





抽象的
钾通道twik相关酸敏钾(TASK)-1通道与其他钾通道一起控制肺动脉低静息音。Src家族酪氨酸激酶(SrcTK)可能控制人肺动脉平滑肌细胞(hPASMCs)钾通道功能,以响应氧紧张的变化,临床使用SrcTK抑制剂已导致部分可逆的肺动脉高压。
本研究旨在确定SrcTK在缺氧诱导的hPASMCs钾通道抑制中的作用。
我们表明SRCTK与任务-1通道共同定位。抑制SRCTK降低钾电流密度并导致相当大的去极化,同时SRCTK的激活增加了贴片夹具中的钾电流。中度缺氧和SRCTK抑制剂降低任务-1通道的酪氨酸磷酸化状态。缺氧还会降低磷酸-SRCTK(TYR419)的水平,并减少任务-1通道和磷酸盐-SRCTK的共定位。对应于此,缺氧减少任务-1在SRCTK抑制之后,并且在分离的灌注小鼠肺中,SRCTK抑制剂增加肺动脉压。
我们建议SRCTK是控制钾通道的关键因素,其作为辅助因子,用于在HPasmcs中设定负静止膜电位和低静息肺血管基调。
在肺动脉中,血管张力部分由肺动脉平滑肌细胞(PASMCs)的静息膜电位调节。在所有可兴奋细胞中,钾离子通道是通过维持静息膜电位接近钾离子平衡电位来调节静息膜电位的主要候选通道。钾通道的激活或抑制导致细胞膜的超极化或去极化,分别导致血管舒张和血管收缩的开始。pasmc中至少有四种钾通道[1].背景或泄漏的钾离子选择性通道在钾离子通道中是例外的,因为它们的活动不受电压控制,它们可能被认为是人类肺动脉平滑肌细胞(hPASMCs)静息膜电位和输入阻抗的主要贡献者[2那3.].
SRC系列酪氨酸激酶(SRCTK)是参与各种蜂窝信号传导和功能的非摄取酪氨酸激酶系列的成员[4.].c-SrcTK由于其n端多酚化区而靶向于质膜。SrcTK的SH1结构域包含Tyr419磷酸化位点,这是c-SrcTK完全激活所必需的[5.].这种机制允许c-SrcTK与离子通道相互作用,调节其性质[6.那7.].当其C末端TYR530磷酸化时,会发生人C-SRCTK的灭活;然后将其与SH2结构域粘合。结晶研究表明,C-末端和SH2结构域之间以及激酶结构域和SH3结构域之间的相互作用,导致C-SRCTK分子呈现覆盖激酶结构域的闭合构型并降低其对基板交互的电位[5.].背景TWIK相关酸敏感钾(任务)-1通道的C-末端含有酪氨酸和丝氨酸激酶的可能磷酸化位点(图S1A)。如果钾通道通常通过SRCTK激活,则该机制的逆转可以解释达克西布治疗后SRCTK抑制后观察到的增加的肺血管间调[8.].
在本研究中,我们研究了SRCTK对钾通道功能和膜电位的功能作用。我们研究了HPASMCS中任务-1通道的缺氧抑制,以确定SRCTK的特定作用。我们证明了SRCTK的特异性抑制导致任务-1和其他钾通道的抑制并导致膜去偏振。此外,缺氧降低了膜中任务-1通道的活性磷酸-SRCTK共定位,同时Srctk的激活增加了任务-1电流,并且这种活化电流被中度缺氧抑制。这表明钾通道功能克定依赖于SRCTK,从而解释了为什么SRCTK抑制可能导致危及生命的肺动量的血管收缩。这是第一个证明SRCTK在静息膜潜力调节中的功能作用的报告,在原发性后热的缺氧抑制中,最后是肺血管基调。
方法
组织捐赠的研究议定书由格拉茨,格拉茨,格拉茨,奥地利格拉茨医务大学的机构审查委员会批准,并符合国家法律和良好临床实践/国际协调会议的指导方针(www.ich.org/).在适当的情况下,每位患者均获得书面知情同意。所有的动物实验都是由动物护理和使用委员会(奥地利BMWF和奥地利格拉茨医科大学)批准的。
原代hpasmc的制备及细胞培养
原发平滑肌细胞从30名接受肺手术的肺癌患者(无肺血管疾病或动脉低氧血症史)或未使用的供肺移植患者的人抵抗肺动脉中分离。在获取肺癌患者的肺动脉时,只使用距离癌组织≥5 cm的动脉。详情请参见在线补充资料。
电生理学
如前所述,hpasmc全细胞膜片钳技术用于测量电流钳下静息膜电位和电压钳下宏观钾电流[3.].协议和解决方案的详细描述在在线补充材料中。
使用市售的Pclamp 9.0软件(Axon Instruments,USA,USA),存储和分析数据。
钙的测量
采用荧光染料fluo-4-AM检测hPAMSCs细胞内钙的变化。协议和解决方案的详细描述在在线补充材料中。
获取的图像被存储,随后使用TillVision软件离线处理(Till Photonics, Munich, Germany)。
对C-SRC,FYN和任务-1的小干扰RNA的转染
商业合成针对c-Src (siC-Src)、Fyn (siFyn)和TASK-1 (siTASK-1)的小干扰rna (sirna) (Eurogentec, Seraing,比利时)。详情请参见在线补充资料。作为阴性对照,使用了不针对任何人类基因产物的非沉默RNA (nsRNA)。hpasmc在盖板或六孔板上生长,使用Effectene转染试剂(Qiagen, Hilden, Germany)转染退火后的siRNA。利用提取的RNA (RNeasy;Qiagen)。
转染48-56小时后进行RNA水平、活细胞钙和电生理测量。为了评估siRNA转染的效率,使用了异硫氰酸荧光素(FITC)偶联的siRNA。只有fitc阳性细胞用于电生理研究。此外,siTASK-1转染通过将细胞与pH值为8.3的浴液进行超融合进行功能控制。
rt - pcr
详细描述在在线补充材料中提供。
定量rt - pcr
采用定量RT-PCR检测c-Src、Fyn和TASK-1在hPASMCs中的表达。要了解更多细节,请参阅在线补充材料。
共免疫沉淀,免疫印迹和免疫协同定位
详情请参见在线补充资料。
HPASMC的缺氧治疗
缺氧对膜片钳和钙成像研究的影响是通过在常氧和低氧灌注池之间切换来研究的。在线补充材料中提供了详细的描述。
隔离,灌注和通风的小鼠肺部
来自成人C57BL / 6只小鼠(Harlan Laboratories,Inc.,Indusapolis,In Usa)的肺部被深入的AAAESTHAESIA和曲面通风,并用Krebs的Henseleit Buffer(NaCl 120 Mmoliberait Buffer(NaCl 120 Mmol)灌注-1, KCl 4.3 mmol·L-1, KH2阿宝4.1.1更易·L-1,Cacl.22.4mmol·L.-1, MgCl21.3mmol·L.-1和葡萄糖13.3mmol·L-1, 5%(重量/体积)羟乙基胺胶(分子量200,000 Da)。肺被安装在一个水加热的房间,允许负压通风的气体含有5.3%的CO2和21.0%o2,与N平衡2.初始稳态稳定状态为15分钟(带克雷布的Hiseleit缓冲液,流量为1 ml·min-1)作为基线。有关详细描述,请参阅在线补充材料。4-amino-5——(4-chlorophenyl) 7 (T.丁)pyrazolo [3,4 - d]嘧啶(PP2;4-氨基-7-苯吡唑[3,4-d]嘧啶(PP3;30 μM)或达沙替尼(100 μM)加入缓冲液15分钟。ΔP.pa显示肺动脉压(P.pa)施用PP2,PP3或Dasatinib后。
解决方案和化学物质
PP2和PP3购自Sigma Chemical Company (St Louis, MO, USA)。Src激活肽来自Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA)。除PP2、PP3和达沙替尼外,所有药物均溶解在实验(浴)溶液中。他们溶解在二甲基亚砜(DMSO)。在这个浓度下,载体本身对离子电流、静息膜电位或P.pa.测试含有药物的溶液的pH值,并用克雷布斯Henseleit缓冲液校正以消除可能的pH诱导效应。NaHCO3.用作缓冲液,调整以导致恒定的pH值为7.37-7.40。
统计分析
数值用平均值±表示sen个单元或测量值。群体差异由阶乘Anova评估后HOC.用Tukey检验分析,或视情况选择不配对和配对t检验。p-值<0.05被认为是显著的。
结果
SRCTK在人肺癌和PASMC的表达
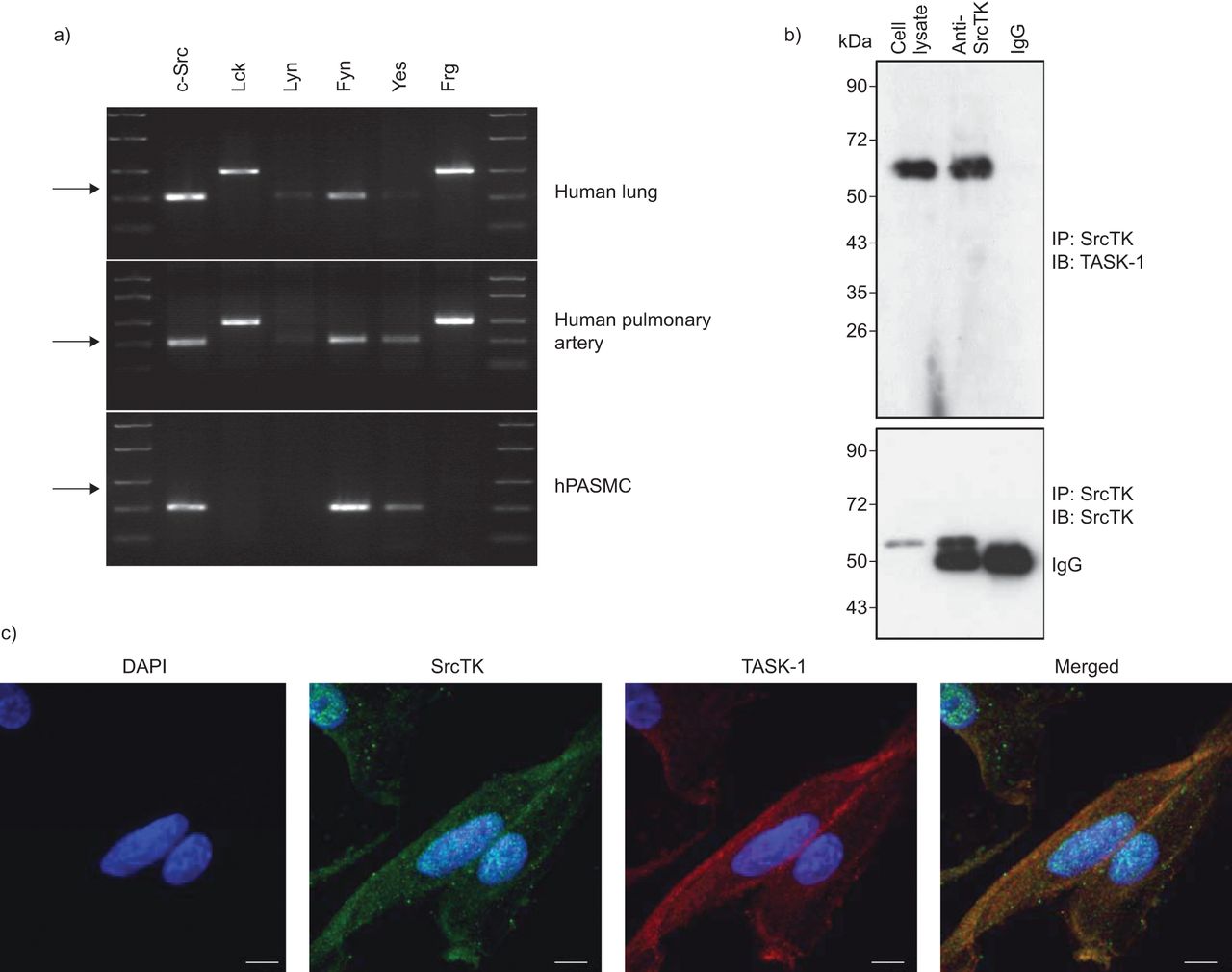
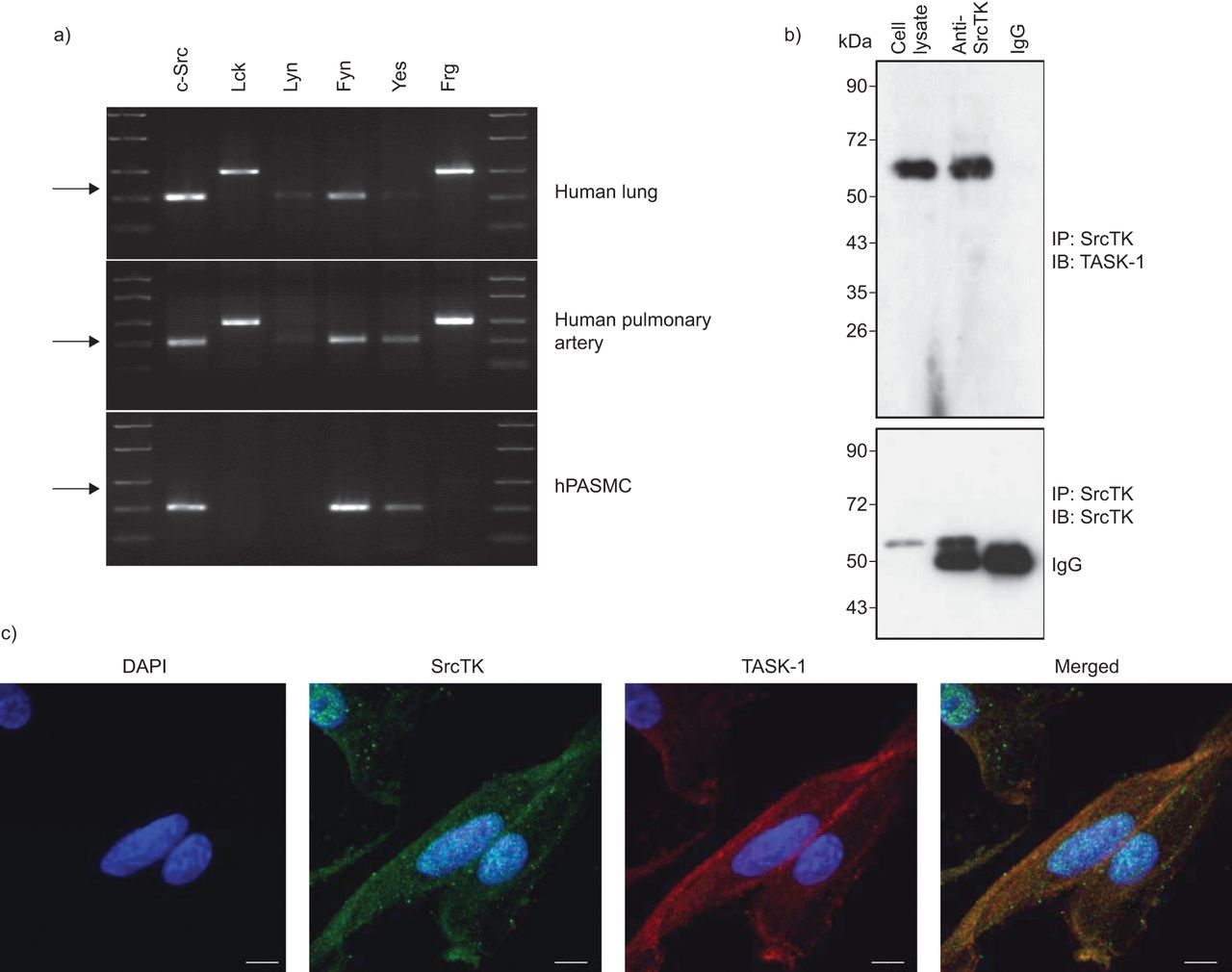
为了分析SRCTK同种型的表达,对人肺组织,人肺动脉和HPASMC进行C-SRC,LCK,Lyn,Fyn,Yes和FRG,RT-PCR(无花果。1A).我们的结果表明,mRNA编码所有研究的亚型在肺组织。初级hPASMCs仅表达c-Src、Fyn和Yes亚型的mRNA。
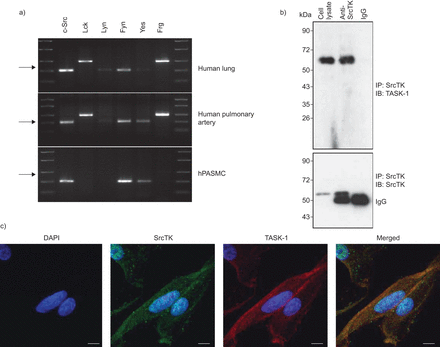
SRC系列酪氨酸激酶(SRCTK)与人类肺动脉平滑肌细胞(HPASMCS)中的TWIK相关酸敏感钾(任务)-1通道共定。a)来自均匀的人肺组织和原发性HPasmc的RNA提取物中的SRCTK的RT-PCR筛选。代表性凝胶说明C-SRC(204bp),LCK(398bp),Lyn(213bp),Fyn(206bp),是(202bp)和FRG(402bp)的mRNA表达。箭头表示200bp。通过来自不同供体肺和原发性高清菌的RNA的至少三种制剂获得相同的结果。B)印迹表示具有抗SRCTK的共免疫沉淀(IP),同种型对照免疫球蛋白(IG)G和细胞裂解物,然后为任务-1(上图)和SRCTK(下图)免疫印迹(IB)。c)荧光免疫抑制表明DAPI核染色(蓝色),SRC系列激酶(红色),任务-1通道(绿色)和在HPASMC的单面共聚焦图像中的重叠图像(合并)。秤杆=20μm。
SrcTK在hPASMC中与TASK-1通道共本地化
SrcTK调节hpasmc中的TASK-1通道活动
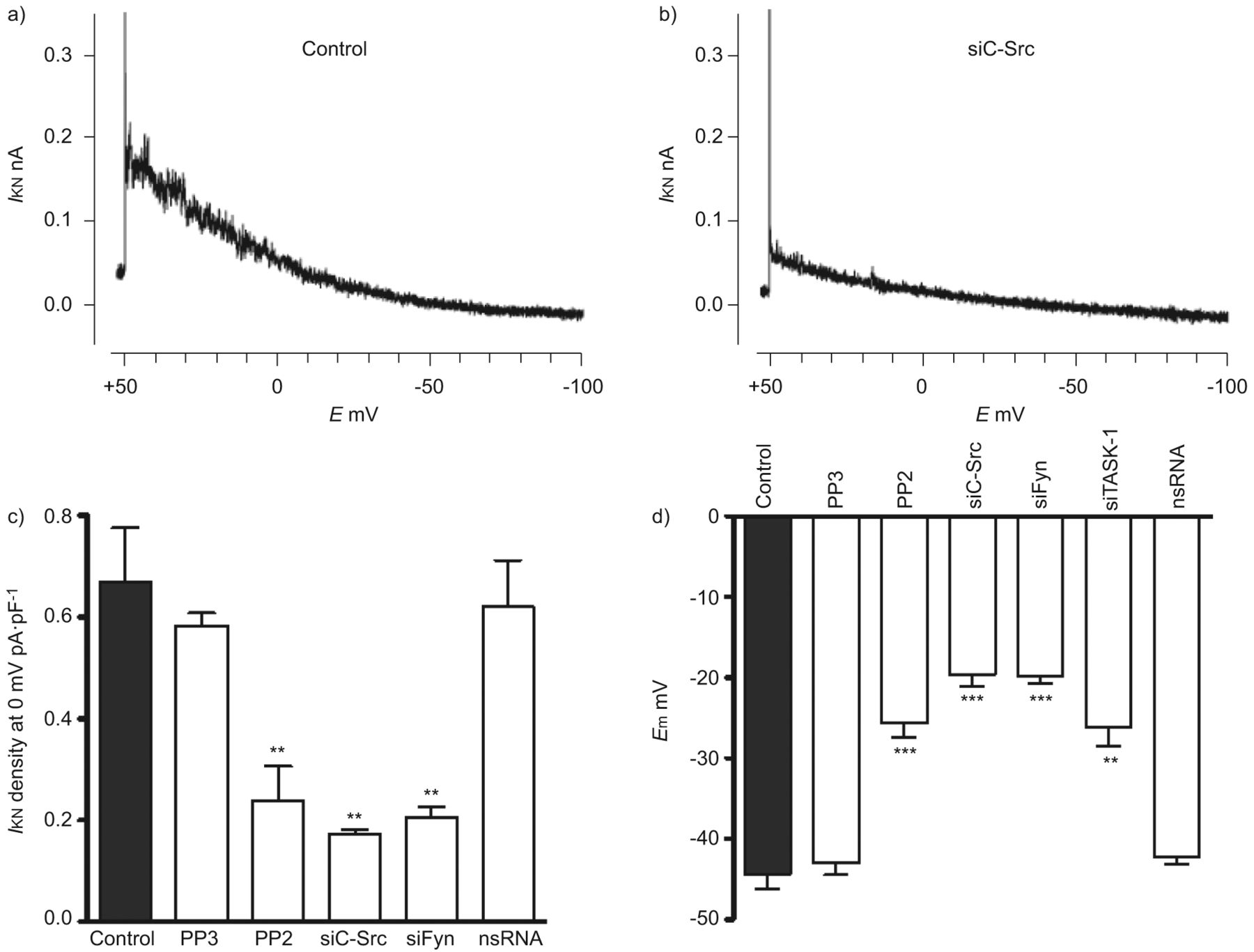
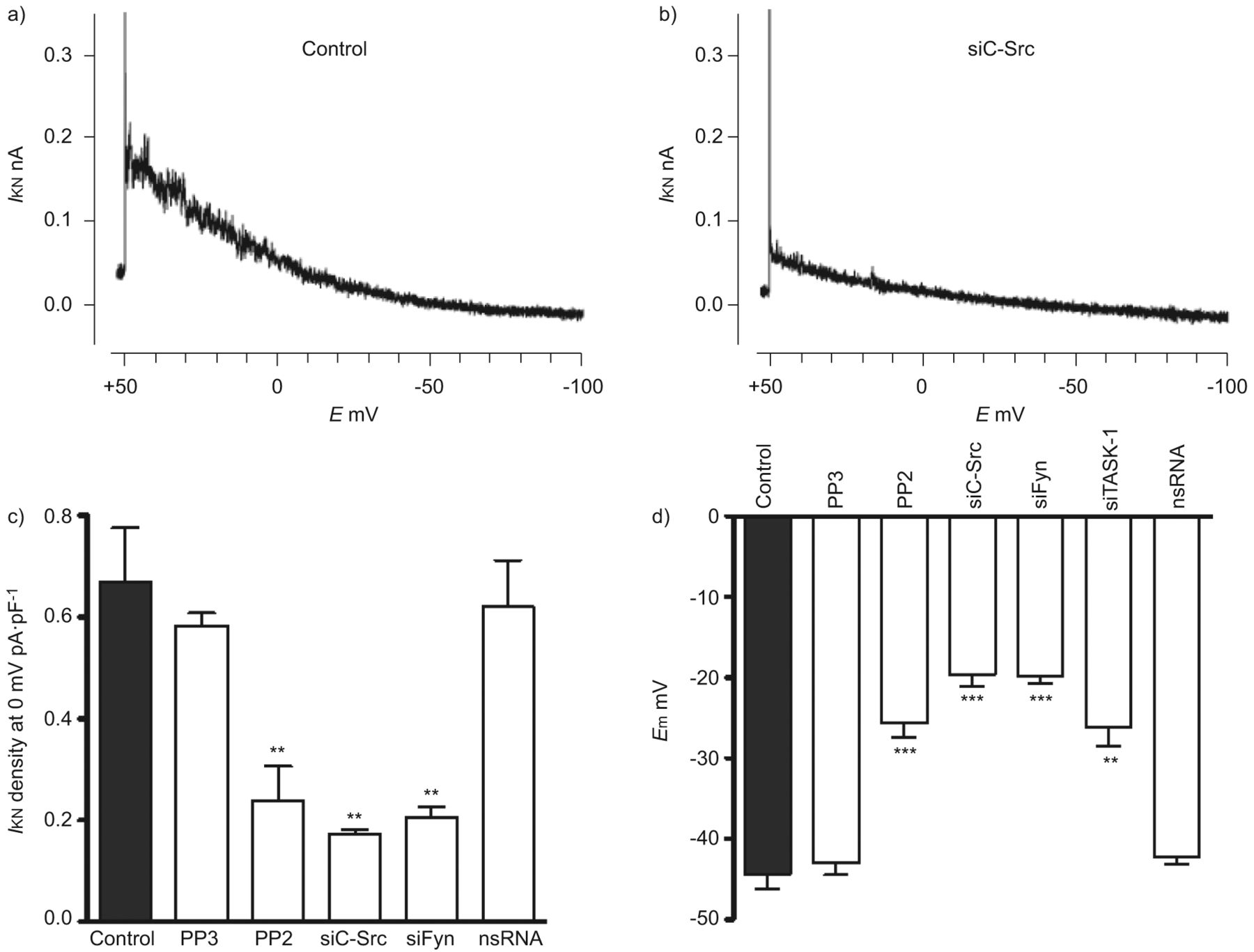
为了进一步确认SrcTK与TASK-1通道相互作用的功能相关性,我们使用了药理学工具和siRNA技术。如前所述,对hpasmc上未失活的TASK-1电流进行膜片钳记录[3.].SRCTK增加任务-1信道活动,通过沉默C-SRC和FIR在原发性HPASMC中确认,导致对控制相比的电流降低(无花果。2A和B.).SrcTK抑制剂PP2显著抑制TASK-1电流密度(0.23±0.06 pA·pF)-1,n = 12)。尽管PP2是SRC系列蛋白酪氨酸激酶的有效和选择性抑制剂,以研究SRC亚型的作用,使用针对C-SRC和FYN的siRNA。用SiC-SRC转染(0.17±0.01 pa·pf-1,n = 4)或Sifyn(0.20±0.02 pa·pf-1, n=8)显著抑制TASK-1电流密度(0.67±0.1 pA·pF)-1, n = 13)。作为对照,使用了c-Src抑制剂PP2的非活性类似物PP3和转染nsRNA。PP3(0.58±0.02 pA·pF)-1, n=4)或nsRNA转染(0.62±01 pA·pF)-1, n=8)对TASK-1 current (无花果。2C).通过定量RT-PCR证实了C-SRC和Fyn的相对程度的C-SRC和FyN的敲击度(图S2)。
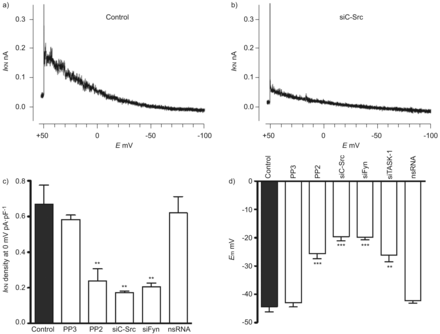
SRC家族酪氨酸激酶(SRCTK)对于初级人肺动脉平滑肌细胞(HPASMC)中的TWIK相关酸敏感钾(任务)-1沟道活性至关重要。任务-1当前的代表录音(一世KNb)转染靶向c-Src的小干扰RNA (siC-Src)的细胞。c)主hpasmc中TASK-1电流密度直方图。经Src家族激酶抑制剂PP2 (1 μM)处理或转染sc -Src和siFyn后,电流密度(一世KN)显着降低。用PP3(1μm; PP2的非活性类似物)转染后,没有减少电流后的电流密度。d)总结SRCTK抑制对静息膜电位影响的直方图(E.mhPASMCs)。转染PP2 (1 μM)或转染siC-Src、siFyn或siTASK-1后,细胞均有明显的去极化现象,但转染nsRNA和PP3后,细胞无明显去极化现象。E.:膜电位。* *: p < 0.01;***:与对照组相比,p<0.001。c和d)中的数据用平均值±表示sen个单元或测量值。
SRCTK抑制去偏振HPASMCS
TASK-1通道在静息膜电位时是活跃的,并在PASMCs中设置负静息膜电位,正如我们之前通过siRNA治疗原发性hPASMCs证实的那样。由于SrcTK抑制也降低了TASK-1电流(无花果。2C),进一步研究SrcTK对原代hPASMCs静息膜电位的生理作用。电生理测量进行hPASMCs显示治疗后明显两极化PP2(-24.1±1.3 mV, n = 39)或转染siC-Src(-19.6±1.5 mV, n = 14)或siFyn(-20±1.1 mV, n = 14)相比与控制(-44.3±1.6 mV, n = 36)或与细胞治疗PP2的不活跃的模拟,PP3(-43.4±1.5 mV, n=4)或转染nsRNA(-43.6±1.5 mV, n=18) (无花果。2D).总之,这些数据进一步加强了任务-1的作用以及SRCTK在原发性腐烂膜电位设置负静息膜电位的重要性。
HPASMCS中SRCTK的缺氧调节
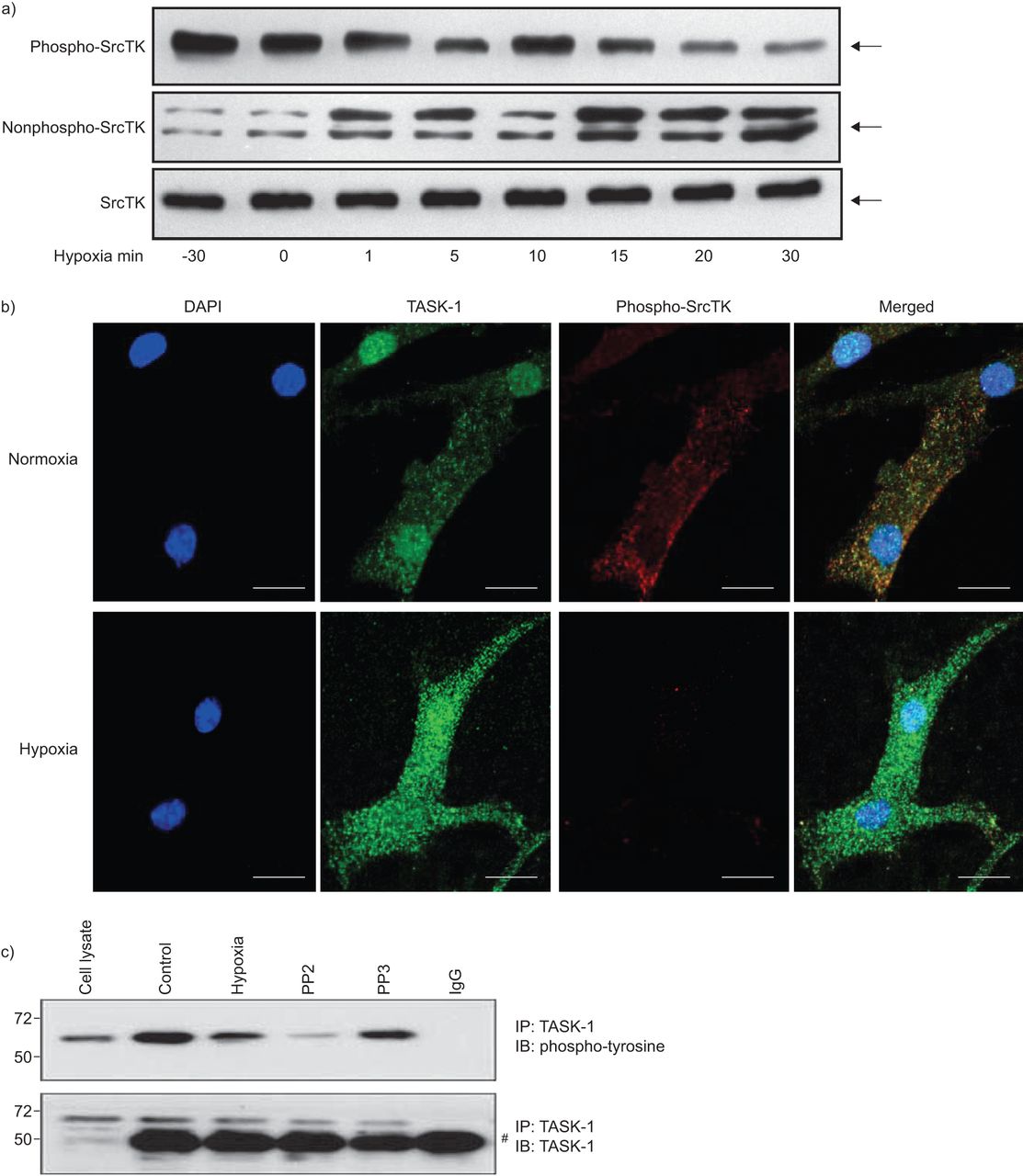
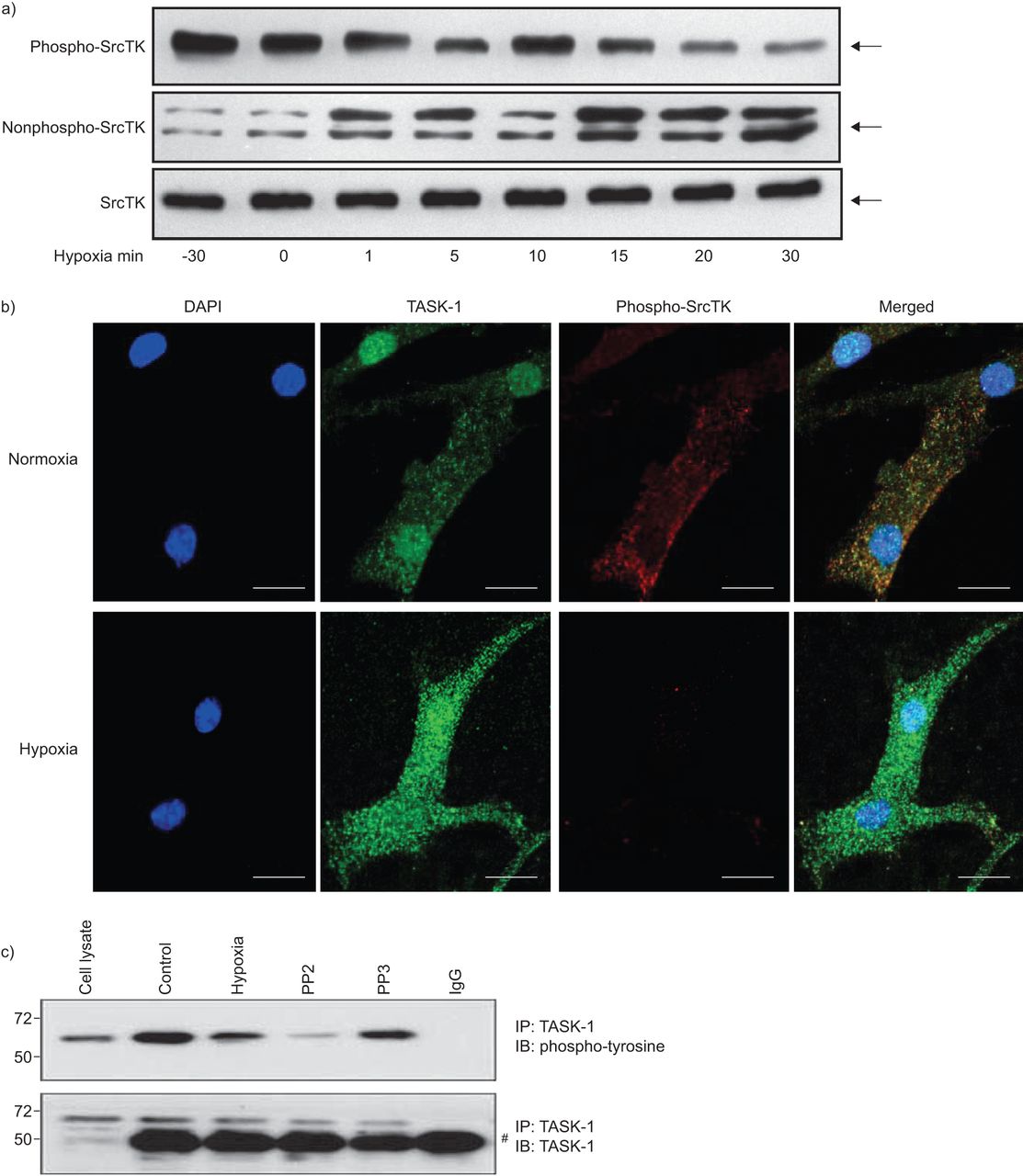
缺氧导致hpasmc的膜去极化,部分原因是TASK-1通道抑制。SrcTK的沉默降低了TASK-1电流,导致膜去极化。SrcTK的活性是由SrcTK在Tyr419位点的磷酸化状态决定的。因此,我们研究了磷酸化(活性;无花果。3A,上图)和非磷酸化(非活性;无花果。3A(中图)缺氧条件下(30 min常氧或0、1、5、10、15、20、30 min缺氧)不同时间点hPASMCs的SrcTK状态。接下来,在常氧和缺氧15分钟后,在hPASMCs中检测背景TASK-1通道和磷酸化- srctk的共定位,并通过共聚焦激光扫描显微镜观察结果(无花果。3B.).下面的小组图3 b清晰显示缺氧时phospho-SrcTK染色减弱。图3C.表明缺氧,以及SRCTK抑制剂PP2的施加,降低任务-1通道的酪氨酸磷酸化状态,而PP3,PP2的非活性类似物,不会改变任务-1磷酸化。图3C.显示不同实验条件下的任务-1的完全保持不变。
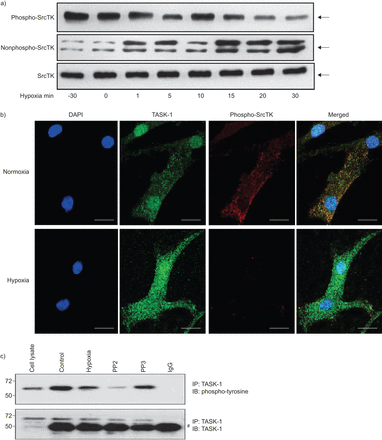
缺氧对原发性人肺动脉平滑肌细胞Src家族酪氨酸激酶(SrcTK)磷酸化的影响a)缺氧降低了phospho-SrcTK (Tyr419)的免疫反应性(60 kDa),而非phospho-SrcTK (60 - 65 kDa)的免疫反应性(60 - 65 kDa)呈时间依赖性增强。蛋白质负载当量由总SrcTK表示(n=4)(其中总SrcTK包括磷酸化SrcTK和非磷酸化SrcTK)。b) hPASMC的单面共聚焦图像显示,缺氧15分钟后,与正常缺氧相比,phospho-SrcTK Tyr419(红色)明显下降。规模酒吧= 20μm。c)缺氧或使用SrcTK抑制剂PP2降低了twik相关酸敏钾(TASK)-1通道的酪氨酸磷酸化状态。下图为不同实验条件下TASK-1总水平不变的情况。知识产权:免疫沉淀反应;IB:免疫印迹。#:免疫球蛋白G (Ig)。
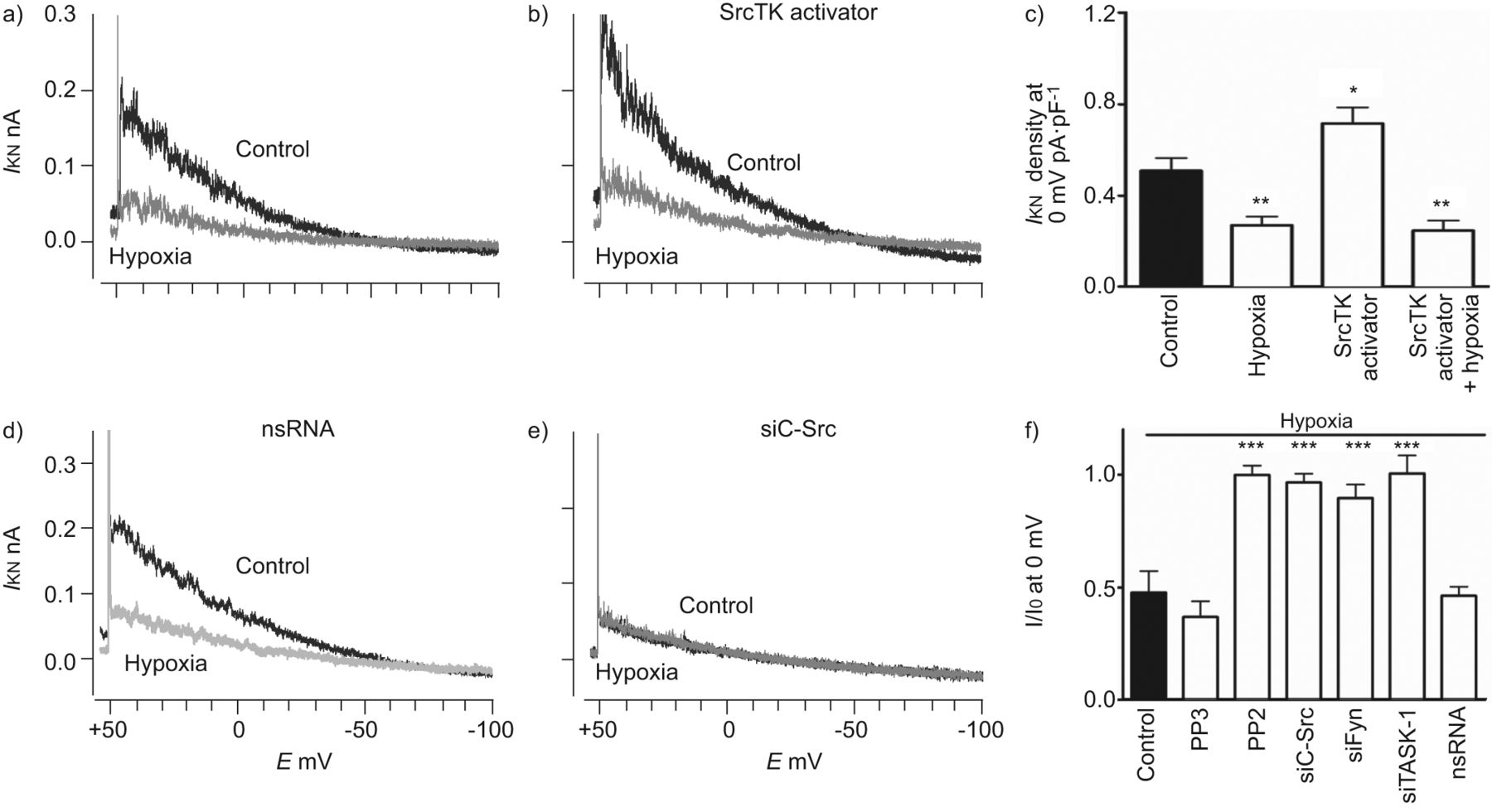
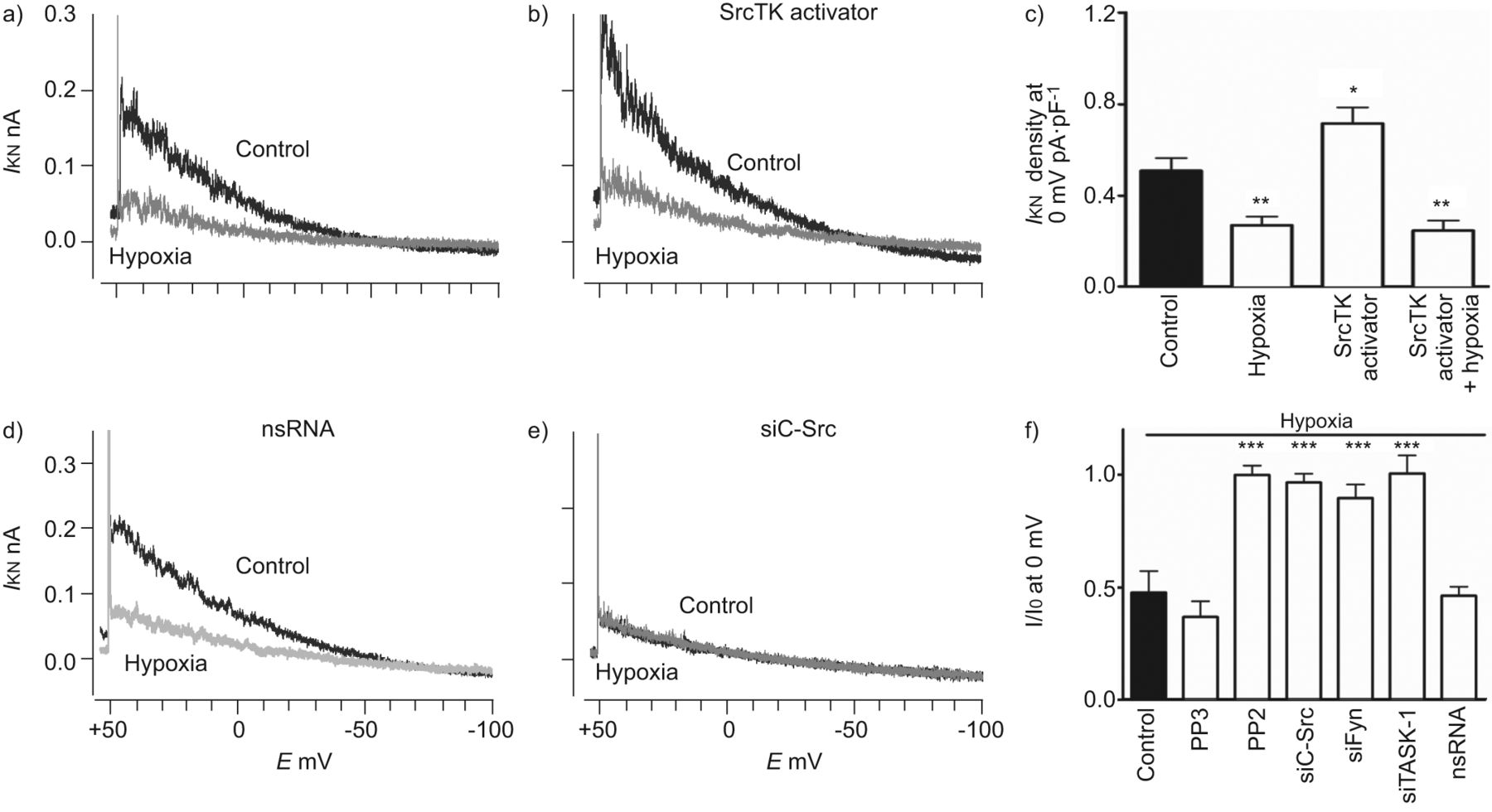
接下来,研究了SRCTK对缺氧抑制任务-1通道的功能作用。具有SRC活化剂肽(EPQYEIPIIPI基)的细胞内透析显着增加了任务-1电流密度(0.72±0.07 PA·PF-1, n=8),对照组(0.51±0.05 pA·pF)-1, n = 10;无花果。4Ab)。缺氧能够抑制控制和激活的电流。图4C.总结了SRCTK活化剂肽和缺氧的影响。由于通过抑制阻滞剂或小干扰(Si)RNA来减少初始任务-1电流(无花果。4E),缺氧对PP2处理的细胞或转染靶向c-Src (siC-Src)或siFyn的siRNA的细胞没有进一步影响。然而,在对照组、pp3处理或非沉默RNA转染的hPASMCs中,缺氧仍能抑制TASK-1电流(无花果。4D.).hpasmc缺氧条件下TASK-1的相对电流见图4 f.
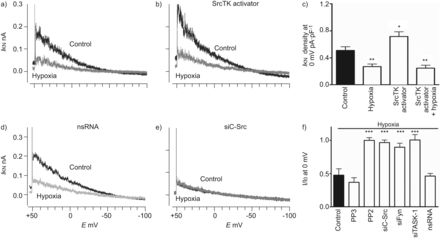
SRC系列酪氨酸激酶(SRCTK)活化剂增加TWIK相关酸敏感钾(任务)-1沟道电流,并且这种效果被一次人类肺动脉平滑肌细胞(HPASMCS)中的缺氧完全抑制。任务-1当前的代表录音(一世KN)控制和在缺氧A)没有或b)与贴剂移液管中的SRCTK活化剂肽(Epqyeipiyl; 1 mm)。c)SRCTK活化剂肽的效果显着增加了与对照相比的任务-1电流的电流密度。这种增加的任务-1电流受到缺氧的显着抑制。任务-1从用D)转染的细胞的当前录像,Nonsilencing(NS)RNA或e)小干扰RNA靶向C-SRC(SiC-SRC)。在后一种情况下,缺氧不能进一步抑制任务-1电流。f)用PP2处理后缺乏进一步的缺氧诱导的任务-1电流抑制,SiC-SRC,SIFYN和SITAMAM-1处理。G)一世KN任务-1的密度在rO-31-8220(PK)c抑制剂),Gö6983(PKC抑制剂),KT5720(PKA抑制剂)和常氧化物下的化合物C(AMP-活化激酶抑制剂)(对照)原发性溶质的缺氧。一世0.:每种细胞中常氧的目前;E.:膜电位。*: p < 0.05;* *: p < 0.01;***:P <0.001与对照相比。
TASK-1通道可以被不同的途径调节,如amp活化蛋白激酶(AMPK) [9.]蛋白激酶(PK)A [3.],pkc [10]和磷脂酶(PL)C [11,对不同激动剂刺激的反应。为了排除这些途径的参与,我们使用不同的抑制剂研究了TASK-1电流的缺氧抑制。这些处理都不影响对照TASK-1电流或其缺氧抑制(图S3和S4g),表明AMPK, PKA, PKC和PLC没有参与。
SrcTK对缺氧诱导hPASMCs细胞内钙增加的影响
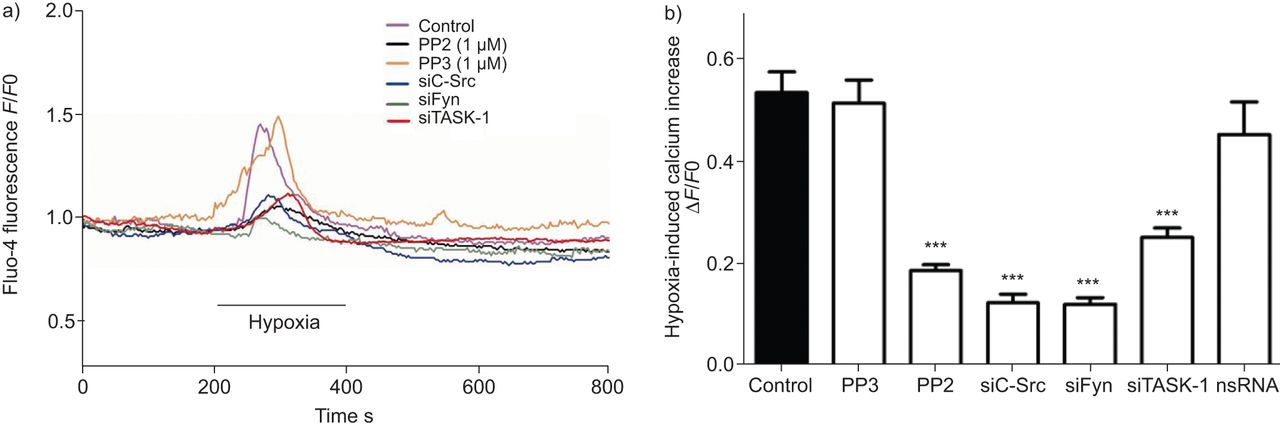
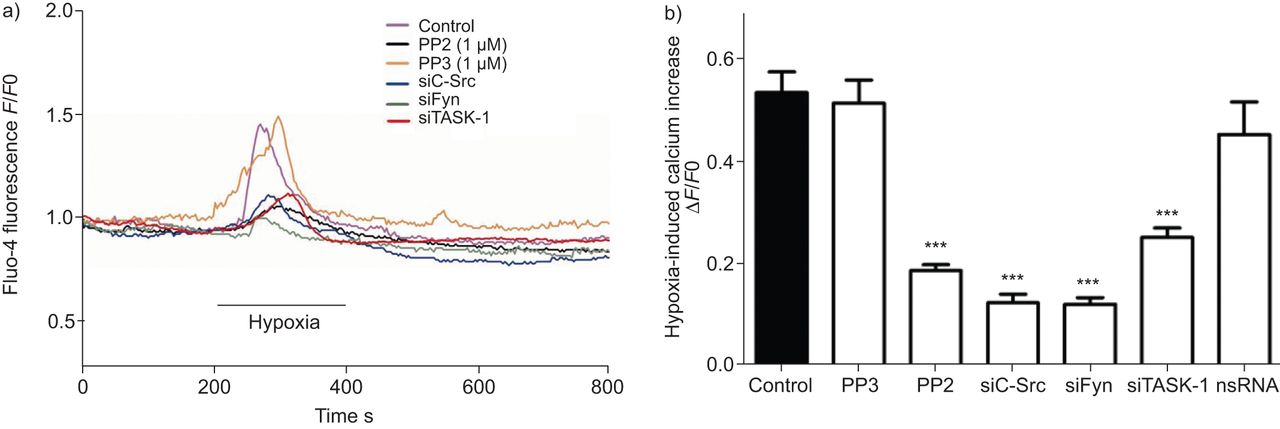
缺氧诱导细胞内游离钙浓度升高[Ca2+]一世是hPASMCs的一个完整和特征特性,它直接与导致血管收缩的下游信号有关。SrcTK在低氧诱导中的作用[Ca2+]一世在hPASMCs中进一步分析。连续监测含氟hpasmc [Ca2+]一世在缺氧(图5).缺氧显著增加[Ca2+]一世对照组(0.54±0.04,n=63), PP3处理组(0.52±0.04,n=17)或nsRNA转染组(0.45±0.06,n=16)。相比之下,PP2(0.18±0.01,n=68)或转染siC-Src(0.12±0.02,n=18)、siFyn(0.12±0.02,n=14)或siTASK-1(0.25±0.02,n=22)后,缺氧诱导的0.54±0.04显著减弱。总结的结果在图5B..
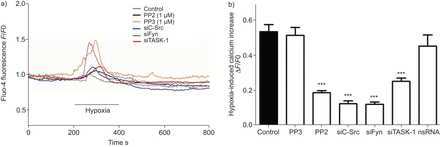
抑制SRC家族酪氨酸激酶衰减初生人肺动脉平滑肌细胞(HPASMC)中细胞内钙浓度的缺氧诱导的增加。a)Fluo-4荧光的代表性记录(F),以及PP2、PP3、靶向c-Src的小干扰RNA (small interfering RNA targeting c-Src (siC-Src) or siFyn处理的细胞。b) PP2、sc - src或siFyn处理后可降低缺氧诱导的细胞内钙含量,但未沉默(ns)RNA或PP3处理后无此变化。F0.:挑战前的荧光。任务-1:TWIK相关酸敏感钾-1。***:与对照组相比,p<0.001。
抑制SrcTK可减弱hPASMCs中的电压门控和钙依赖的钾电流
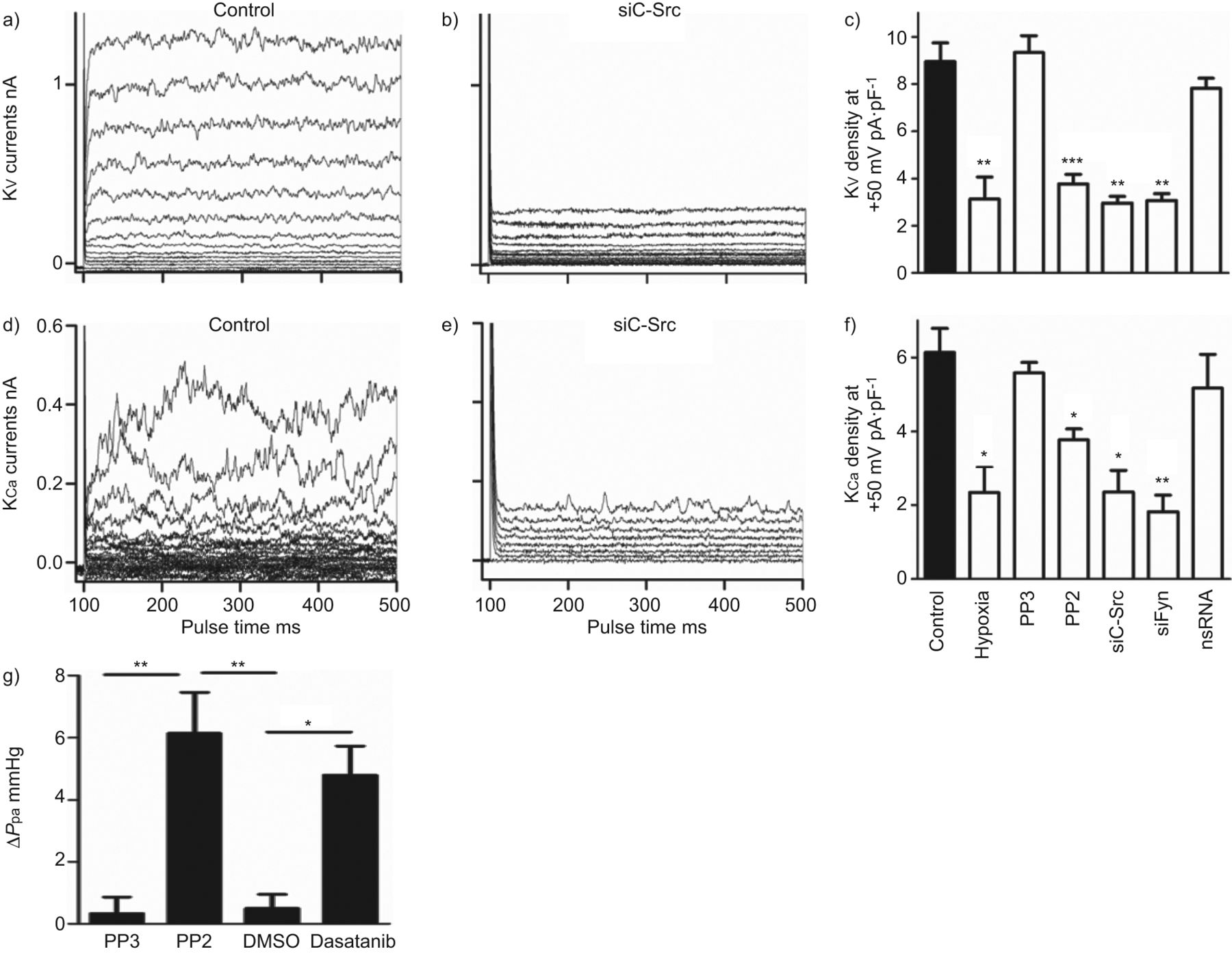
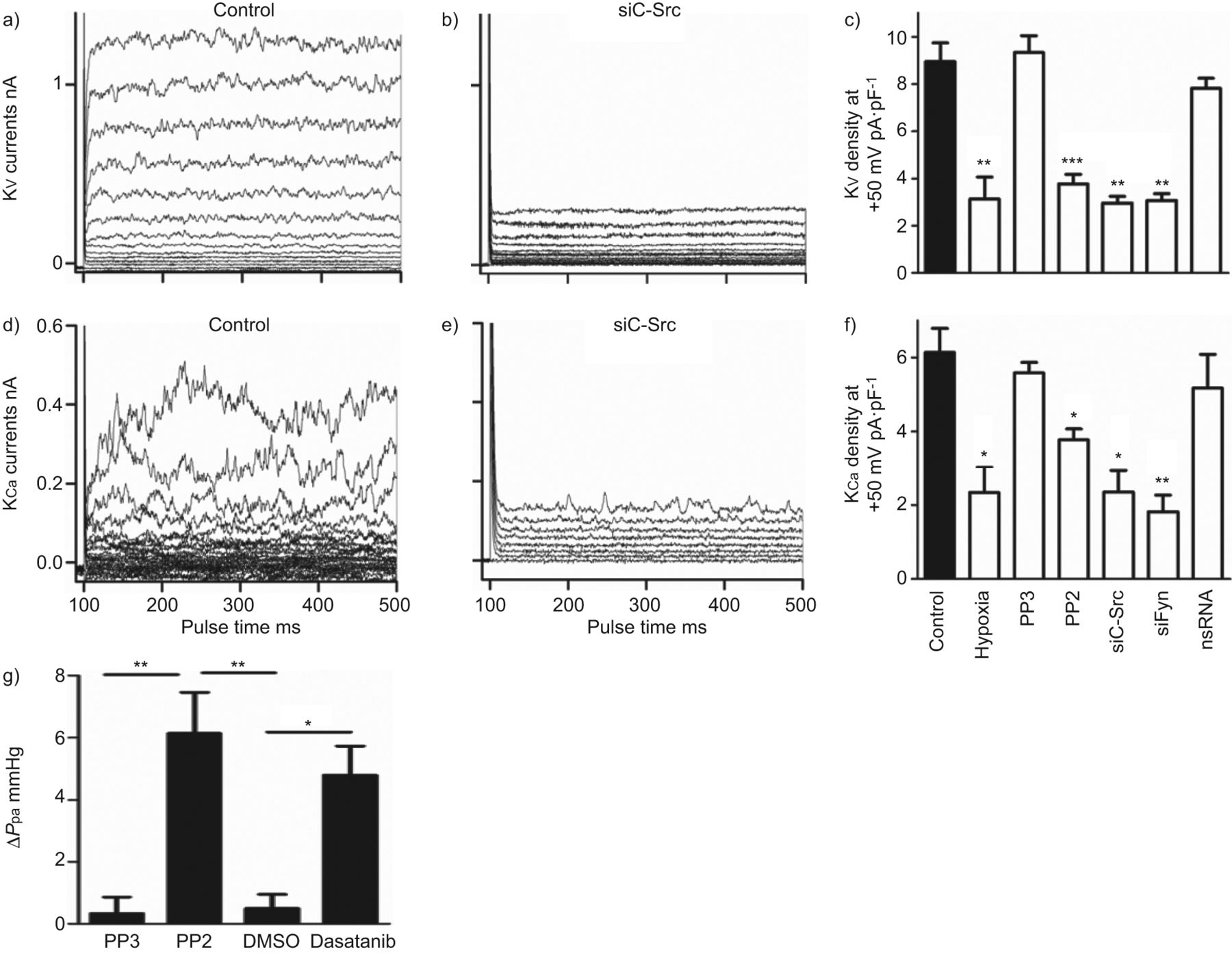
SrcTK抑制对全细胞钾电流(电压门控)的影响V.)和钙依赖(K加利福尼亚州)钾电流)。K代表V.控制中的录音 -图6)和经sc - src治疗后图6 b,表明沉默c-Src显著降低KV.电流。记录K的实验也得到了类似的结果加利福尼亚州控制电流(图6 d)和c-Src沉默后(图6 e)在初级素材中。
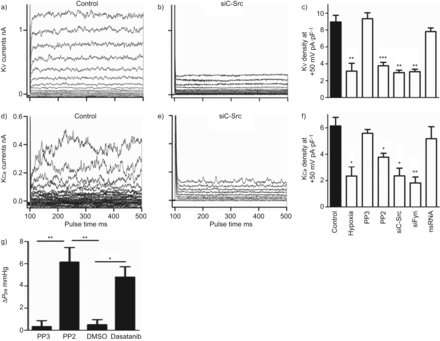
抑制Src家族酪氨酸激酶降低整个细胞的电压门控(KV.)和钙依赖(K加利福尼亚州)初级人肺动脉平滑肌细胞中的钾电流,增加肺动脉压(P.pa)在孤立的,灌注的小鼠肺模型中。A,D)K的代表痕迹V.和K加利福尼亚州在对照细胞和B,E)用小干扰RNA靶向C-SRC(SiC-SRC)后的电流。C,f)总结缺氧,PP3,PP2,SiC-SRC,Sifyn或非ilcening(NS)RNA的效果的直方图V.和K加利福尼亚州电流密度。g)概述变化的直方图(Δ)P.pa从PP2, PP3,二甲基亚砜(DMSO)和达沙替尼存在的小鼠肺中分离的。*: p < 0.05;* *: p < 0.01;***:P <0.001与对照相比。
进一步评估SrcTK对K激活的作用V.和K加利福尼亚州研究了PP2,PP3和用SiC-SRC治疗,Sifyn或NSRNA的效果,并与缺氧的效果进行了研究,并进行了比较(图6 c).用PP2处理溶液(3.8±0.4Pa·PF-1,n = 16)或SiC-SRC(2.9±0.3 PA·PF-1, n=5)或siFyn(3.1±0.3 pA·pF .-1,n = 5)显着降低了kV.电流与对照相比(8.9±0.8 PA·PF-1分别为,n = 24)。缺氧也有类似的效果(图6 c).PP3(9.3±0.7 pA·pF)-1,n = 4)或用NSRNA转染(7.8±0.4 PA·PF-1, n=9)没有改变电流(图6 c).当K加利福尼亚州目前被记录(图6 ef).仅缺氧(2.3±0.5 pA·pF)-1, n=16), PP2(4±0.3 pA·pF-1, n=17), sc - src(2.35±0.6 pA·pF .-1, n=5)或siFyn(1,81±0.4 pA·pF .-1, n=5)与对照组相比电流降低(5.7±0.6 pA·pF)-1,n = 12),而pp3(5.58±0.3 pa·pf-1, n=5)或nsRNA(5.17±0.9 pA·pF-1, n=8)均无显著影响(图6 f).
抗SRCTK抑制剂的肺血管收缩
为了描述SrcTK在肺血管张力中的作用,我们使用了离体灌注小鼠肺,其中PP2显示了显著的增加P.pa(6.3±1.3 mmHg, n=4)与非活性类似物PP3(0.6±0.2 mmHg, n=4)相比,有趣的是,慢性髓系白血病二线治疗药物达沙替尼(dasatinib,也是一种有效的SrcTK抑制剂)也显示了类似的增加(5.5±0.2 mmHg, n=3)P.pa对比溶剂DMSO(0.4±0.4 mmHg, n=4)。图6G.显示总结ΔP.pa在PP2,PP3,DMSO和Dasatinib之前和之后。
讨论
本研究的主要结果是:1)SRCTK系列,C-SRC和FYN的两个成员在原发性氢氨症中高度表达;2)任务-1通道和SRCTK在HPASMC的质膜中共定;3)任务-1频道的活动需要SRCTK;4)抑制SRCTK DepoLarises HPasmcs;5)缺氧降低了任务-1的酪氨酸磷酸化水平;6)缺氧降低了SRCTK的活性磷酸化状态,抑制SRCTK活化剂促进的任务-1电流;7)SRCTK抑制显着衰减缺氧诱导的细胞内钙升高;8)KV.和K加利福尼亚州SrcTK抑制降低了电流;9) PP2或达沙替尼对SrcTK的抑制可导致SrcTK显著升高P.pa孤立的灌注小鼠肺。
平滑肌细胞的膜电位是控制肺血管基调的重要因素。在休息时,PASMC的膜电位约为-50 mV [2那3.那12].这是由这些细胞通过钾通道排出的钾维持的。抑制或激活肺血管平滑肌细胞钾通道的药物分别引起去极化或过极化。肺动脉钾通道的功能、控制和表达是一个持续关注的问题,因为钾通道表达的减少,如。钾电压门通道,振动瓶相关的亚家族,成员5(kV.1.5) (13那14,或钾通道功能障碍引起膜去极化[15[加上[CA的增加2+]一世,导致增殖增加,凋亡减少,最终导致肺动脉高压的发病[16].
在hpasmc中,钾通道抑制引起去极化,随后钙通过电压门控(l型)钙通道流入,导致肺血管阻力增加。急性缺氧可通过多种机制引起血管收缩。它通过抑制钾通道使pasmc去极化,导致如上所述的钙内流[17],导致钙从肌肉网上易钙释放,并通过储存通道进行随后的再收集[18那19,增加钙通过l型钙通道流入pasmc,不受膜电位影响[20.]还促进钙敏化[21],从而增加肺血管阻力[22].负责缺氧诱导的细胞溶质钙的增加的大多数钙来自脂肪外,但其中一些被从内部储备释放,例如肌肉网状术[1].由PP2、sc - src或siFyn (图5)继发于缺乏缺氧抑制钾电流,因为抑制钾通道已经由SRCTK抑制引起。在枯萎的SRCTK活性存在下消除了缺氧反应(图4和6.).
一些研究表明,酪氨酸激酶可能作用于钾通道。然而,这些研究是在细胞系或异种表达系统中进行的,使用的是广泛的抑制剂,但没有显示出这些发现的生理作用[7.那23那24].我们之前的研究表明,背景的两个孔域TASK-1通道设置了人类PASMCs的膜电位,它可以被PKA、PLC、PKC和AMPK通路通过不同激动剂(如内皮素)的丝氨酸-苏氨酸磷酸化而调控[10],Treprostinil [3.],等。此外,缺氧通过在HPasmcs中可逆地抑制任务-1信道而去极振荡膜电位[3.].此外,k加利福尼亚州和KV.通道也可能对膜电位有贡献,特别是当它们受到环磷酸腺苷和环GMP等药物的刺激时。因此,我们的结果表明,SrcTK活性对于PASMCs的低生理张力和低肺血管阻力是必不可少的。
缺氧抑制这些通道的机制目前未知。然而,在本研究中,特异性抑制内源性SRCTK在后溶液中的任务-1电流降低。同样,当用Srctk活化剂透析HPASMC时,观察到任务-1电流的显着增加,表明任务-1电流需要SRCTK活性,脱磷酸化降低了增加的电流和磷酸化。其他几个钾渠道,包括k加利福尼亚州和KV.,此前已被证明是由srctk介导的酪氨酸磷酸化调控的[25].我们的观察证实了这一点,但证明了ScrTK活性对这些通道的功能的关键作用,并表明TASK-1的活性需要SrcTK活性。
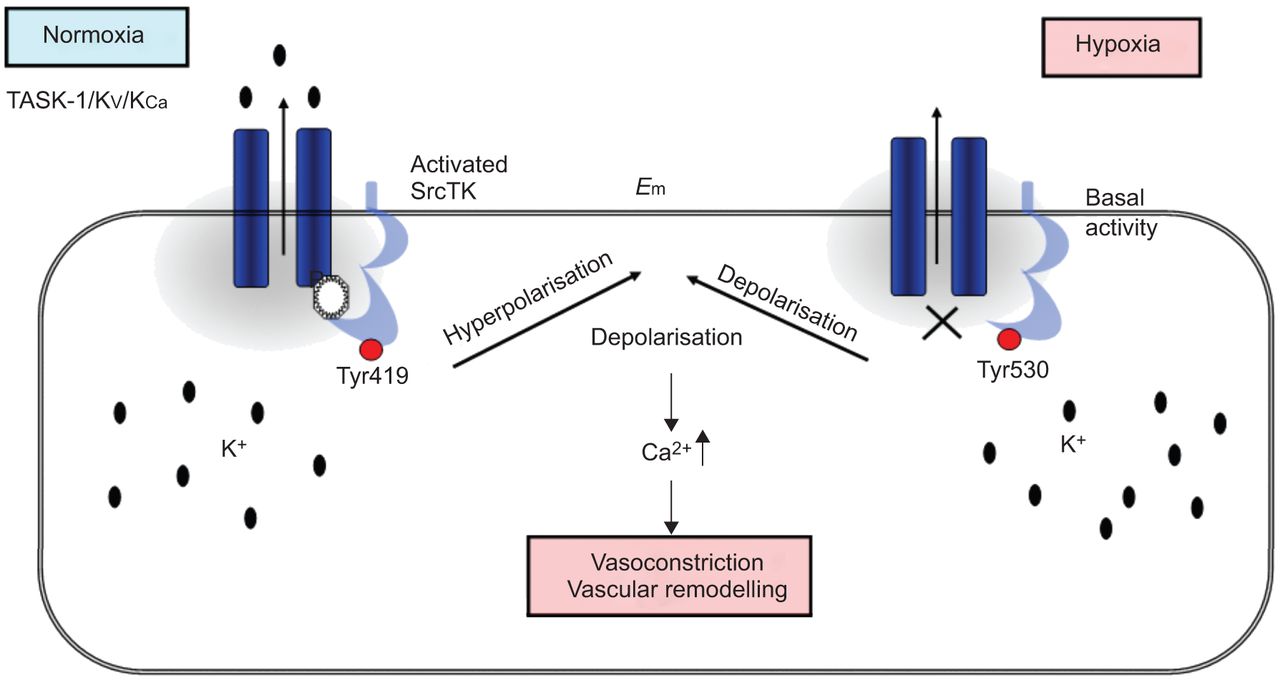
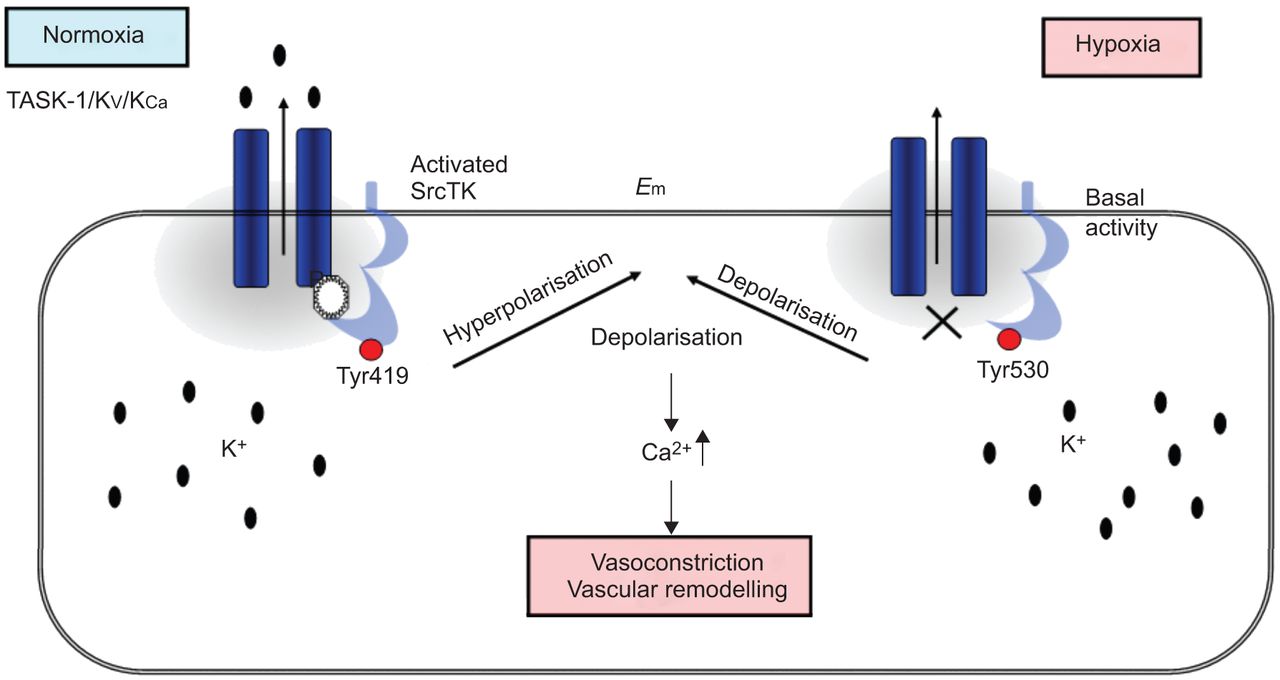
在目前的研究中,我们表明,SrcTK抑制减少当前任务1和任务1的缺氧抑制中扮演着关键角色渠道,可能通过减少磷酸化SrcTK Tyr419和离解task 1和SrcTK,虽然这个链接的分子调控尚未确定(图7).
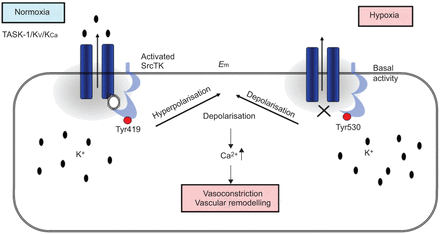
在人肺动脉平滑肌细胞(hPASMCs)中twik相关酸敏钾(TASK)-1通道和c-Src之间的相互作用方案。在常氧条件下,磷酸化Src(活性Src, Tyr419位点磷酸化)与TASK-1通道结合,产生功能性TASK-1通道。在hpasmc中,活跃的TASK-1通道维持负静息电位。缺氧时,磷酸Src(活性Src)减少。封闭的TASK-1导致去极化和细胞内钙水平升高。E.m:静息膜电位。Src家族酪氨酸激酶;K.V.:电压门控钾电流;K.加利福尼亚州:依赖钙的钾电流。
K最近的研究n和同事(26那27通过PP2的SRCTK抑制脱氧诱导的肺血管收缩,抑制大鼠肺动脉和分离的PASMC中的rhO激酶。他们发现PP2降低了PASMC [CA的缺氧诱导的增加2+]一世与我们的观察结果很一定。鉴于PP2的抑制结果和任务-1,K上的SIRNAV.和K加利福尼亚州我们在此描述的电流以及缺氧对这些电流的类似效果,这使得SRCTK磷酸化被抑制在导致电流抑制的序列中。但是,在他们的实验中,Kn和同事(26那27]注意到缺氧增加了TYR419的SRCTK磷酸化而不是降低,因为我们在此报告。这些结果的差异可能是由于物种,细胞培养条件,循环次数和缺氧下的研究的严重程度和严重程度,持续时间或时间点的差异。
观察到的增加P.pa在PP2和达沙替尼抑制SrcTK后,表明SrcTK在调节肺血管张力方面具有功能性作用。这对严重肺动脉高压患者意味着什么尚不清楚。T条件下等.[28]描述了16例原发性肺动脉高压(根据目前的分类,特发性肺动脉高压)患者的c-Src水平下降。这可能与钾通道活性降低有关,并可能有助于血管收缩和其他病理机制,从而导致肺动脉高压。在此,我们报告了TASK-1降低,全细胞钾电流和K加利福尼亚州C-SRC抑制在HPASMCS中的电流。这符合显示减少k的观察结果V.1.5活性导致肺动脉高压[13].相比之下,最近的一项研究ourboulin等.[29]报道了三例肺动脉高压患者肺样本中磷-Src和总Src水平升高。
最近的临床观察突出了酪氨酸激酶在PAH病理生理学中的潜在重要性。慢性髓性白血病(CML)是由组成型活性BCR-ABL酪氨酸激酶引起的。抑制该激酶的伊马替尼是CML的一线治疗[30.].伊马替尼也是血小板衍生生长因子受体的有效抑制剂[31]这被认为是它可能改善一些PAH患者的血液力学的原因[32].Dasatinib是酪氨酸和丝氨酸 - 苏氨酸激酶抑制剂,其在抑制BCR-Abl激酶时作为伊马替尼的效力325倍体外,并诱导CML中较高的细胞源性应答率[33].然而,Dasatinib效果抑制SRC系列,这可能是单一突出的达替尼靶向蛋白激酶系列,包括C-SRC和FYN,并且据报道据报道是PAH [8.那34那35].停用达沙替尼后,PAH倾向于迅速消退[34,提示其主要原因可能不是明显的细胞增殖,而是慢性血管收缩。可以推测,达沙替尼引发的肺动脉高压可能与我们的发现有关,即针对c-Src和Fyn的siRNA降低了钾通道电流,导致hpasmc去极化。很明显,要弄清不同激酶在PAH的病因学和治疗中的作用,还有很多工作要做。
本研究的局限性
虽然目前的研究结果表明TASK-1通道和SrcTK在hPASMCs的质膜中是共定位的,这进一步得到了共免疫沉淀研究的支持,但我们不能排除Src也可以间接作用于钾通道通过其他SRC下游分子。此外,涉及急性缺氧诱导的钾通道抑制的机制可能与慢性暴露于缺氧和/或达司替尼的钾通道的机制不同。最后,改变了P.pa在PP2和/或达斯替尼可能与这种精确提出的机制无关。
结论
综上所述,我们证明SrcTK在TASK-1和其他钾通道的控制中具有重要作用,并且它在hpasmc中设置了负静息膜电位。缺氧诱导的TASK-1电流抑制和细胞内钙离子升高依赖于SrcTK,这一事实强调了SrcTK和TASK-1通道关联的生理相关性。更好地描述SrcTK在钾通道调控方面的多功能作用可能有助于我们理解肺动脉高压的病理生理学。
致谢
非常感谢M. Schloffer和P.Blümel(实验性麻醉,麻醉,格拉茨医疗大学,格拉茨,格拉茨,奥地利的实验性麻醉和密集护理部门)的优秀技术援助。
脚注
这篇文章有补充资料可从www.www.qdcxjkg.com.
有关编辑评论,请参阅第3页.
支持声明
这项研究的部分资金来自格拉茨医科大学(C. Nagaraj分子医学博士项目)。E.K. Weir由VA医学研究基金和RO1 HL 65322(美国国立卫生研究院,Bethesda, MD)资助。
感兴趣的语句
H. Olschewski和A. Olschewski的兴趣陈述www.www.qdcxjkg.com/site/misc/statements.xhtml
- 已收到2011年12月6日。
- 公认2012年4月3日。
- ©2013人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}