





文摘
在这项研究中,我们调查是否DNA双链断裂(双边带)导致慢性阻塞性肺疾病(COPD)的发病机制。
我们immunofluorescence-stained肺组织样本来自慢性阻塞性肺病患者无症状吸烟者和非吸烟者的双边带标记。
双边带疫源地的数量(磷酸化组蛋白2 ax(γH2AX)磷酸化ATM(共济失调毛细血管扩张突变)衬底和磷酸化p53-binding蛋白1疫源地)每个细胞在肺泡I和II型细胞和内皮细胞在慢性阻塞性肺病患者高于在无症状吸烟者和非吸烟者。II型的肺组织细胞含有较高数量的γH2AX疫源地每个细胞有更高比例的II型细胞p16表达INK4a(p16)磷酸化核因子(NF) -κB和白介素6 (IL)和肺泡壁细胞caspase-3表达活跃。II型细胞含有较高数量的γH2AX疫源地每个细胞有较高的p16的表达,磷酸化NF-κB, il - 6。一半的肺泡壁细胞表达active-caspase-3包含γH2AX疫源地。II型细胞染色阳性8-hydroxy-2-deoxyguanosine包含更多的γH2AX疫源地比II型细胞,每个细胞染色阴性。
总之,双边带,至少部分由氧化应激引起的,似乎对COPD的发病机制通过诱导细胞凋亡,细胞衰老和炎性反应。
慢性阻塞性肺疾病(COPD)的特点是有毒环境刺激的持续异常炎症反应,最常见的香烟。最近的证据表明,COPD的发病机制涉及氧化应激、细胞凋亡和细胞衰老,以及炎症(1]。这些多个pathobiological流程在慢性阻塞性肺病被认为是与一代的互动反馈循环,导致肺泡破坏,气道重构和无效的组织修复1]。虽然吸烟直接导致炎症、氧化应激、细胞凋亡和细胞衰老,只有少数的吸烟者开发临床上重要的慢性阻塞性肺病,炎症和肺功能下降继续即使戒烟(1]。这表明存在的具体机制,负责对吸烟所造成的破坏,持续的炎症和慢性,渐进的性质在COPD患者的肺结构错乱。我们目前的研究来测试我们的假设,肺实质细胞的DNA损伤,改变了行为背后COPD-specific机制。
吸烟是最重要的环境污染物,导致DNA损伤(2- - - - - -4]。DNA双链断裂(双边带)是最致命形式的由吸烟造成的DNA损伤(5),如果不修理他们可以导致细胞凋亡,细胞衰老,促炎症反应和肿瘤形成6- - - - - -8]。当双边带诱导,组蛋白2 (H2A) X变得迅速在丝氨酸磷酸化139年由ATM(共济失调毛细血管,突变)/ ATR(共济失调毛细管扩张和Rad3-related)和依赖dna的蛋白激酶。这个修改后的形式,称为γH2AX,很容易被染色和抗体作为一个可靠的、敏感的指标双边带(7- - - - - -10]。因为每个双边带对应于一个γH2AX焦点,immunofluorescence-based化验,允许可视化的离散核γH2AX疫源地≥100倍比其他方法更敏感的检测双边带,如pulsed-field凝胶电泳和碱性彗星试验(7- - - - - -10]。γH2AX然后新兵DNA修复蛋白,包括p53-binding蛋白质(53 bp) 1、双边带的网站和启动DNA损伤信号转导决定是否将修复DNA;如果修复是不可能的,细胞凋亡、衰老或炎性反应将被激活6,11,12]。因此,持久性γH2AX焦点被视为表明一些DNA损伤仍未修理的。
在目前的研究中,我们为γH2AX疫源地进行免疫荧光染色,双边带的存在的一个标志,确定双边带的强度损伤肺部的慢性阻塞性肺病患者。结果显示存在更多γH2AX疫源地的肺泡I和II型细胞和内皮细胞的慢性阻塞性肺病患者的肺部比non-COPD吸烟者和非吸烟者的肺,和γH2AX疫源地的更高的数字与细胞凋亡有关,细胞衰老,炎性表型变化和DNA氧化。这些结果表明,DNA损伤,至少在某种程度上,引起氧化应激导致慢性阻塞性肺病的分子发病机制通过诱导细胞凋亡,细胞衰老和炎性反应。
方法
人类的肺组织样本
研究的协议符合赫尔辛基宣言和批准获得东京女子医科大学的机构审查委员会,日本东京(批准号1783)。肺组织块从慢性阻塞性肺病患者获得吸烟者(COPD吸烟;n = 14)在肺减容手术(n = 13)或局部的肺癌肺切除术(n = 1),吸烟者没有慢性阻塞性肺病(non-COPD吸烟者;n = 10),和非吸烟者(n = 10)在肺切除局部的肺癌。每个组织块固定于10%福尔马林,嵌入在石蜡和切成3部分μm厚。所有的慢性阻塞性肺病和non-COPD吸烟者停止吸烟≥3个月前的手术。病人的临床信息所示表1。
免疫荧光染色
Deparaffinised组织部分是两倍或三倍使用初级γH2AX抗体免疫荧光染色,53 bp1,磷酸化ATM / ATR衬底,p16INK4a(p16)裂解(主动)caspase-3磷酸化核转录因子(NF) -κB、白介素6 (IL) 8-hydroxy-2-deoxyguanosine (8-OHdG),表面活性剂蛋白(SP) - c,水通道蛋白(AQP) 5和CD31(有关详细信息,请参阅在线补充材料)。
我们检查了≥40随机选择的微观领域每个幻灯片和一个奥林巴斯BX60荧光显微镜数字化(奥林巴斯光学有限公司,日本东京)配备了100×物镜,和视觉计算的数量γH2AX疫源地,磷酸化53 bp1疫源地和磷酸化ATM衬底焦点在SP-C-positive细胞,每个细胞的数量γH2AX焦点在AQP5-positive细胞,每个细胞的数量和每个细胞在CD31-positive细胞γH2AX疫源地。我们也算γH2AX疫源地的数量每细胞SP-C-positive细胞是否表达p16,磷酸化NF-κB, il - 6或8-OHdGγH2AX疫源地的数量每个细胞在肺泡壁细胞是否表达了积极caspase-3。
细胞培养和辐照
正常的人类肺微血管内皮细胞与10-Gy x射线辐照剂量(有关详细信息,请参阅在线补充材料)。
豚鼠暴露于香烟烟雾中
Hartley-strain豚鼠暴露在空气或十香烟的烟每周5天10周(有关详细信息,请参阅在线补充材料)。
统计分析
统计分析使用Excel X(美国雷德蒙的微软公司(Microsoft Corp .),佤邦)与Statcel 3插件(OMS、东京、日本)。临床、细胞培养和动物实验数据提出了意味着±扫描电镜、和人类组织学数据中位数。不同的临床资料进行分析通过方差分析,如果结果是显著的,Tukey-Kramer测试用作多重比较事后测试。人类组织学差异Kruskall-Wallis进行分析的数据测试,如果结果是显著的,使用Steel-Dwass多重比较检验。不同的细胞培养和动物实验数据进行分析的数据未配对t。斯皮尔曼等级相关试验的相关性进行分析。的假定值< 0.05被认为是具有统计学意义。
结果
增加的双边带COPD患者的肺
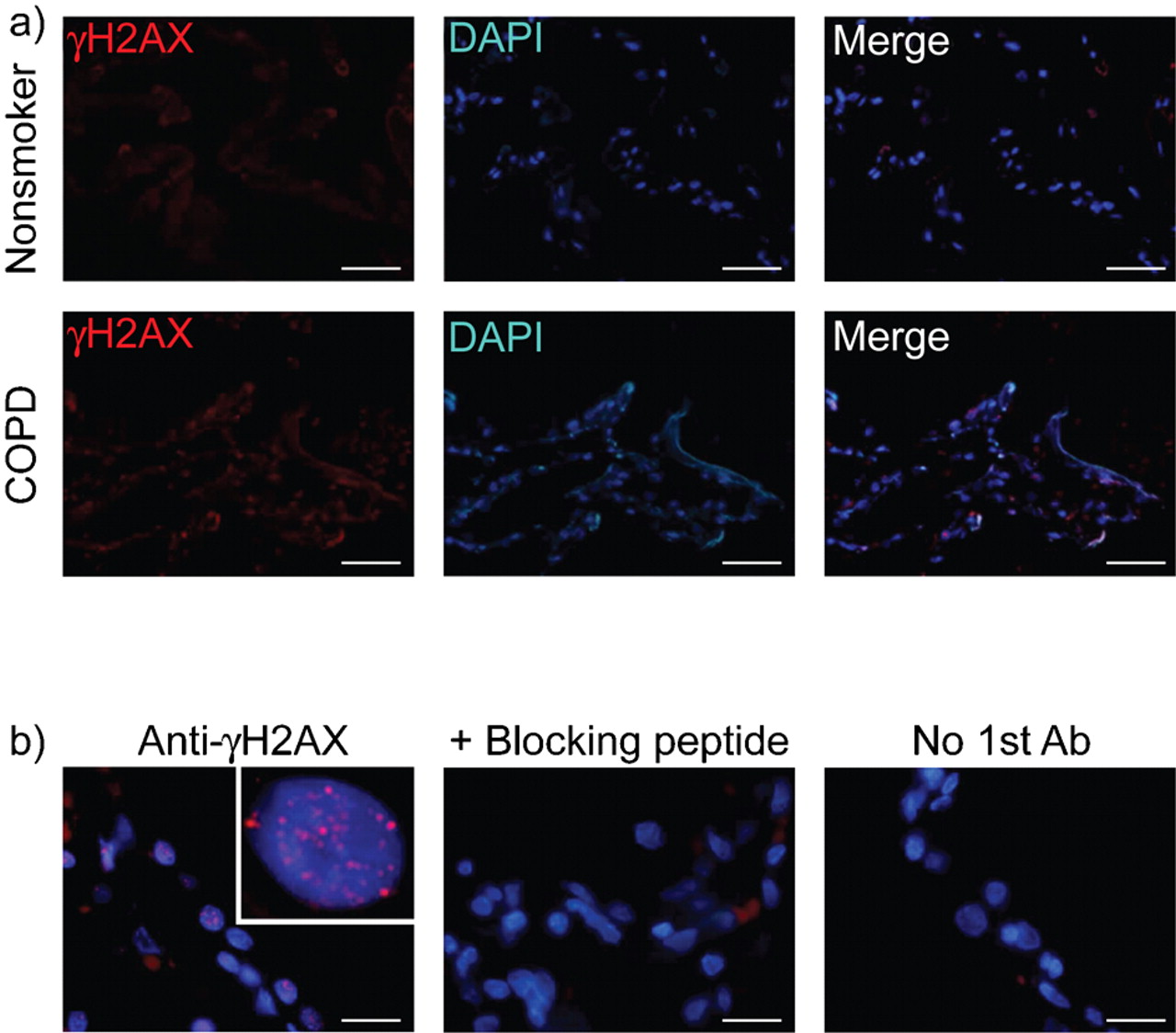
一个γH2AX集中反映了成百上千γH2AX蛋白质分子集中在一个双边带(9]。所示图1一个,免疫荧光染色γH2AX表明许多肺泡壁细胞的细胞核在肺部的慢性阻塞性肺病患者包含多个γH2AX疫源地。没有信号可见anti-γH2AX抗体时省略或抗体pre-incubated阻断肽,从而验证γH2AX的特异性信号(图1 b)。所示图2 b,γH2AX疫源地co-localised与其他双边带标记,包括磷酸化和磷酸化ATM / 53 bp1 ATR基质(13,14),从而确凿的有效性使用γH2AX作为生物标志物来衡量双边带的水平。
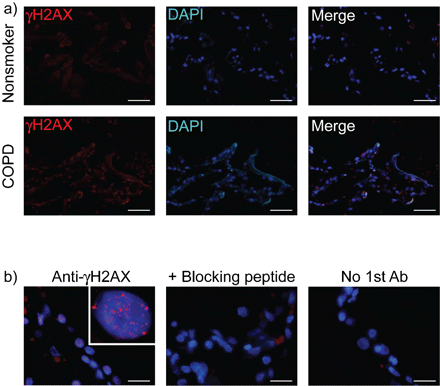
)代表的图像anti-phosphorylated组蛋白2 ax(γH2AX) immunofluorescence-stained部分肺组织获得从无症状不吸烟者和慢性阻塞性肺疾病(COPD)患者。核DNA与4复染色,6-diamidino-2-phenylindole (DAPI)。酒吧= 50μm规模。b)特异性γH2AX免疫荧光染色。离散γH2AX焦点在肺泡壁细胞的细胞核在COPD患者的肺不再可见pre-incubated抗体时阻断肽应用程序之前的二次抗体或anti-γH2AX抗体的问题却被忽略了。阿瑟:抗体。酒吧= 20μm规模。
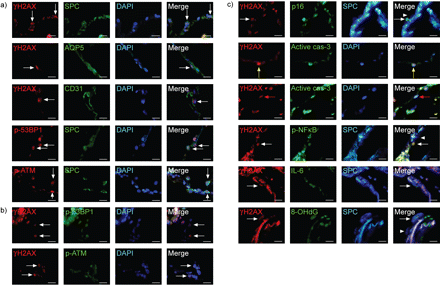
代表图像双重和三重immunofluorescence-stained部分肺组织获得慢性阻塞性肺疾病患者。双链断裂的)双重免疫荧光染色法标记磷酸化组蛋白2 ax(γH2AX)磷酸化p53-binding蛋白1 (p-53BP1)或磷酸化共济失调毛细血管,突变/共济失调毛细管扩张和Rad3-related衬底(p-ATM)和一个特定类型的细胞标记表面活性剂蛋白(SP) - c,水通道蛋白(AQP) 5、CD31。核DNA与4复染色,6-diamidino-2-phenylindole (DAPI)。白色箭头表示double-immunopositive细胞。b)双重免疫荧光染色γH2AX和p-53BP1和γH2AX p-ATM。核DNA与DAPI counter-stained。白色箭头指示co-localisation。γH2AX c)三重免疫荧光染色,SP-C和p16INK4a(p16)磷酸化核factor-κB (p-NF-κB)、白介素(IL) 6或8-hydroxy-2-deoxyguanosine (8-OHdG)和双重免疫荧光染色γH2AX和活跃的半胱天冬酶3 (cas),其次是DNA与DAPI对比染色。白色箭头表示triple-immunopositive细胞。白色箭头指示γH2AX SP-C-positive细胞没有污点或p16,γH2AX或p-NF-κB或γH2AX或8-OHdG。活跃cas-3-positive的细胞核染色广泛(黄色箭头)或局部γH2AX(红色箭头)。酒吧= 20μm规模。
定量分析显示更多的γH2AX焦点在AQP5-positive I型细胞,每个细胞SP-C-positive II型细胞和CD31-positive内皮细胞在慢性阻塞性肺病患者比non-COPD吸烟者和不吸烟者(无花果2和3得了)。同样,磷酸化53 bp1疫源地的数量和磷酸化ATM / ATR衬底疫源地每个细胞在慢性阻塞性肺病II型细胞明显高于吸烟者比non-COPD吸烟者和非吸烟者(无花果2和3 a和e)。然而,I和II型细胞和内皮细胞的non-COPD吸烟者和非吸烟者被发现含有类似数量的γH2AX疫源地,磷酸化53 bp1疫源地和磷酸化ATM / ATR每细胞基质疫源地(图3)。γH2AX疫源地的数量每细胞II型密切相关的细胞的数量每细胞磷酸化53 bp1疫源地(r = 0.83)和磷酸化ATM / ATR衬底疫源地每个细胞(r = 0.78) II型细胞(图S1)。这些结果表明存在更严重的DNA损伤肺泡I和II型细胞和内皮细胞的慢性阻塞性肺病患者比non-COPD吸烟者和非吸烟者。
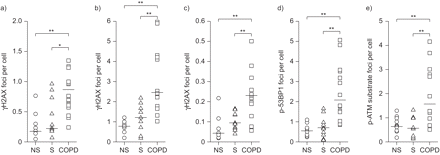
定量分析肺肺泡壁细胞的DNA双链断裂的慢性阻塞性肺病(COPD)患者和无症状吸烟者和非吸烟者(NS)。ax)磷酸化组蛋白2(γH2AX)焦点/细胞水通道蛋白五阳性I型细胞,b)表面活性剂蛋白(SP) -C-positive II型细胞和c) CD31-positive内皮细胞,和d)的数字磷酸化p53-binding蛋白1 (p-53BP1)焦点和e)磷酸化共济失调毛细血管扩张突变(p-ATM)每细胞基质疫源地SP-C-positive II型细胞。-:中位数。*:p < 0.05。* *:p < 0.01。
组织层面分析双边带和细胞凋亡之间的关系,细胞衰老,炎症和氧化应激
与以前的研究结果一致(15- - - - - -21),我们发现肺部的慢性阻塞性肺病患者含有高百分比的肺泡壁细胞表达活跃caspase-3(细胞凋亡的一个标志)(图4),II型细胞表达p16(衰老的标志)(图4 b)、II型细胞磷酸化NF-κB表达(图4摄氏度)和il - 6 (图4 d)炎性表型变化的(标记)和II型细胞表达8-OHdG(氧化应激的标记)图4 e)比non-COPD吸烟者和非吸烟者的肺。由于双边带破坏强烈诱导细胞凋亡,细胞衰老和促炎细胞因子的生产(6,11,12,我们调查是否存在γH2AX双边带的一个标志,是与细胞凋亡有关,细胞衰老在II型细胞和炎性表型变化。当所有的受试者包括在组织水平的相关性分析,与II型肺组织细胞含有较高数量的γH2AX疫源地含有高百分比的肺泡壁细胞表达活动caspase-3 (图5)和更高比例的II型细胞表达p16 (图5 b)、II型细胞磷酸化NF-κB表达(图5度)和II型细胞表达il - 6 (图5 d)。这些结果表明,更严重的肺组织中DNA损伤与更多的II型细胞发生凋亡,衰老,炎性表型变化和DNA氧化。
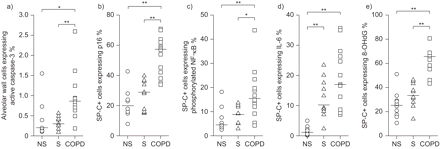
定量分析细胞凋亡、细胞衰老、炎性表型变化和氧化应激在肺部的慢性阻塞性肺病(COPD)患者和无症状吸烟者和非吸烟者(NS)。面板显示的百分比)肺泡壁细胞表达活性caspase-3和II型细胞的百分比,b) p16表达INK4a(p16), c)磷酸化核转录因子(NF) -κB, d)白介素(IL) 6和e) 8-hydroxy-2-deoxyguanosine (8-OHdG)。-:中位数的值。SP:表面活性剂的蛋白质。*:p < 0.05。* *:p < 0.01。

组织层面分析磷酸化组蛋白的数量之间的关系2 ax(γH2AX)焦点/细胞和细胞的百分比呈阳性标记细胞凋亡,细胞衰老,炎性表型变化和氧化应激在整个集团的主题。面板显示数字之间的正相关性γH2AX焦点在II型细胞,每个细胞的百分比)肺泡壁细胞表达活性caspase-3之间γH2AX疫源地的数量在II型细胞,每个细胞的百分比II型细胞p16表达b)INK4a(p16), c)磷酸化核转录因子(NF) -κB, d)白介素(IL) 6和e) 8-hydroxy-2-deoxyguanosine (8-OHdG)。慢性阻塞性肺病:慢性阻塞性肺疾病。
细胞水平分析双边带和细胞凋亡之间的关系,细胞衰老,炎症和氧化应激
接下来,我们进行了细胞水平分析研究DNA损伤是否直接相关的细胞凋亡,细胞衰老和炎性表型变化。II型细胞的慢性阻塞性肺病患者的分析显示,细胞中含有较高数量的γH2AX疫源地有较高的p16的表达(图6)、磷酸化NF-κB (图6 b)和il - 6 (图6 c)。几乎所有的II型细胞包含≥21γH2AX焦点表达p16和磷酸化NF-κB。此外,大约一半的肺泡壁细胞表达活跃caspase-3包含γH2AX焦点(焦染色模式),这表明,DNA损伤引起的细胞凋亡,而其余部分表现出扩散γH2AX染色模式,这表明DNA碎片是发生在细胞凋亡(图6 d)[22]。这些发现表明,DNA损伤直接与诱导细胞凋亡,细胞衰老和慢性阻塞性肺病患者的肺部炎性表型变化。
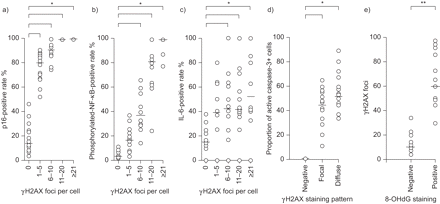
细胞水平分析磷酸化组蛋白的数量之间的关系2 ax(γH2AX)焦点/细胞和细胞的比例,积极彩色标记的细胞凋亡,细胞衰老,炎性表型变化和氧化应激在慢性阻塞性肺疾病(COPD)患者。百分比的II型细胞p16表达了)INK4a(p16), b)磷酸化核转录因子(NF) -κB和c)白介素6 (IL)根据γH2AX疫源地,每个细胞的数量。d)比例的活跃caspase-3-positive肺泡壁细胞表现出没有γH2AX染色,焦γH2AX染色和扩散γH2AX染色(见文本的解释)。e)数量的γH2AX焦点在II型细胞,每个细胞表达或不表达8-hydroxy-2-deoxyguanosine (8-OHdG)。平均值获得的个体慢性阻塞性肺病病人所示。- - - - - -:中位数。*:p < 0.05。* *:p < 0.01。
由于氧化应激是双边带的主要原因之一(23),我们试图确定是否存在相关性DNA氧化和γH2AX疫源地的存在。肺部的组织层面分析的对象作为一个整体与II型显示肺组织细胞含有较高数量的γH2AX疫源地每个细胞有更高比例的II型细胞表达8-OHdG (图5 e)。此外,细胞水平分析肺部的慢性阻塞性肺病患者单独显示,II型细胞表达8-OHdG包含γH2AX疫源地明显多于II型细胞不表达8-OHdG (图6 e)。这些结果表明,氧化应激是至少部分负责DNA双边带在COPD患者的肺实质细胞。
在体外双边带对细胞凋亡的影响,细胞衰老和炎症
确认DNA双边带直接导致细胞凋亡,细胞衰老和炎性反应,我们辐照正常的人类肺微血管内皮细胞与单个10-Gy x射线剂量。这一剂量γH2AX疫源地,每个细胞的数量增加3倍,每个细胞磷酸化53 bp1疫源地6倍的数量,从而验证双边带的感应X-irradiation(图S2)。X-irradiation还增加了肺微血管内皮细胞的百分比,caspase-3彩色的积极活跃,p16和磷酸化NF-κB, il - 6分泌的数量由肺微血管内皮细胞(图S2)。这些结果表明,DNA双边带直接引起细胞凋亡,细胞衰老和肺微血管内皮细胞的炎性反应。我们也把肺泡II型细胞样的上皮细胞系A549细胞对博来霉素,是诱发单链断裂和双边带(24]。A549细胞p21表达但没有p16 (25]。我们发现,博来霉素治疗也增加了γH2AX的数量和每个细胞磷酸化53 bp1疫源地,A549细胞的百分比,彩色积极活跃caspase-3, p21和磷酸化NF-κB,由A549细胞的il - 6分泌量(图S3)。
增加了双边带的香烟烟雾诱发肺气肿豚鼠模型
我们也决定是否使豚鼠暴露于香烟烟雾导致的肺泡壁细胞DNA双边带肺气肿。组织学检查肺组织样本豚鼠暴露于香烟烟雾的10周显示放大的肺泡气空间和Lm值(平均领空大小)是45%高于不吸烟对照组(p < 0.01)(图S4)。肺泡壁细胞的肺香烟smoke-exposed组包含两倍γH2AX疫源地每个细胞的肺的肺泡壁细胞对照组(意味着±扫描电镜2.05±0.26与1.06±0.17;p < 0.01)(图S4)。这些结果表明,双边带发生气肿的豚鼠肺的肺泡壁细胞暴露于香烟烟雾。
讨论
Immunofluorescence-based化验γH2AX提供敏感、高效、可再生的方法,测量双边带的数量(7- - - - - -10]。持久以来γH2AX焦点在最初的诱导DNA双边带表明一些仍未修理的损害,疫源地的持久性是广泛使用的DNA损伤的生物标志物在不同组织(7- - - - - -10]。在目前的研究中,我们发现显著增加数字γH2AX疫源地的肺泡壁细胞,包括I型和II型细胞,内皮细胞的慢性阻塞性肺病吸烟者比non-COPD吸烟者和非吸烟者。γH2AX疫源地的存在在II型细胞与细胞凋亡有关,细胞衰老,炎性表型变化和氧化应激。这项研究的结果也显示,γH2AX焦点与DNA损伤修复co-localised蛋白质,即。磷酸化ATM / ATR基质和磷酸化53 bp1的数字γH2AX疫源地每个细胞都密切相关的数字磷酸化53 bp1疫源地和磷酸化ATM / ATR每细胞基质疫源地,从而证明的有效性γH2AX作为DNA损伤的生物标志物。综上所述,本研究的结果表明,DNA损伤,特别是双边带至少部分地是由于氧化应激引起的,导致慢性阻塞性肺病的分子发病机制通过诱导细胞凋亡,细胞衰老和炎性反应。
这项研究的结果提供的证据表明,双边带是肺泡壁细胞的显著特征,包括I型和II型细胞和内皮细胞,慢性阻塞性肺病患者。这人类肺组织研究中获得的证据支持我们的动物研究的结果在豚鼠显示明显高于数字γH2AX疫源地的肺的肺泡壁细胞与香烟烟雾诱发肺气肿豚鼠肺部sham-exposed动物(图S4)。我们的研究结果是一致的与他人(26- - - - - -28),检测DNA损伤形式的微卫星不稳定性(MSI)和杂合性丢失(LOH)痰液细胞和支气管肺泡液体(BALF)细胞获得慢性阻塞性肺病患者。香烟中含有许多genotoxins,包括苯并(a)芘、亚硝胺、醛和氧化剂,诱发各种形式的DNA加合物(5]。多个基因位点的DNA损伤积累,有时导致双边带,大部分细胞毒性损伤;这些双边带触发γH2AX疫源地的形成打破网站(29日]。本研究的结果显示更高数量的γH2AX疫源地,每个细胞在慢性阻塞性肺病的吸烟者肺泡壁细胞,但不是non-COPD吸烟者比不吸烟者。这一发现印证了他人的研究成果(26- - - - - -28),他发现MSI和LOH痰细胞和BALF细胞的COPD吸烟但不是non-COPD吸烟者。他们的发现,和自己一起,强烈建议吸烟者肺部的慢性阻塞性肺病的DNA损伤是放大和/或仍未修理的,这导致DNA损伤逐渐累积在肺部。
目前COPD的发病机制的理论表明,肺泡破坏是由几个pathobiological过程之间的相互作用,包括炎症、细胞凋亡、细胞衰老和氧化应激(1]。通过仔细分析组织和细胞水平的相关性,我们发现DNA损伤与细胞凋亡、细胞衰老和慢性阻塞性肺病患者的肺部炎性表型变化,从而凸显出DNA损伤的分子之间的联系pathobiological过程认为是参与肺泡的破坏在慢性阻塞性肺病。
由于这是一项观察性研究人类的肺组织标本,是不可能建立一个因果关系之间的DNA损伤和细胞凋亡,细胞衰老和炎性表型变化。一些DNA损伤的观察到在这个研究可能是由于细胞凋亡,细胞衰老和炎症,而不是他们的事业。然而,证实DNA损伤,特别是双边带,是一个强烈的诱导细胞凋亡,细胞衰老和炎性反应在各种类型的细胞和组织6- - - - - -12]。事实上,许多研究表明,ATM /γH2AX-mediated信号转导通路的激活反应未修理的双边带引起细胞凋亡和细胞衰老,从而消除DNA-damaged细胞组织,防止其致癌的转换(6,9]。此外,小说ATM /γH2AX-mediated促炎症信号转导通路,以应对双边带最近发现激活NF-κB和CCAAT / enhancer-binding protein-β转录活性和刺激分泌的促炎细胞因子,il - 6等,进而在反馈回路自加强衰老增长逮捕[11,12]。在目前的研究中,我们表明,双边带X-irradiation培养引起的肺微血管内皮细胞诱导炎性反应,如NF-κB磷酸化和il - 6生产,以及caspase-3激活和p16表达,这表明双边带的直接原因是促炎症反应、细胞凋亡和细胞衰老(图S2)。我们还表明,治疗与博来霉素A549细胞,诱导γH2AX焦点的形成,造成NF-κB磷酸化和il - 6生产,以及caspase-3激活和p21表达(图S3)。我们最近还发现持久的克拉拉细胞DNA损伤引起重复注射的老鼠与萘和bromo-2-deoxyuridine引起气道上皮细胞衰老伴随着p38增殖蛋白激酶(MAPK)端依赖气道炎症反应30.]。这些线的证据,虽然不是完全确凿,支持假设DNA损伤细胞凋亡中起着决定性作用,细胞衰老和炎性反应在慢性阻塞性肺病患者的肺。然而,未来的研究在COPD动物模型将需要显示这个观点是否正确。
氧化应激是双边带的主要原因之一23]。在目前的研究中,我们展示了一个关联γH2AX疫源地和8-OHdG的存在,表明氧化应激负责双边带在肺部的慢性阻塞性肺病患者。它提出了氧化应激导致细胞凋亡,细胞衰老和炎症在慢性阻塞性肺病1,31日),最近的证据表明,氧化应激诱发炎症通过多种机制涉及氧化c-Jun激酶激活,激活p38-MAPK, NF-κB和激活蛋白1,氧化抑制组蛋白脱乙酰酶活性(31日]。我们的发现在这个研究表明氧化DNA损伤的机制也可能导致持续的炎症慢性阻塞性肺病。
本研究的结果表明,γH2AX焦点的形成也与il - 6生产由II型细胞。这项发现支持了最近的证据表明,“危险信号”从受伤的细胞引起免疫反应32),il - 6是由DNA-damaged分泌细胞,是一个主要因素在炎性表型(11,12,33]。它已经表明,il - 6参与了COPD的肺和系统性炎症(34)和更高的血清il - 6水平与低用力呼气量在1 s [35]。超表达的il - 6在老鼠身上发现了导致肺气肿和气道纤维化的发展36]。鉴于这些发现,我们表明,il - 6可能是一个关键的中介将DNA损伤与炎症反应在慢性阻塞性肺病。
这项研究有一些局限性。首先,慢性阻塞性肺病患者吸烟pack-yr历史超过1.6倍non-COPD吸烟者,尽管两组之间的差异没有统计学意义(表1由未配对t检验)(p = 0.12)。这可能对于吸烟强度不匹配,然而,不大可能占γH2AX疫源地的更高的数字每个细胞在慢性阻塞性肺病吸烟者,因为之间没有明显的相关性被发现吸烟pack-yrs和γH2AX疫源地的数量每个细胞在I型细胞(r = 0.16, p = 0.58), II型细胞(r = 0.001, p = 0.99)或内皮细胞(r = 0.18, p = 0.22)。其次,它仍然是不确定DNA损伤早期pathobiological事件在慢性阻塞性肺病,因为大多数的慢性阻塞性肺病患者纳入本研究先进COPD肺减容手术。进一步研究评价慢性阻塞性肺病γH2AX疫源地的数量在所有阶段,在主体与不同程度的吸烟暴露和慢性阻塞性肺病患者不同的药物将使我们更好地阐明COPD的DNA损伤。第三,这项研究可能高估了的DNA损伤的肺non-COPD吸烟者和非吸烟者,因为共病癌症可能提升γH2AX疫源地的形成在周围肺组织通过旁观者效应(37]。最后,它仍然未知是否nonparenchymal细胞DNA损伤,如气道上皮细胞和肺动脉内皮细胞,在慢性阻塞性肺病患者也更严重。
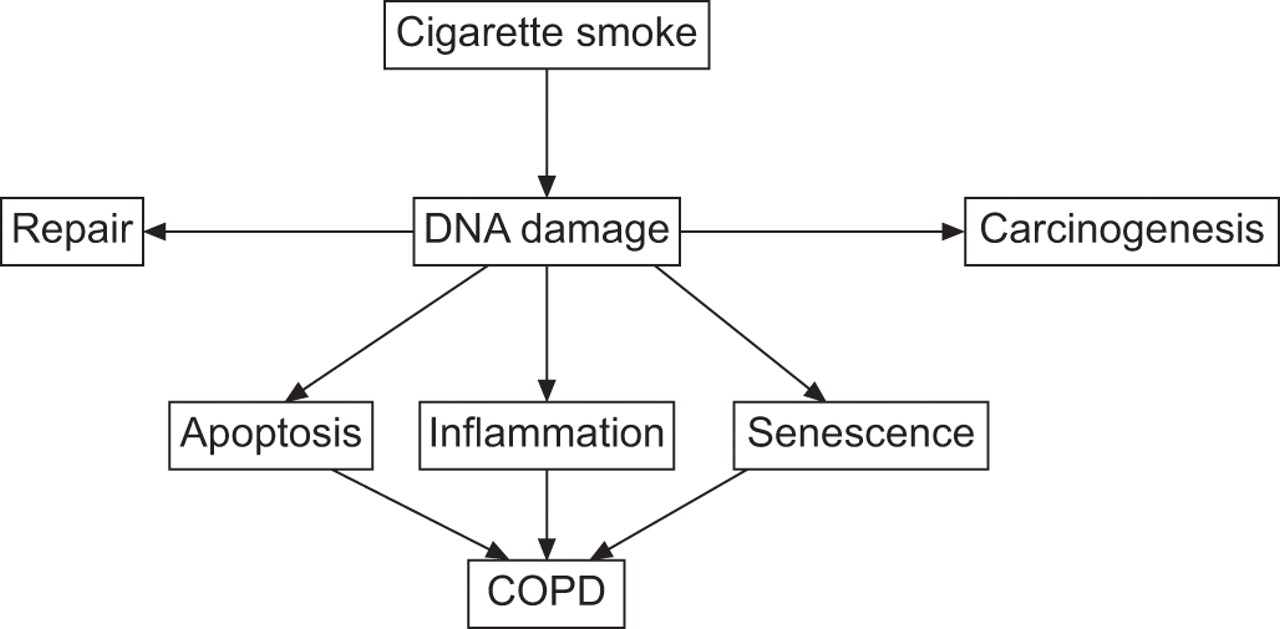
基于本研究的结果,我们假设,细胞凋亡,细胞衰老和炎症,这被认为是慢性阻塞性肺病的pathobiological过程(1),至少部分归因于DNA损伤(图7)。COPD的发病机制的传统理论表明,激活炎症的吸入香烟烟雾和其他污染物过程中扮演着重要的角色在气道壁增厚,肺泡破坏,空域扩张和血管重塑38]。我们假设DNA损伤分子机制背后的慢性阻塞性肺病似乎表明几个重要问题的答案,传统的理论并没有解决。第一个问题是,为什么慢性阻塞性肺病需要几十年的时间来开发?答案基于我们的DNA损伤假设会造成的DNA损伤长期吸烟需要经过几十年的积累在慢性阻塞性肺病发展之前,通过类比肺癌的发展。第二个问题是,为什么停止吸烟后炎症持续存在?答案是,它可能会持续,因为吸烟引起的DNA损伤持续戒烟之后很久,正如前面报告(3,4]。第三个问题是,为什么糖皮质激素对慢性阻塞性肺病的炎症?几乎没有影响答案可能是糖皮质激素不修复DNA损伤。最后,为什么有些吸烟者慢性阻塞性肺病而其他人不发展,,为什么慢性阻塞性肺病吸烟者比non-COPD吸烟者更容易患肺癌?最后两个问题的答案是,由于吸烟更大的对DNA损伤的易感性可能由基因决定更容易吸烟导致肺癌,所以吸烟者更容易受到DNA损伤可能倾向于COPD和肺癌。然而,目前的研究结果并不回答所有这些问题;纵向研究需要,包括更多的慢性阻塞性肺病患者不同阶段疾病的严重性。
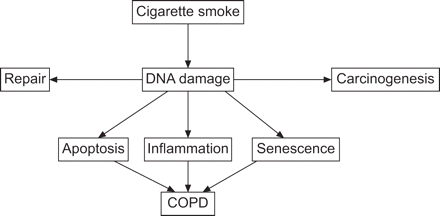
DNA损伤发病的机制的假说在吸烟者慢性阻塞性肺病(COPD)。吸烟会引起DNA损伤,至少部分通过氧化应激。如果不修理,DNA损伤凋亡、细胞衰老和肺泡壁细胞的炎性反应,所有这一切为COPD的发展作出贡献。未修理的DNA损伤也促进致癌作用。
两个独立的调查人员最近提出了一个体细胞突变假说的慢性阻塞性肺病,这与我们的假设的不同之处在于需要“体细胞基因突变”来解释增强炎症反应在慢性阻塞性肺病。2003年,一个nderson和Bozinovski(39)提出收购体细胞突变基因编码p53, Ras、表皮生长因子受体和PTEN香烟致癌物引起的(磷酸酶tensin同系物)可能导致COPD的发病机制导致异常的炎症反应。2009年,Tzortzaki和Siafakas(40)提出收购体细胞突变,如MSI和LOH,可能会导致改变肺上皮细胞屏障层,反过来误解的宿主免疫系统“异物”,导致异常的免疫反应和细胞毒性CD8 +细胞的克隆扩张,最终导致细胞凋亡或坏死。虽然我们的DNA损伤假说和体细胞突变假说需要进一步实验支持,我们认为这两种假说提供了一种新的,以前被忽视的遗传损伤在COPD的发病机制中的作用。
总之,目前的研究结果有力地表明,DNA损伤是慢性阻塞性肺病的分子发病机制的基础。DNA损伤假说可能有助于更好地理解COPD的发病的机制,针对新药物,如药物以防止DNA损伤和调节响应DNA损伤,导致慢性阻塞性肺病的pathobiological过程。
确认
作者感谢m . Shino和y Sugimura(第一医学系的东京女子医科大学、东京、日本)的技术援助。
脚注
可以从本文的补充材料www.www.qdcxjkg.com
支持声明
这项工作是支持的补助金从教育部科学研究,科学和文化,日本,由卫生部、劳动和福利日本调查棘手的疾病。
感兴趣的语句
没有宣布。
- 收到了2011年3月22日。
- 接受2011年8月29日。
- ©2012人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}