼吸道微生物群组成肺移植设置是强大到足以被视为生态失调,并通过特定的辣子鸡的代表,体现包括下面列出的,持久性相关的异常的潜在宿主炎症概要文件(70年,71年]。此外,移植后闭塞性细支气管炎综合征的发病还与宿主的相互作用,通过pathogen-driven炎症触发和/或受损宿主先天反应影响细菌间隙(72年,73年]。
研究使用文化无关的技术,识别微生物群失调患者的肺移植的报道经常明确变形菌门和/或厚壁菌门的优势,与微生物的假单胞菌和葡萄球菌属(68年,74年],Burkholderiaceae家庭(75年]。这些细菌,这可能代表超过70%的球微生物群落,通常与炎性反应有关,而类似规模的拟杆菌门的代表,主要是由于大量的普氏菌,而不是与改造宿主基因表达谱(70年]。这些发现表明,microbiome-host移植肺内的交互影响先天免疫过程。未来的研究应该将这些模式与长期的同种异体移植物的结果和移植排斥反应发生的风险。
教训其他人体器官系统:肠道
肠道微生物的研究开创了微生物研究领域和更先进的比呼吸道微生物。首先,它现在使用新一代测序技术,使微生物群落在更深入的理解通过微生物基因或全部基因组的研究76年],metatranscriptomics,包括RNA序列(见术语盒3)。第二,计划像人类微生物组77年)和MetaHIT (78年)项目,由美国国立卫生研究院(美国)和欧盟委员会(European Commission),分别让深描述人类肠道微生物组的健康和疾病状态。结果,我们现在知道,人类胃肠道(GI)束港口的一个最复杂和丰富现有的100万亿多个微生物,微生物群落微生物基因的数量超过了100倍人类胃肠道细胞的数量。虽然稳定在年龄、肠道微生物的组成和功能是受很多因素的影响,包括遗传学和出生时曝光相关交付,年龄、地理位置、饮食、吸烟和医学治疗(79年]。第三,尽管也有很多潜在的可变性来源可以显著影响胃肠道微生物群研究的结果,一个全球性的努力已经定义的最佳实践和协议来比较不同胃肠道微生物群的研究,meta-analyse和提取新的知识。协议的努力,国际人类微生物组的标准项目,可在线(www.microbiome-standards.org)。第四,肠道微生物群不仅影响到胃肠道,它还可以影响身体的许多功能,包括处理和收集从我们的饮食营养,先天和适应性免疫系统反应的影响(80年,81年]。因此,胃肠道微生物群的变化可以有利于胃肠道非胃肠道疾病的发展。例如,现在大量的文章链接功能和代谢胃肠道疾病,如炎症性肠病、肠易激综合症或肥胖,肠道微生物变化(82年- - - - - -85年),但也报道之间的关系改变肠道微生物组和神经障碍(如。自闭症)[86年- - - - - -89年)和呼吸道疾病(如急性呼吸窘迫综合征患者发生感染性休克(90年])。第五,艾滋病毒流行教会我们,男同性恋者往往有明显的粪便微生物群组成,增加微生物的丰富性和多样性,以及浓缩的普氏菌菌群,却能合成更多形成独立于他们的艾滋病毒状况91年]。hiv - 1感染与减少细菌丰富有关,尤其是在主题与次优的CD4 + T细胞计数在抗逆转录病毒治疗(91年]。最后,干预旨在修改肠道微生物的组成已经成功的在特定的胃肠道疾病。粪便微生物群移植越来越被认为是一种有效和安全的干预患者艰难梭状芽胞杆菌感染,和不同中心报道与这种治疗成功率> 90%92年]。这种方法要复杂得多在炎症性肠病,在粪便移植成功率约为13% (93年]。细菌修改的影响肠道微生物群的健康受试者的呼吸道微生物和各种呼吸道疾病的患者,以及潜在的间接影响通过改变宿主的免疫反应(及其应对粪便移植)到目前为止还没有得到妥善解决。当前的知识,包括早期的有益和有害的肠道微生物群的变化,及其与过敏性呼吸道疾病的关系,最近审查(94年),现在gut-lung轴提供了一个广泛的研究可能性,我们将在下面进行讨论。
盒3其他系统术语
国际人类微生物组的标准:标准操作程序旨在优化人类微生物领域的数据质量和可比性。
粪便移植:从一个健康人粪便细菌的移植过程变成一个收件人。
未来的呼吸道微生物研究
从上面的讨论中,参与者在车间同意以下九个具体方面需要特别的解决未来呼吸道微生物研究:
正常模式:研究在健康受试者进行到目前为止已清楚地表明,在呼吸系统有丰富的微生物群,包括壁厚菌门的微生物,拟杆菌和变形菌门的门,并显示密切相似的口咽的微生物群。病毒和真菌正常模式仍然需要被定义,然而。呼吸道微生物群的微生物组成慢性呼吸道疾病的变化,但这些变化的时机和分布只是部分。
多样性在抽样程序:有广泛共识,最好的抽样程序取决于正在解决的问题。痰液可能是一个适当的方法研究呼吸系统疾病有明显的支气管组件,考虑到它可以获得广泛的病人和不需要侵入性程序,但更可靠的信息外围支气管和肺泡表面需要侵入性的样品(即。落下帷幕,公安局,支气管或肺活检)。同样,在胃肠道的研究中,粪便现在收集的大量研究和地方活检用于回答特定问题的患者数量限制。在任何情况下,这些测量仍需要平行的传统微生物研究,因为尽管测序提供了一个通用的图片细菌的组成社区,临床微生物文化提供有意义的信息等呼吸道病原体的作用嗜血杆菌和假单胞菌疾病,仍然没有一个等价的微生物分析。
标准化:有迫切需要标准化协议用于分析呼吸道微生物,包括采样、处理和生物信息学方法。建立联盟和网络研究这个话题会促进标准化,因此,从不同的群体共享的结果的可能性。
Non-cultivable和/或非致病性细菌:16 s rRNA基因分析显示高相对丰富和non-cultivable微生物的特定模式(一般代表的变形菌门)在支气管和肺样本来自COPD患者,IPF, CF和支气管扩张。特定物种的角色以往被视为非致病性需要解决在这些不同的临床情况。
损失的多样性:丧失多样性与慢性阻塞性肺病疾病严重程度有关,IPF和CF,它也被描述在这些疾病的发作。类似的观察肠道据报道,表明下降的一般模式的多样性相关的微生物组成代表特定的辣子鸡可能发生在人类疾病,但这些微生物变化的时序动态广泛未知。驱使这损失的细菌多样性,包括种间竞争的影响,抗生素暴露和宿主免疫反应,必须定义。
与宿主的相互作用:microbiome-host交互的数据是不完整的肠道疾病和呼吸道疾病的几乎不存在。未来的研究应该解决当地的和系统性的微生物群落的影响,因为重要的远程效应可以通过释放介质施加在血液中。因此,解剖宿主交互的复杂的相互作用在身体不同的网站,如肺、肠道和皮肤,代表着一个重大的挑战在未来微生物研究,但有可能帮助澄清在几个慢性呼吸道疾病发展的决定因素。为了正确评估这一点,新研究应该包括研究微生物的多样性在同一个主机上几家网站;纵向维度;评估本地主机和系统性免疫;最后应该证明微生物的影响模式在呼吸道疾病的发病机制通过微生物移植动物模型。
细菌RNA和宏基因组:16 s rRNA基因分析之后,研究的一个新阶段的微生物开始鸟枪测序DNA和RNA分析。这些技术需要实现呼吸道微生物的研究,因为他们将提供功能信息,没有在16 s rRNA基因分析。此外,16 s rRNA基因不能区分活的和死的细菌,和死亡的细菌的DNA坚持多久呼吸道样本是未知的。
病毒和真菌:病毒的作用,包括大量的噬菌体感染细菌和真菌在呼吸道健康和疾病不能通过16 s rRNA基因分析、目标和需求调查。病毒之间的相互作用,真菌和细菌已经略微评估迄今为止,但是初步结果显示明确的指出影响无菌微生物群在变形菌门。
干预措施:通过益生菌和细菌微生物群的补充和调制等价物尚未探索的呼吸道疾病,但它是一个潜在的富有成果的研究领域。益生菌是否直接针对肺实质或恢复正常上呼吸道或肠道微生物群,能产生有利影响的呼吸道疾病仍有待确定。
披露的信息
确认
作者感谢动量®后勤支持组织的研讨会。
脚注
利益冲突:披露可以找到与这篇文章www.qdcxjkg.com
本文的摘要巴塞罗那呼吸网络研讨会在巴塞罗那举行6月3日,2016年。《会饮篇》是由Menarini无限制的经费支持,阿斯利康,基耶西,葛兰素史克和诺华和部分由Fundacio雷蒙解放军Armengol,洋底de Investigacion疗养地和PI15/02042 15/00167。
- 收到了2016年的10月25日。
- 接受2017年2月8日。
- 版权©2017人队











{kind=link}
{kind=link}
![Key factors determining the respiratory microbiome: microbial immigration, microbial elimination and the relative reproduction rates of its members. In healthy subjects, the microbiome is determined mainly by immigration and elimination. In severe lung diseases, however, regional growth conditions are a main determinant of microbiome composition. Reproduced from [102] with permission.](http://www.qdcxjkg.com/content/erj/49/4/1602086/F2.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
![Bacterial load (16S copy number·mL−1 of bronchoalveolar lavage (BAL)) in patients with idiopathic pulmonary fibrosis (IPF), chronic obstructive pulmonary disease (COPD) and healthy controls. Patients with IPF (red; n=64) had a significantly higher bacterial burden than subjects with COPD (green; n=17) and the healthy control subjects (blue; n=27) (p=0.006 and p=0.0007, respectively). The box signifies the 25th and 75th percentiles, and the median is represented by a short line within the box. Reproduced from [53] with permission.](http://www.qdcxjkg.com/content/erj/49/4/1602086/F3.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}