





摘要
慢性粘液高分泌(CMH)导致哮喘的发病率和死亡率,在严重的类固醇耐药性疾病患者中,目前的治疗方法仍然无法控制。由促炎细胞因子如白介素(IL)-1β驱动的气道上皮细胞和气道平滑肌细胞(ASMCs)之间的交叉信号改变,提供了影响CMH的潜在机制。本研究通过比较哮喘和控制性ASMCs之间il -1β诱导的基因表达谱以及随后的旁分泌对气道上皮粘液产生的影响,研究了CMH的机制在体外.
应用微阵列分析和ELISA分析了il -1β治疗的哮喘患者和健康供体ASMCs。气液界面(ALI)培养的CALU-3和原代气道上皮细胞用确定的候选细胞进行处理,并评估粘液产生情况。
全身的il - 1的βCCL20与轻度哮喘患者和健康对照组相比,中度哮喘患者ASMCs的表达和蛋白释放增加.IL-1β诱导降低MIR146A哮喘源性ASMCs的表达。减少MIR146A表达得到验证在活的有机体内在16名哮喘患者的支气管活检中与39个健康捐献者过表达miR-146a-5p可抑制ASMCs中CCL20的释放。CCL20治疗ali培养的CALU-3和原代气道上皮细胞诱导粘液产生,而哮喘患者痰液中CCL20水平与CMH水平升高相关。
ASMCs产生的CCL20的升高,可能是由抗炎miR-146a-5p表达失调引起的,可能有助于哮喘患者粘液的产生。
摘要
ECCL20水平升高导致哮喘患者粘液分泌增多http://ow.ly/m7F830ko3Rb
简介
哮喘是一种慢性炎症性疾病,影响全球3亿人[1].慢性粘液分泌过多(CMH)可导致哮喘的发病率和死亡率[2],目前的治疗方法仍然无法控制。迫切需要确定新的治疗靶点。
尽管炎症期间气道上皮层黏液分泌增加[3.], CMH的潜在机制仍有待阐明。气道上皮细胞向纤毛细胞或杯状细胞的分化受粘膜下层其他结构细胞的指导。上皮层与气道平滑肌细胞(ASMCs)之间的相互作用可能调节粘液的产生,因为哮喘患者气道平滑肌质量增大[4].
长期以来,ASMCs一直被认为具有被动作用,但越来越多的证据表明,这些细胞在CMH的炎症过程中发挥着重要作用,提供了细胞因子和趋化因子的活跃来源通过许多途径[5].
其中一个在哮喘中被改变的炎症途径是炎症小体,这是一种多蛋白复合体,在激活促炎细胞因子中起着重要作用,如。白细胞介素(IL)-1β由原形态转化为活性状态[6].中性粒细胞性哮喘中炎性小体的活性增强[7],导致痰中活性IL-1β水平升高。
IL-1β是一种强促炎信号分子,其下游介质与粘液的产生有关[8].然而,IL-1β对气道结构细胞(尤其是ASMCs)促炎反应的影响以及CMH在哮喘中的潜在作用尚不清楚。
在这项研究中我们发现CCL20而且MIR146A比较哮喘和健康ASMCs之间的基因表达谱在体外对IL-1β的反应重要的是,CCL20已被证明通过与其唯一已知的受体CCR6结合,在上皮培养物中诱导MUC5AC的表达[9].此外,在小鼠模型中,抗ccl20治疗显著减少了病毒诱导的粘液产生[10].此前,miR-146a中的单核苷酸多态性(SNP)已被证实与哮喘和其他促炎症疾病的存在有关[11,12].有趣的是,我们最近发现,在慢性阻塞性肺疾病(COPD)中,支气管活检中miR-146a-5p的低表达与CMH呈负相关[13],强调了miR-146a-5p作为呼吸系统疾病中粘液调节的调节因子。基于已知的CCL20和miR-146a-5p在黏液产生中的作用,我们随后研究了ASMCs产生的这些因子如何影响气道上皮细胞中的黏液产生。
方法
人体组织
原代人ASMCs如前所述获得[14,15],并获得了悉尼大学和澳大利亚悉尼的参与医院(康科德遣返总医院、悉尼西南地区卫生服务中心和皇家阿尔弗雷德医院)的伦理批准。所有患者或其近亲均提供书面知情同意。病人的特征概要见表1.
微阵列处理与分析
从哮喘患者(n=3)和健康对照(n=3)中分离ASMCs,并按照前面描述的方法培养(数据集A) [14,15].细胞用10 ng·mL处理−1IL-1β(研发系统,明尼阿波利斯,MN,美国)8小时[16].根据制造商的说明,将细胞总mRNA分离、标记并在genchip人类基因1.0 ST阵列(Affymetrix, Santa Clara, CA, USA)上运行(基因表达综合标识GSE63383) [4].为了进行独立验证,来自哮喘患者(n=2)和健康供体(n=4)的ASMCs被培养并以同样的方式用IL-1β处理(数据集B)。根据制造商的说明,样本被标记并在人类U133Plus 2.0阵列(Affymetrix)上运行。微阵列分析概述补充材料.
路径分析
利用基因集富集分析(GSEA)软件2.0.14 (http://software.broadinstitute.org/gsea).蛋白质网络分析采用STRING版本10 (https://string-db.org)对重叠基因的研究。GSEA还用于研究数据集A中调节的通路,使用BioCarta数据库(www.biocarta.com).转录因子结合位点分析使用g:Profiler (https://biit.cs.ut.ee/gprofiler).
候选微阵列验证
在转录(实时定量PCR)和翻译(ELISA)水平上对微阵列结果进行了验证补充材料.
支气管活检的定量处理MIR146A表达式
采集呼吸健康受试者的支气管活检[17]和现患哮喘的病人[18,19有可逆性证明和气道对组胺的高反应性(刺激性剂量导致组胺在1 s内用力呼气量下降20%(采用30秒潮动呼吸)<32 mg·mL−1).这项研究对39名健康受试者和16名哮喘患者进行了分析,他们都不吸烟,目前也没有吸入皮质类固醇。病人的特征概要见补充表S1.所有研究方案都得到了格罗宁根大学医学中心(格罗宁根,荷兰)医学伦理委员会的批准,所有受试者都提供了书面知情同意。RNA被分离和测序描述在补充材料.
miR-146a-5p预测靶点
为了识别miR-146a-5p的下游靶点,我们使用了一个公开的微阵列数据集(基因表达综合转染miR-146a-5p mimic (5 nM)的人肝脏Huh7.5.1细胞的标识符GSE79340)与阴性对照(n=3)比较。
功能分析
在使用miR-146a-5p (100 nM)模拟物或使用RNAiMAX (Invitrogen, Carlsbad, CA, USA)模拟对照转染前,将永生ASMCs培养到80-90%的融合并去除血清。24 h后,10 ng·mL处理细胞−1IL-1β或0.1%牛血清白蛋白(对照)。24小时收集无细胞上清,ELISA法检测IL-8水平。
在气液界面(ALI)培养人肺上皮细胞系CALU-3和原代气道上皮细胞,处理浓度为10 ng·mL−1CCL20 48小时,粘液用阿利新蓝染色评估补充材料.
CMH的定义
为了定义CMH,哮喘患者被要求回答一份临床问卷:“你在上周咳痰的频率是多少?”“(22].这个问题有七个可能的答案:1)从不,2)有时,3)偶尔,4)经常,5)大部分时间,6)经常,7)总是。1)有应答者为无CMH, 2)和3)有应答者为中度CMH, 4) -7)有应答者为重度CMH。在有痰液可用的哮喘患者中,有n=80人回答了问卷并进行了分析。
统计数据
使用Prism版本6 (GraphPad, La Jolla, CA, USA)进行统计检验和图表绘制。p值<0.05被认为有统计学意义。
结果
ASMCs对IL-1β的反应
为了评估IL-1β是否参与了ASMCs和气道上皮细胞之间的异常交叠,从而导致哮喘气道中的CMH,我们首先检查了IL-1β治疗后的基因调控.将哮喘患者(n=3)和对照组(n=3)合并,以获得足够的能量来确定IL-1β对基因表达的影响(数据集A)。基因表达分析发现,与基线相比,IL-1β治疗后,408个基因上调,143个基因下调(翻倍变化>±2,错误发现率(FDR) <0.05) (补充表S2).图1一个和b分别说明了IL-1β和火山图显著改变的基因。
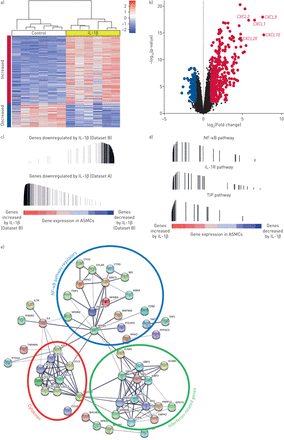
白细胞介素-1β对气道平滑肌细胞(ASMCs)的治疗IL-1R: IL-1受体;GSEA:基因集富集分析;罗斯福:错误发现率。a) ASMCs经IL-1β处理8 h后基因改变的热图和b)火山图(倍增变化>±2,FDR <0.05)。c) GSEA:比较两个独立数据集,ASMCs中IL-1β处理上调和下调基因的富集(GSEA FDR <0.05)。d) GSEA: ASMCs中NF-κB、IL-1R和TID通路相关基因的富集,IL-1β处理上调了相关基因(GSEA FDR <0.05)。(c)和(d)中的彩色条表示基于在ASMCs中不同表达对治疗的排序基因;竖条表示正在测试的途径中涉及的基因的运行GSEA富集评分和位置(在排序的基因列表中)。e)蛋白质相互作用分析。
为了验证这些发现,我们研究了另一个独立数据集,来自n=2个哮喘患者和n=4个健康来源的ASMC培养物,使用10 ng·mL处理−1IL-1β治疗8小时(数据集B)。基因表达分析发现,与基线相比,IL-1β治疗期间有377个基因上调,98个基因下调(倍变化>±2,FDR <0.05)。两个数据集的GSEA显示,在IL-1β处理后,两个数据集中有255个显著上调的基因和111个显著下调的基因在同一方向上核心富集(图1 c).
数据集A的通路分析显示,由IL-1β增加的大部分通路是促炎通路,包括NF-κB、IL-1受体(IL-1R)和TID(调节干扰素(IFN)信号通路的伴随物)通路(图1 d).蛋白质网络分析(使用STRING 10.0版本)显示,ASMCs中增加的IL-1β表达特征在蛋白质-蛋白质相互作用中富集,表明识别的基因可能具有相似的功能(图1 e).三个不同的集群被确定:ifn相关基因,NF-κB信号相关基因和促炎细胞因子。NF-κB和IFN调节因子1 (IRF1)被鉴定为连接多个蛋白网络簇的中心枢纽蛋白,表明这些蛋白在ASMCs中IL-1β刺激过程中起着中心作用(图1 e).IL-1β上调的前10个基因中的6个形成了一个清晰的个体细胞因子集群,其中包括CXCL家族蛋白CXCL8(一种先前与哮喘中性粒细胞性气道炎症相关的细胞因子[23]), CXCL10 (ifn调节的肥大细胞迁移相关细胞因子[24)和CCL20 (CCR6的趋化剂+未成熟的树突状细胞、t辅助细胞17和中性粒细胞)。
使用g:Profiler对数据集A和数据集B之间重叠的基因进行转录因子结合分析,发现上调的NF-κB基因富集(FDR 7.84×10−10), RelA (NF-κB复合体的组成部分;罗斯福7.18×10−13)和IRF1 (FDR 1.72×10−13)转录因子结合位点,而ETF的下调基因被富集(FDR 5.57×10−7)和EGR1(早期生长反应1;罗斯福5.64×10−5)转录因子结合位点。这些结果再次证实了NF-κB和IRF1是ASMCs中IL-1β信号的关键调控因子。
重要的是,CCL20已被证明在上皮培养物中诱导MUC5AC的表达[10,25].此外,在小鼠模型中,抗ccl20治疗显著减少了病毒诱导的粘液产生。基于其在粘液产生中的已知作用,我们选择CCL20进行进一步的功能研究。
ASMCs中IL-1β诱导的CCL20蛋白释放
为了验证微阵列的研究结果,对数据集A中的哮喘(轻度和中度)和健康来源的ASMCs进行10 ng·mL刺激后测量CCL20 mRNA表达−1IL-1β作用8小时。IL-1β显著增加了CCL20的表达,支持微阵列结果(图2一个).哮喘源性ASMCs与健康源性ASMCs及轻、中度哮喘患者ASMCs mRNA表达无差异。接下来,我们在蛋白质水平上证实了我们的发现,并观察到IL-1β在24小时后显著增加了ASMCs中CCL20的释放(图2 b).与健康对照组和轻度哮喘患者相比,中度哮喘ASMCs患者的CCL20水平明显升高。基线时ASMCs释放的CCL20水平与先前报道的上皮细胞释放的CCL20水平相当[20.].
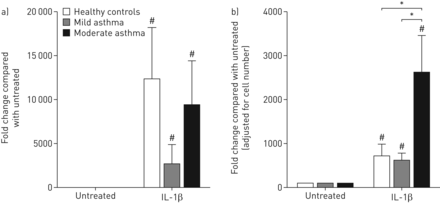
白介素(IL)-1β诱导气道平滑肌细胞(ASMCs)产生ccl20mrna。将ASMCs培养至与生长培养基汇合,静置72 h后用0.1%牛血清白蛋白(对照)或10 ng·mL处理−1IL-1β作用8小时(mRNA)或24小时(蛋白质)。a) CCL20 mRNA水平(健康对照组n=4,轻度哮喘组n=3,中度哮喘组n=3)。b)测定无细胞上清液中CCL20蛋白水平(健康对照组n=4,轻度哮喘组n=3,中度哮喘组n=3)。数据以平均值±表示扫描电镜.对配对和未配对样本分别采用配对t检验和t检验进行统计分析。*: p<0.05,与健康对照组比较;#: p<0.05,与IL-1β处理比较。
MIR146A在哮喘中降低,并调节CCL20
与健康来源的ASMCs相比,在哮喘患者中,IL-1β对CCL20蛋白的调控存在差异,我们研究了其他IL-1β诱导的基因。
该分析是在数据集a中IL-1β处理调节的基因子集上进行的(倍增变化>±2,FDR <0.05)。只有一份记录,MIR146A与健康对照的ASMCs相比,IL-1β治疗后哮喘源性ASMCs的基因表达增加明显较小(翻倍变化>±2,FDR <0.05) (图3一).MIR146A是miR-146a-3p和miR-146a-5p的前体转录本,后者是一种著名的抗炎microRNA (miRNA),在许多炎症疾病中被确定为调节失调[26].以确定是否MIR146A哮喘患者的表达改变在活的有机体内我们研究了其在16例哮喘患者和39例健康对照者支气管活检中的表达。MIR146A与健康对照相比,哮喘支气管活检中表达减少在体外结果(图3 b).
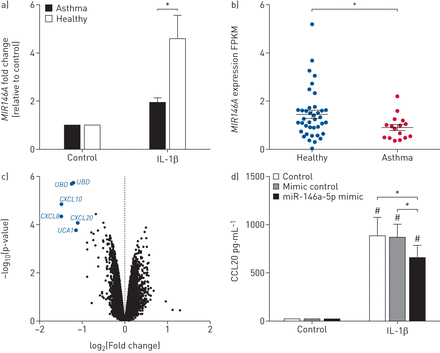
miR-146a-5p在气道平滑肌细胞(ASMCs)中的作用IL:白介素;FPKM:每千基转录本每百万映射读的片段数。a)用IL-1β治疗哮喘患者(n=3)和健康对照组(n=3) ASMCs 8 h的芯片结果。b)MIR146A哮喘患者和健康对照者支气管活检中的表达。c)与阴性对照相比,转染miR-146a-5p mimic (5 nM)的Huh7.5.1细胞的火山图(n=3)。d)与模拟对照组相比,在miR-146a-5p (mimic)过表达存在或不存在的情况下,经IL-1β处理的ASMCs中的CCL20水平(n=5)。数据以平均值±表示扫描电镜.对配对和未配对样本分别采用配对t检验和t检验进行统计分析。*: p < 0.05;#: p<0.05,处理与对照组比较。
确定…的功能MIR146A,我们关注已知的抗炎成熟转录物miR-146a-5p,并使用公开的过表达miR-146a-5p的Huh7.5.1细胞基因表达数据集研究该转录物的直接和间接靶标。基因表达分析发现五个基因(克服各种(泛素D),CXCL10,CXCL8,CCL20而且UCA1(尿路上皮癌相关1)),而miR-146a-5p过表达时没有上调的基因(翻倍变化>±2,FDR <0.05)。火山分布图如图所示图3 c并给出了一个重要基因的表补充表S3.其中一个被下调的基因,CCL20,已知由miR-146a-5p调节[27].因此,我们研究了miR-146a-5p是否负向调节il -1β诱导的ASMCs中CCL20蛋白释放。在不朽的ASMCs中过表达miR-146a-5p模拟物导致il -1β诱导的CCL20释放显著下调(图3 d).
CCL20受体CCR6存在于气道结构细胞中,CCL20诱导ALI中生长的CALU-3细胞产生粘液
在确定了CCL20作为一种中介因子,在哮喘和健康来源的ASMCs中表达差异后,我们下一步想要了解在哮喘气道中增加的CCL20的功能后果。首先,为了确定气道结构细胞是否能够对CCL20产生反应,我们研究了其独特的受体CCR6在气道细胞中的表达。最初,我们对人原代ASMCs、永生ASMCs和CALU-3细胞进行了实时PCR,结果显示CCR6信使rna (补充图S1).CCR6在人支气管切片的免疫组化染色证实了在ASMCs和气道上皮上的表达(补充图S2).
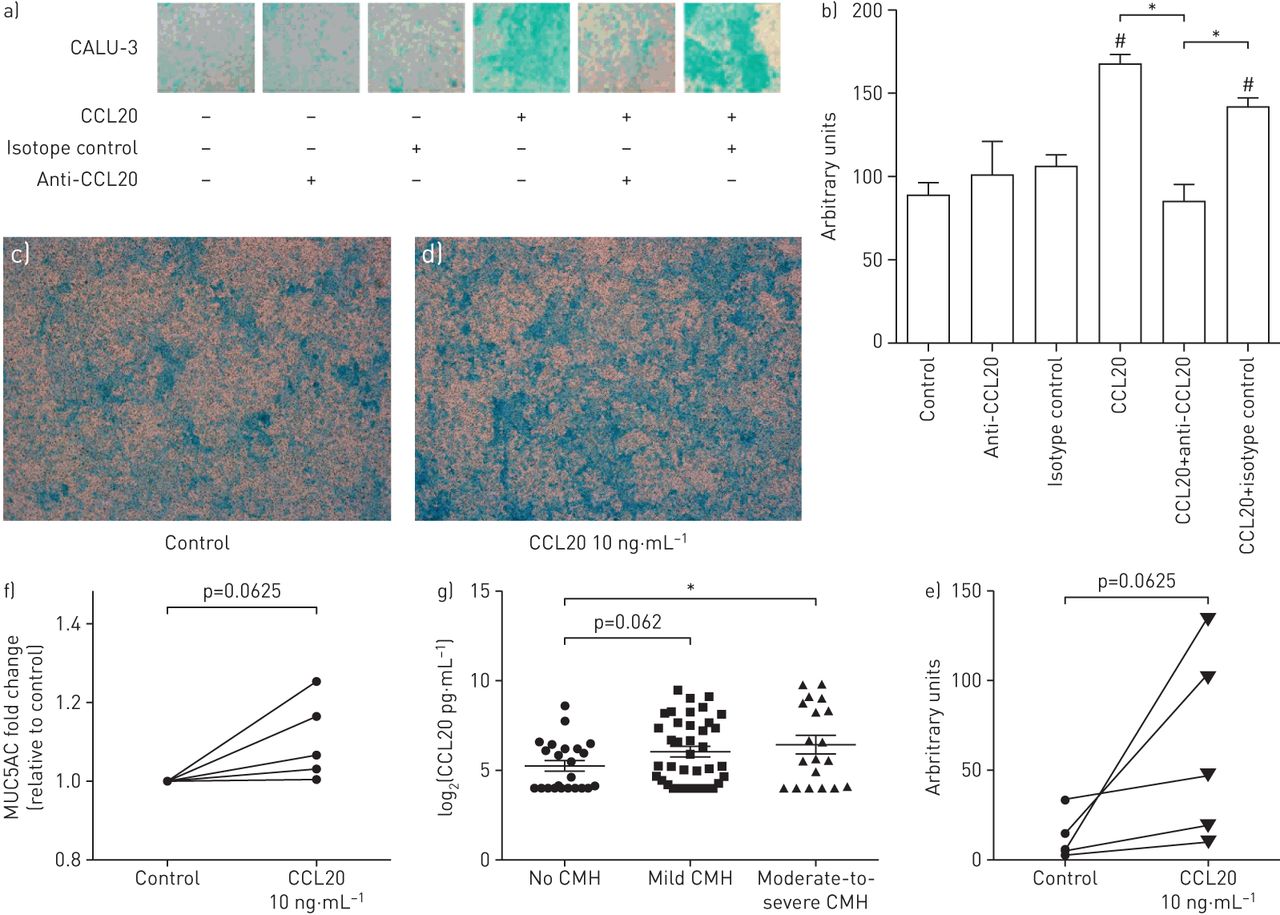
此前,在小鼠模型中,抗CCL20治疗显著减少呼吸道合胞病毒感染时粘液的产生,并且CCL20已被证明可诱导液体培养中MUC5AC的表达[9,10].为了确定CCL20是否能直接促进分化上皮细胞模型中黏液的产生,在ALI中培养CALU-3细胞,让其分化为黏液产生细胞,然后在基底外侧用生理相关水平的CCL20处理。CCL20处理CALU-3细胞48小时增加了粘液的产生,Alcian blue染色(图4一).ccl20特异性抗ccl20抗体显著减少CALU-3细胞中ccl20诱导的粘液产生,而同型对照无显著影响(图4 b).阿利新蓝染色发现CCL20处理ali分化的原代人气道上皮细胞培养物也有相同的趋势(p=0.0625) (图4 c- e)。在ALI根尖清洗中测定的MUC5AC蛋白水平也被发现在CCL20处理后增加(图4 f).
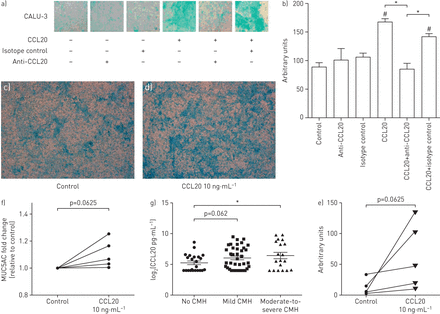
CCL20对气液界面(ALI)生长的CALU-3和原代气道上皮细胞黏液产生的影响。CMH:慢性粘液分泌过多。a)第5天用完全DMEM(对照)、兔抗人CCL20抗体同型对照(10ng·mL)处理的ALI中生长的CALU-3细胞的阿利新蓝染色代表性图像−1CCL20,兔抗人CCL20抗体+10 ng·mL−1CCL20和同型对照+10 ng·mL−1CCL20处理48 h (n=3)。b) CALU-3细胞Alcian blue染色的密度分析。c - e)在第28天用c) PBS或d) 10 ng·mL处理的ALI中生长的初代气道上皮细胞的阿利新蓝染色代表性图像−1CCL20处理48 h, e)密度分析(n=5)。f) ALI洗涤液中的MUC5AC蛋白测定(n=5)。g)无CMH、轻度CMH或中度至重度CMH患者痰中CCL20水平。数据以平均值±表示扫描电镜.对配对和未配对样本分别采用配对t检验和t检验进行统计分析。*: p < 0.05;#: p<0.05,与对照组比较。对ALI培养的原代气道上皮细胞匹配样本进行Wilcoxon分析。
哮喘患者痰液中CCL20水平与痰液高分泌相关
先前的研究表明,间充质因子可以穿过基底固有层,并在肺液中发现[28].这种现象被认为在哮喘患者中增强,因为有文献记载上皮层的渗漏特性[29],允许运输到粘膜层,在那里它可能诱导粘液的产生。为了确定CCL20水平是否与哮喘患者的粘液生成有关,我们调查了伴有和不伴有CMH的哮喘患者的痰液中CCL20的水平。在纳入的80例患者中,24例为无CMH, 32例为轻度CMH, 19例为中重度CMH。痰中CCL20蛋白水平在中度至重度CMH与无CMH比较显著升高(p<0.05),轻度与无CMH比较有升高趋势(p=0.062) (图4 g),进一步支持了CCL20在粘液产生中的作用。由于吸烟可能是一个混杂因素,该分析只在不吸烟的患者中重复进行,与不吸烟的患者(n=21)相比,中度至重度CMH患者的CCL20痰液水平也显著升高(n=11)。
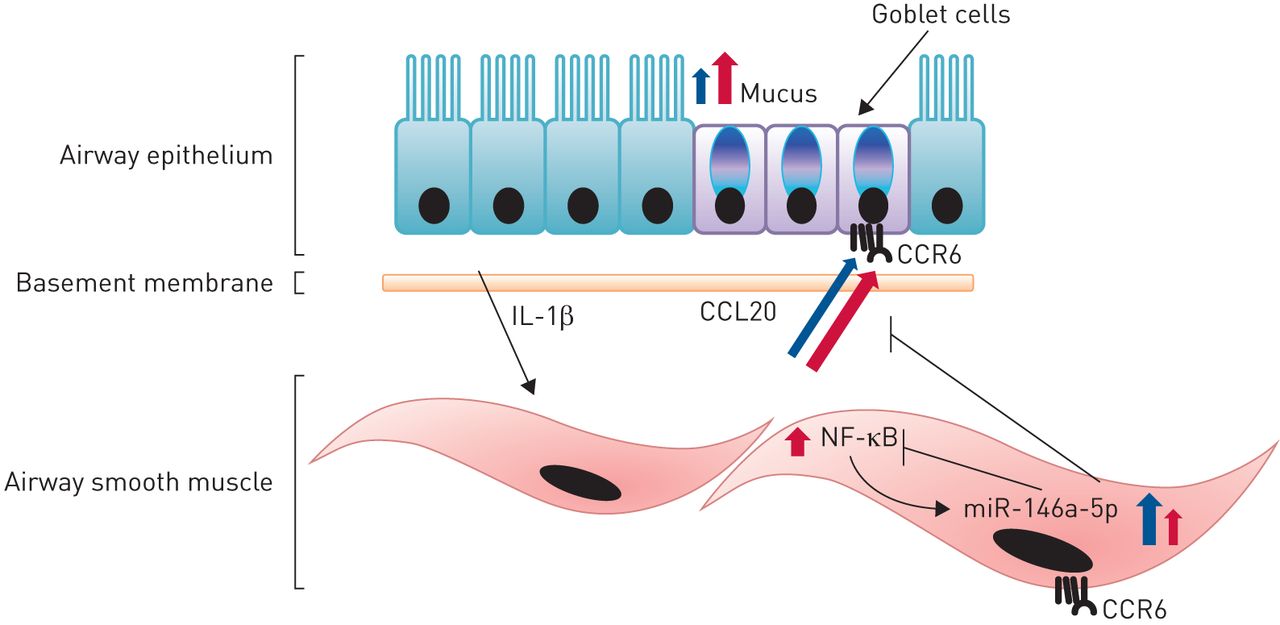
我们目前的研究结果表明,气道上皮细胞在受损伤后产生的IL-1β可诱导哮喘ASMCs中增加的气道平滑肌块产生CCL20。这种CCL20可以通过与CCR6结合作用于气道上皮细胞,导致粘液产生增加。NF-κB激活诱导的miR-146a-5p表达可抑制CCL20的产生。然而,这种miR-146a-5p的诱导在哮喘ASMCs中较低(图5)
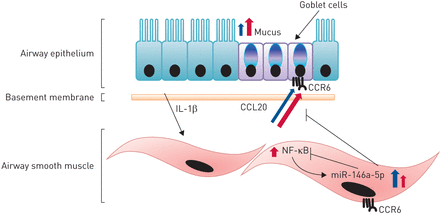
哮喘气道中气道上皮细胞与气道平滑肌细胞(ASMCs)的相互作用。IL:白介素。受损的气道上皮细胞产生的基底侧分泌的IL-1β诱导ASMCs产生CCL20,在哮喘性ASMCs中增加。随后,CCL20可以通过结合其唯一已知的受体CCR6作用于气道上皮细胞,导致粘液产生增加。NF-κB激活诱导的miR-146a-5p表达可抑制CCL20的产生。哮喘ASMCs中miR-146a-5p诱导较低,导致CCL20抑制减弱。红色箭头:哮喘;蓝色箭头:健康
讨论
在这项研究中,我们比较了哮喘和健康ASMCs之间的基因表达谱在体外对NLRP3炎性小体下游介质IL-1β的响应。通过这项分析,我们提供了全基因组的证据,证明ASMCs对IL-1β的活性形式有反应,IL-1β的水平在哮喘患者的痰中升高[7,在气道中提供炎症趋化因子和细胞因子的来源。此外,我们观察到增强的表达CCL20而且MIR146A在ASMCs中对IL-1β的响应较低MIR146A在哮喘asmc中。此外,与轻度哮喘患者和健康对照组相比,IL-1β诱导中度哮喘患者ASMCs中CCL20蛋白分泌增加更强。有趣的是,过表达miR-146a-5p后,CCL20的释放减少,这提供了证据,表明该miRNA可能是哮喘患者ASMCs中CCL20产生的失调抑制剂。重组CCL20可直接诱导分化的气道上皮细胞产生黏液,提示CCL20可能参与哮喘的CMH。我们的观察证实了这一发现的重要性,痰中的CCL20水平与哮喘患者CMH水平的增加有关。目前的研究强化了这一假设,即气道平滑肌在炎症和CMH中不是被动的旁观者,而是关键的驱动因素[30.,31].
此前已发现miR-146a-5p可调节toll样受体2刺激后皮肤角质形成细胞中CCL20的产生,与本研究的结果一致[27].然而,这种炎症细胞因子的抑制不仅限于CCL20,因为miR-146a-5p的过表达导致众所周知的NF-κ b调控的促炎症细胞因子UBD、CXCL10和CXCL8的减少。先前的研究已经确定miR-146a-5p作为一种抗炎miRNA,通过靶向IL-1R下游信号分子IL-1受体相关激酶1和肿瘤坏死因子受体相关因子6降解NF-κB信号,抑制NF-κB信号,这是NF-κB通路激活的关键基因[32].
在这项研究中,我们发现归纳MIR146A与健康对照相比,哮喘ASMCs对IL-1β的响应较低,这可能是这些细胞中CCL20分泌增加的原因。值得注意的是,我们验证了较低的值MIR146A用哮喘患者和健康对照的支气管活检在哮喘患者中的表达。在循环CD4的人类炎症细胞中也有类似的发现+和CD8+重症哮喘患者的t细胞中miR-146a-5p的表达低于健康对照组[33].减少的一个可能的理由MIR146A在哮喘患者中可能存在SNP (rs2910164),这是已知的影响前期和成熟水平MIR146A成绩单(34].这种SNP先前与哮喘和其他促炎症疾病的存在有关[11,12].有趣的是,我们最近发现,在COPD中,支气管活检中miR-146a-5p的低表达与CMH呈负相关[13],强调miR-146a-5p是呼吸系统疾病中粘液调节的一致调节因子。
尽管我们观察到来自哮喘患者ASMCs的CCL20分泌增加,但我们没有发现哮喘患者和健康对照组之间的CCL20基因表达有显著差异。此外,我们观察到miR-146a-5p过表达72小时后CCL20基因表达下降。我们假设,只有当il -1β诱导的miR-146a-5p的增加开始抑制CCL20的表达时,才会导致哮喘组和对照组之间的差异,哮喘源性ASMCs中miR-146a-5p水平不足。由于没有miR-146a-5p的抑制,本研究中所选择的8小时时间点可能过早,无法检测哮喘患者ASMCs和健康对照组之间CCL20基因表达水平的差异。
CCL20被确定为未成熟树突状细胞的关键趋化因子[35],最近被描述为一种抗菌蛋白[36]及粘液分泌调节剂[9,25].总的来说,CCL20的功能在本质上似乎是促炎的,在包括哮喘在内的许多炎症性呼吸道疾病中,CCL20在痰液中的表达上调[20.,37], copd [38和囊性纤维化[39].有趣的是,CMH至少在一部分患者中是所有这些疾病的一个特征[40- - - - - -42].在目前的研究中,我们发现CCL20促进了ali分化的气道上皮细胞系粘液的产生。此外,哮喘患者痰液中CCL20水平与粘液产生之间有直接联系。值得注意的是,我们小组的数据显示吸入皮质类固醇治疗增加了痰中CCL20的水平[20.],解释了为什么目前的抗炎疗法无法恢复粘液的过度分泌。
本研究的主要优势是采用多学科方法,通过包括微阵列分析在内的大规模筛选方法来识别新的基因靶点,然后对候选基因进行功能询问在体外模型。这项研究有一些局限性,因为我们无法确定哮喘患者痰液中CCL20水平的来源。除ASMCs外,包括气道上皮细胞在内的许多细胞类型也产生这种趋化因子[43].尽管如此,哮喘气道中增加的敏感性和肌肉体积为CCL20提供了一个潜在的存储库。此外,哮喘患者气道平滑肌块由于气道的重塑而更接近上皮层,这可能会增加上皮细胞与ASMCs之间的相互作用。的测量MIR146A在支气管活组织检查中反映的是其在哮喘气道混合细胞群中的表达,而不是仅反映在气道平滑肌中的表达。最后,我们发现重组CCL20增加黏液蛋白表达并不能直接证明气道平滑肌来源的CCL20驱动上皮黏液的产生在活的有机体内.在未来的研究中,我们将使用上皮细胞和平滑肌共培养物来进一步支持我们目前的发现。
总之,在本研究中,我们发现了一种导致哮喘患者产生粘液的新途径,其中,由于miR-146a-5p抑制减弱,从增强的气道平滑肌中释放的增加的CCL20可能导致哮喘气道上皮细胞产生的粘液增多。
补充材料
脚注
本文的补充资料可从www.qdcxjkg.com
作者贡献:A. Faiz参与项目设计,微阵列分析,在体外细胞工作,写作和校对手稿。C.J.维尔穆伦,C-J。Xu和M. Van den Berge参与了miRNA分析、撰写和校对手稿。N.H.T. ten Hacken, G.G. King, C.S. Farah, A.J. Halayko, T.H. Lee和J.P.T. Ward提供了不朽的ASMC样本或痰样本,并参与了手稿的校对。G. Tjin帮助进行免疫组化分析。J.K. Burgess, M. Weckmann, J.L. Black和B.G. Oliver参与了项目设计,为项目提供资金,撰写和校对手稿。
利益冲突:M. Van den Berge报告了Teva、Chiesi和GlaxoSmithKline在提交的工作之外的研究资助(支付给大学)。j·l·布莱克报告了国家卫生和医学研究委员会在进行研究期间的拨款。j.k Burgess报告了国家卫生和医学研究委员会在进行研究期间的拨款。
支持声明:本工作得到澳大利亚国家卫生和医学研究理事会(NHMRC)的支持(赠款570867)。a . Faiz获得了澳大利亚研究生奖和Longfonds初级调查者奖(4.2.16.132JO)的支持。jk Burgess获得了NHMRC职业发展奖学金(1032695)和由格罗宁根大学和欧盟资助的罗莎琳德·富兰克林奖学金。J.L. Black获得了NHMRC高级首席研究奖学金(571098)的支持。B.G. Oliver获得了NHMRC职业发展奖学金(1026880)的支持。
本文的补充资料可从www.qdcxjkg.com
- 收到了2018年2月11日。
- 接受2018年5月25日。
- 版权所有©ERS 2018











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}