





抽象
成纤维细胞生长因子(FGF)23的循环水平与全身炎症和慢性肾脏疾病的死亡率增加有关。α-Klotho, FGF23受体,在慢性阻塞性肺疾病(COPD)表达下调。然而,FGF23和klothol介导的FGF受体(FGFR)激活是否描绘了COPD的病理生理机制尚不清楚。我们推测FGF23可能增强气道炎症通过Klotho的独立FGFR4激活。
使用等离子体和慢性阻塞性肺病和对照患者支气管活组织检查,并从COPD患者以及鼠COPD模型分离的原代人支气管上皮细胞FGF23和它的效果进行了研究。
血浆FGF23水平在COPD患者显著升高。呼吸道上皮细胞于香烟烟雾和FGF23的曝光导致白介素1β释放显著增加通过磷脂酶Cγ/活化的T细胞的信令的核因子的Klotho-独立FGFR4介导的激活。此外,Klotho细胞基因敲除小鼠发展慢性阻塞性肺病和显示气道炎症和肺部升高FGFR4表达,而Klotho细胞的过度表达导致气道炎症的衰减。
慢性阻塞性肺病患者吸烟通过下调Klotho和激活气道上皮FGFR4诱导气道炎症。抑制FGF23或FGFR4可能是一种新的COPD抗炎策略。
抽象
成纤维细胞生长因子23和吸烟诱导的FGFR4信号导致的气道炎症和肺气肿的发展http://ow.ly/j4tZ30jWyGF
介绍
慢性阻塞性肺疾病(COPD)代表全球领先的死亡原因的第三位;暴露于香烟烟雾是负责大部分的COPD病例,是全球领先的避免过早死亡的原因[1]。多项研究显示慢性阻塞性肺病(COPD)气道上皮的炎症变化以及吸烟引起的炎症变化,包括杯状增生和促炎细胞因子表达增加[2-4]。
成纤维细胞生长因子(FGF)23是一种由骨细胞分泌的27-kDa蛋白,在维持血清磷酸盐稳态中起重要作用[五-7]。在慢性肾病,FGF23的血浆浓度升高,并强烈慢性肾脏疾病进展,心血管事件和死亡率的风险[相关联8,9]。迄今为止,还没有被报道对肺FGF23的直接影响。然而,已经表明的Klotho表达在气道从COPD患者[减小10]。Klotho的,存在于膜结合和可溶形式,并作为一个共受体FGF23单跨膜蛋白,防止氧化性损伤的肺泡上皮[11-13]。此外,缺乏Klotho的小鼠发展与扩大肺泡腔,与肺气肿一致的老化表型14]。
FGF受体(FGFR的)是受体酪氨酸激酶的亚家族,并包括四个亚型(FGFR1-4)15]。在其典型的靶器官,如肾脏和甲状旁腺,FGF23信号通过FGFR1和需要的Klotho。通过结合到FGFR1 / Klotho的,FGF23激活的Ras /促分裂原活化蛋白激酶信号传导途径[16]。在不存在的Klotho的,FGF23可以激活FGFR4和随后诱导磷脂酶Cγ(PLCγ)/活化的T细胞(NFAT)信令的核因子。这种机制介导,其包括在心肌细胞肥大性生长的诱导和在肝细胞中的炎性细胞因子产生的增加[FGF23的病理生理学动作17,18]。
我们假设香烟烟雾有助于提高生产和COPD FGF23信号的激活。在这里,我们检查香烟烟雾是否减少Klotho以及增加FGFR4表达,从而导致促炎PLCγ/ NFAT在人初级支气管上皮细胞(BEC中)信令。我们推测,FGF23可以在肺部炎症增强通过FGFR4的激活和Klotho的下调。
方法
请参阅补充材料进一步的方法细节。
研究批准
临床研究是根据赫尔辛基宣言的原则进行,并批准了迈阿密大学的机构审查委员会(迈阿密,FL,USA)。书面知情同意从每个参与者包括在研究之前收到。人的气道是从器官捐献者,其肺部被拒绝移植获得。机构审查委员会批准的研究同意,由迈阿密大学或LifeCenter西北(华盛顿州贝尔维尤,美国)的生活联盟器官恢复机构获得,符合赫尔辛基宣言。所有动物方案都得到了机构动物护理和使用委员会在迈阿密大学和阿拉巴马大学伯明翰分校(伯明翰,AL,USA)的批准。
Interleukin-1β6和8个ELISA和mRNA评估
使用超灵敏白介素(IL)-1β和-6 ELISA自Invitrogen(赛默飞世尔科技,科学,格兰德岛,NY,USA)。胡man BECs were stimulated with FGF23 and/or cigarette smoke (25 ng·mL-1和/或4香烟24 h)和100µL基底外侧介质(稀释)是用于测量。使用TaqMan探针(Life Technologies/Applied Biosystems, Carlsbad, CA, USA)进行定量PCR检测基因表达。
统计数据
与棱镜版本分析数据5(的GraphPad,拉霍亚,CA,USA)和显示为平均值±扫描电镜利用t检验和方差分析或秩和检验H-测试用单向ANOVA与适当后试验为至少三个独立的实验。显着性为p <0.05接受。
结果
血浆FGF23水平与COPD的个体增加
我们的血浆样本中测得FGF23从28个人:18名COPD患者和10年龄匹配的非COPD患者。COPD was defined as forced expiratory volume in 1 s (FEV1)/用力肺活量<0.70。后支气管扩张肺功能1应用FEV法,% pred法测定气流阻塞严重程度1%预计≥50%和≤1去年指示中COPD急性加重轻度至中度慢性阻塞性肺病(九个人)和FEV1%预计<50%,而过去一年内≥2COPD急性加重表示严重慢性阻塞性肺病(九个人)。临床特征,包括年龄,性别,吸烟史和FEV1% pred,在补充表S1。正如所料,支气管扩张剂后FEV1当与对照组相比均COPD组%预计是显著降低。
轻度至中度慢性阻塞性肺病患者的FGF23血浆水平高于非慢性阻塞性肺病患者(59±12)与42±10 pg·毫升-1;p = 0.004) (图1一个)。然而,与对照组相比,重度慢性阻塞性肺病患者的FGF23没有显著增加(51±18)与42±10 pg·毫升-1;p值= 0.26)。因此,我们专注于轻度至中度慢性阻塞性肺病进一步的实验。我们发现主动吸烟者和非吸烟者之间没有差异(补充图S1A)。
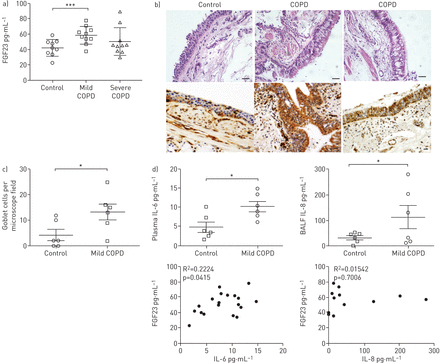
成纤维细胞生长因子(FGF)23水平在伴有杯状细胞增生和促炎细胞因子水平升高的轻度至中度慢性阻塞性肺疾病(COPD)患者中升高。IL:白介素;支气管肺泡灌洗液。a)非COPD和COPD患者FGF23血浆水平分为轻度至中度(“轻度”)和重度COPD。b) 1名对照组(左栏)和2名轻度至中度COPD患者(中、右栏)代表性支气管内活检的苏木精(上排)和抗fgf23(下排)染色。比例尺:40µm。c)每个视野杯状细胞的定量显示,与非COPD受试者相比,轻度至中度(“轻度”)COPD显著增加。每组n=6。d)对照组和轻度至中度(“轻度”)COPD患者血浆IL-6水平(左侧面板)和BALF IL-8水平(右侧面板),以及显示其与FGF23水平相关性的点图。每组n=6。 *: p<0.05; ***: p<0.005.
轻度至中度组FGF23水平明显升高,支气管内活检显示为BEC上皮化生(图1 b,上排)。从控制和这些COPD供体气管切片显示上皮细胞层的不同FGF23染色(图1 b,下排)。如所预期的,当与对照组相比,COPD小组显著增加杯状细胞数(图1 c)。
我们观察到血浆IL-6水平增加显著(图1 d支气管肺泡灌洗液(BALF) IL-8水平(图1 d(右上角)在慢性阻塞性肺病患者中也升高。皮尔逊相关系数的测定表明,血浆FGF23水平与血浆IL-6水平呈显著正相关(R2= 0.2224其中p = 0.0415)(图1 d,左下小图),但血浆FGF23水平和BALF IL-8水平之间没有相关性显著(R2=0.01542, p=0.7006 (图1 d右下图)。组合,这些数据表明,COPD患者具有增加的FGF23血浆水平,这与肺和循环炎性细胞因子水平升高一致。
FGF23和香烟烟雾诱导IL-1β分泌的BEC从患有COPD的个体
确定直接影响FGF23结合香烟,我们从四个不同的暴露主要bec孤立患有慢性阻塞性肺病(COPD-BECs)香烟烟雾和FGF23和评估的il - 6的表达,8和1β定量实时PCR。总的来说,我们观察到细胞因子表达有相当大的异质性。IL-6 mRNA随着FGF23和/或香烟烟雾的高变异性增加(图2一个);IL-8与FGF23和/或香烟烟雾被显著增加,但增幅较少变化和更温和(图2 b)。IL-1βmRNA水平没有显著单独增加了FGF23,反而导致更大的刺激都超过五倍海拔组合(图2 c)。COPD-BEC中任刺激的曝光,然而,引起了IL-1β,当两种刺激物合并,将其进一步升高的基底外侧分泌显著增加(图2 d)。这些发现表明,结合香烟,FGF23可以直接目标COPD-BECs IL-1β诱导表达和释放。
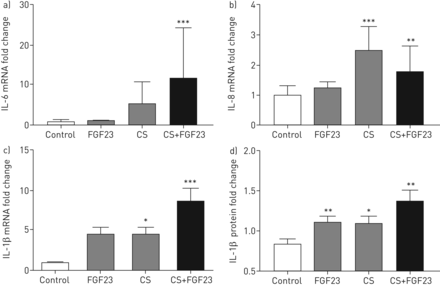
效果香烟烟雾(CS)和成纤维细胞生长因子(FGF)23对慢性阻塞性肺病(COPD)支气管上皮细胞(BEC中)促炎细胞因子的表达。a) Interleukin (IL)-6 and b) IL-8 mRNA levels in COPD-BECs 24 h after stimulation with CS (4 cigarettes) and/or FGF23 (25 ng·mL-1)。C)IL-1βmRNA和d)IL-1β分泌的蛋白水平在COPD从-的BEC基底外侧媒体与FGF23,CS或两者刺激后。数据被表示为mRNA或倍数变化与来自六个不同的肺n = 3的独立实验的蛋白表达。*:p < 0.05;* *:p < 0.01;***:P <0.005,与对照组相比。
FGF23激活FGFR4和PLCγ/钙调磷酸酶/ NFAT信号在COPD-的BEC,导致增加的IL-1β分泌
如其他组织所示,FGF23可以诱导的磷酸化PLCγ[19]和细胞外信号调节激酶(ERK)[20.]根据的Klotho的不存在或存在。为了确定是否FGF23可以激活FGFR4和随后PLCγ/钙调磷酸酶/ NFAT信号,并引起所述肺部的炎症反应,如我们已经在肝中最近显示[18),我们分析了FGFR4绑定PLCγFGF23治疗后。
在COPD-BECs,刺激FGF23 +香烟引起的磷酸化PLCγ,符合前提,这些影响可能由Klotho-independent FGFR4信号。然而,单独的FGF23并没有导致ERK的磷酸化,这一效应显然与香烟烟雾暴露(图3一左面板)。
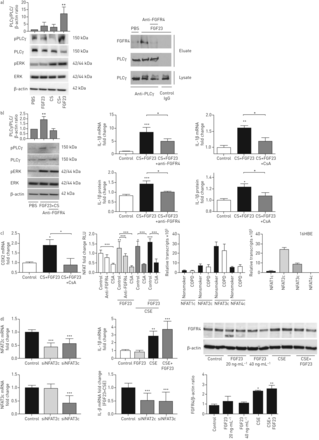
成纤维细胞生长因子(FGF)FGF受体4(FGFR4)的23个诱导激活/磷脂酶Cγ(PLCγ)/活化的T细胞(NFAT)在慢性阻塞性肺疾病(COPD)支气管上皮细胞(BEC中)信令的核因子。p:磷酸化;ERK:细胞外信号调节激酶;CS(E):香烟烟雾(提取物);IL:白介素;环孢素A:环孢菌素A;COX:环氧合酶;RLU:相对荧光素单位;SI:小干扰。一)左面板:代表性免疫印迹和光密度分析(pPLCγ/总PLCγ/β肌动蛋白之比)示出了FGF23加PLCγ的CS诱导的磷酸化(FGF23和CS依赖性)和ERK(纯粹CS依赖性)。 n=3. Right panel: immunoblot of co-immunoprecipitation with FGFR4 using an anti-PLCγ antibody shows activation of PLCγ after FGF23 stimulation in COPD-BECs, which could be inhibited by pre-incubation with anti-FGFR4. b) CS plus FGF23 induces PLCγ and ERK phosphorylation as seen in (a), but anti-FGFR4 only inhibits PLCγ phosphorylation. Both anti-FGFR4 and CsA, a calcineurin inhibitor, inhibit CS plus FGF23-mediated induction of IL-1β mRNA and secreted protein levels. c) CsA also attenuates induction of COX2 mRNA. FGF23, CSE alone and both activate NFAT, as assessed by a luciferase-based reporter gene assay in 16HBE cells, which can be blocked by CsA. Anti-FGFR4 inhibits FGF23-induced NFAT activation. The right two bar graphs indicate mRNA levels of NFAT isoforms in both human BECs from nonsmokers and COPD patients as well as 16HBE cells. d) siRNAs targeting NFAT isoforms NFAT2c and NFAT3c were transfected into 16HBE cells. siNFAT2c reduces the expression of NFAT2c and NFAT3c, whereas siNFAT3c is specific for NFAT3c (left panel). When 16HBE cells were stimulated with FGF23 (20 ng·mL-1),CSE(5%)或两个24 h, IL-1β增加(中间面板)。最后,用CSE + FGF23(右图)刺激siNFAT2c (2c和3c亚型的表达下调)和siNFAT3c转染后的效果进行评估。CSE和/或FGF23也导致16HBE细胞中FGFR4蛋白表达显著上调。所有的n=3个独立的实验(在COPD-BECs病例中来自三个不同的肺)。*:p < 0.05;* *:p < 0.01;* * *:p < 0.005。
为了验证这一发现,内生PLCγ免疫沉淀反应,洗出液免疫印迹分析anti-FGFR4抗体。与洗出液控制细胞相比,水平co-purified FGFR4高架在FGF23-treated COPD-BECs,而总PLCγ表达水平持平。使用fgfr4特异性阻断抗体(抗fgfr4)进行预处理[17)抑制co-immunoprecipitation FGFR4和PLCγ(图3一右面板)。COPD-BEC中的至FGF23和香烟烟雾暴露组合也导致PLCγ的磷酸化,这是通过阻断抗FGFR4(图3 b左面板)。香烟的烟雾,但不是FGF23本身的刺激,引起在ERK磷酸化的显着增加,这已经在BEC中[先前显示21]。然而,使用抗fgfr4预孵育的ERK磷酸化没有发生变化(图3 b左面板)。这些结果表明,FGF23信令在支气管上皮主要发生通过PLCγ而非ERK这种效果,并且激活由FGFR4介导的。
以确定是否FGFR4 /PLCγ/钙调磷酸酶/ NFAT信号级联介导的IL-1β表达FGF23的影响,我们预孵育COPD-的BEC具有抗FGFR4接着用FGF23和香烟烟雾刺激。抗FGFR4的mRNA显著降低隆起如IL-1β的以及分泌水平(图3 b,中图)。建立FGF23和NFAT活化之间的联系,我们所用的广泛使用的神经钙蛋白抑制剂环孢菌素A(CSA)[17,18],并表明环孢素也抑制了FGF23和香烟烟雾诱导的IL-1β反应(图3 b右面板)。此外,支气管上皮内的NFAT靶基因环氧化酶(COX) 2的mRNA水平[22,与FGF23和香烟烟雾共同刺激后增加。当细胞经CsA (图3 c左面板)。此外,我们使用了BEC线16HBE和转染这些细胞具有NFAT报告基因荧光素酶构建。在NFAT活性的增加与FGF23,当细胞用抗FGFR4和环孢素A预温育将其显著抑制刺激后观察。此外,香烟烟雾提取物和香烟烟雾提取物加FGF23也诱导NFAT活化,这可通过CSA(被阻塞图3 c,中间左图)。
我们发现NFAT2c和NFAT3c是COPD-BECs、人BECs和16HBE细胞(图3 c,中右面及右面)。为了进一步验证fgf23诱导的NFAT激活的特异性,在16HBE细胞(图3 d左面板)。因为这些16 hbe细胞表现出类似IL-1β响应作为主要的细胞(图3 d,上中间面板),我们刺激对照,siNFAT2c和siNFAT3c转染16HBE细胞与香烟烟雾提取物和FGF23。siNFAT2c下降都NFAT2c和NFAT3c,而siNFAT3c是亚型特异性。用“SI控制”转染的细胞中,香烟烟雾提取物和FGF23诱导的IL-1β反应相比在siNFAT2c钝和siNFAT3c转染的细胞(图3 d,下中板)。这说明NFAT2c和NFAT3c都是fgf23诱导的信号转导介质。此外,香烟烟雾提取物和/或FGF23导致16HBE细胞FGFR4蛋白表达水平显著升高(图3 d右面板)。结合起来,我们的结果表明,FGF23直接激活FGFR4 / PLCγ/钙调磷酸酶/ NFAT bec的信号,从而诱导IL-1β。
暴露于香烟烟雾可下调慢性阻塞性肺病患者的Klotho表达,并上调BECs中的FGFR4水平
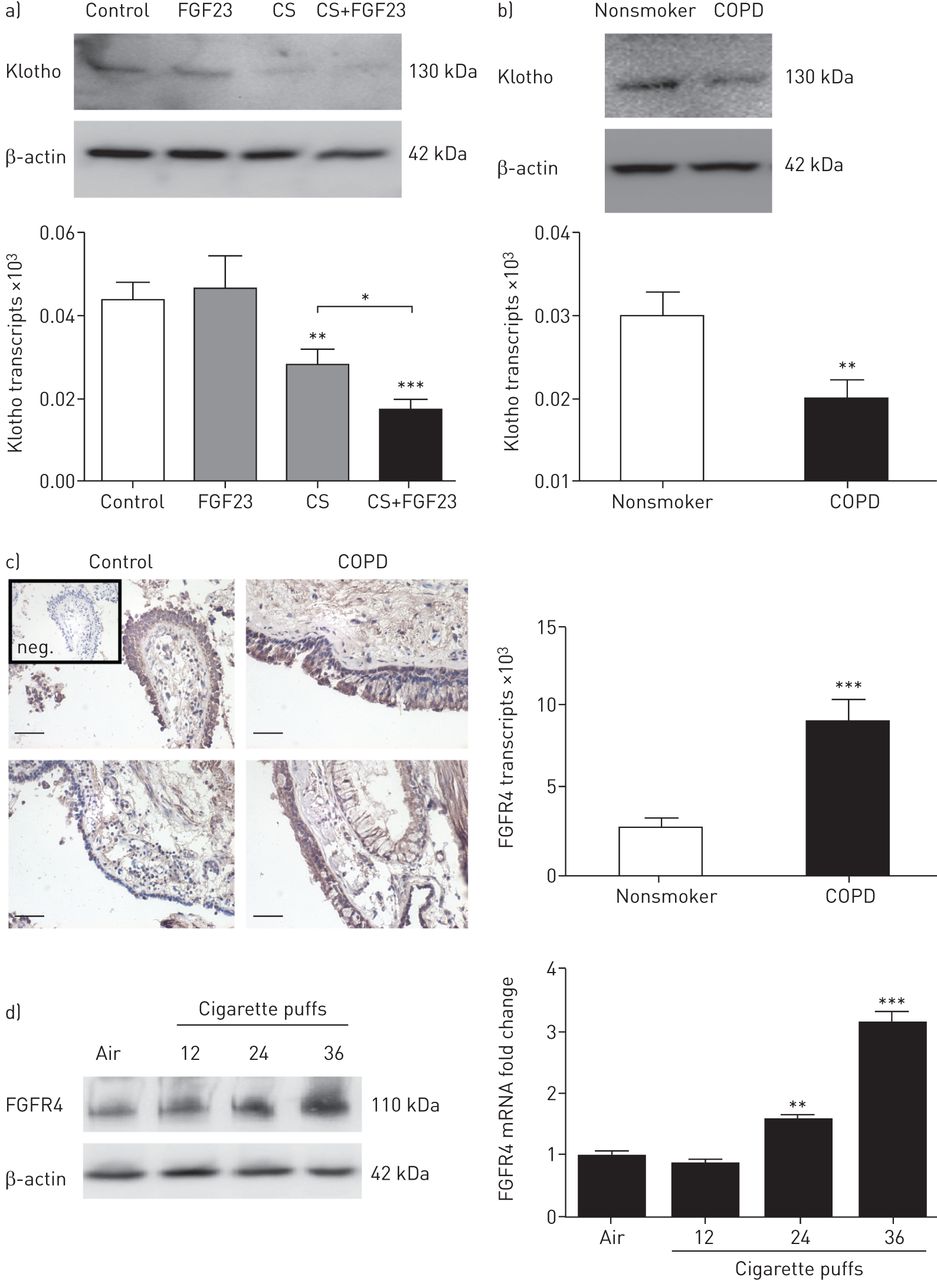
由于的Klotho表达在COPD气道下降[10],我们分析了一个主空气 - 液体界面BEC培养系统的Klotho表达。慢性阻塞性肺病,建筑物能源效益守则于香烟烟雾的暴露引起的Klotho蛋白和mRNA水平的显著下降,而FGF23单独无影响(图4一)。与非吸烟者和非COPD肺,Klotho细胞mRNA和蛋白水平得出人类的BEC相比,慢性阻塞性肺病,BEC中均显著减少(图4 b)。这些数据表明,在慢性阻塞性肺病-BEC中,Klotho的表达降低和由香烟烟雾进一步减小,表明这两个COPD和香烟烟雾可以作为促进在肺中的Klotho-FGF23独立信号因子。
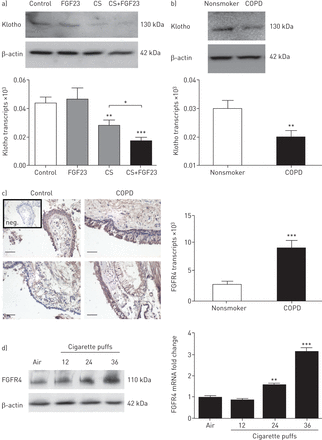
香烟烟雾(CS)曝光调节的Klotho的表达和成纤维细胞生长因子(FGF)受体4(FGFR4)的慢性阻塞性肺病(COPD)的气道上皮细胞。BEC:支气管上皮细胞。a) Representative immunoblot showing downregulation of Klotho protein and bar graph indicating mRNA levels from COPD-BECs after treatment with CS, FGF23 or both stimuli for 24 h. b) Representative immunoblot and Klotho mRNA levels of human BECs from nonsmokers compared with COPD-BECs. n=3 independent experiments from six different lungs. c) Left: immunohistochemistry for FGFR4 and counterstaining with haematoxylin of endobronchial biopsies from two representative control patients and two COPD patients, compared with a negative control (inset: “neg.”). Scale bar: 50 μm. Right: bar graph showing increased FGFR4 mRNA expression in COPD-BECs compared with control lungs. d) Immunoblot analysis of FGFR4 protein levels (representative blot from one COPD patient, tested in three different COPD lungs) and quantitative real-time PCR of FGFR4 mRNA levels in COPD-BECs 24 h after exposure to 2, 4 and 6 cigarettes (1 cigarette=6 puffs). All n=3 independent experiments from three different lungs. *: p<0.05; **: p<0.01; ***: p<0.001.
我们测量FGFR4表达在肺,作为FGFR4先前已经显示介导的Klotho-FGF23独立在心脏和肝脏[信令17,18]。石蜡包埋支气管活检用于苏木染色和免疫组化FGFR4。FGFR4染色定位于支气管上皮细胞,并出现在COPD患者支气管组织活检(待提高图4 c左面板)。为了更好地量化FGFR4表达,FGFR4 mRNA水平确定,并表现出慢性阻塞性肺病,BEC中一个显著增长与控制人的BEC相比(图4 c右面板)。
接下来,COPD-BECs暴露在越来越多的香烟烟雾中。24小时后检测到FGFR4蛋白和mRNA水平呈剂量依赖性上调(图4 d)。总之,在FGFR4支气管上皮细胞中表达,并在COPD患者和暴露于香烟烟雾上调。
Klotho细胞缺乏导致增加FGFR4激活和炎症
由于缺乏Klotho细胞基因小鼠模型开发升高血清FGF23的水平,我们想确定的Klotho独立FGF23信令和相关的炎症发生在这些动物的肺。如图所示由他人先前[12],我们观察了Klotho低形态小鼠(KL- / -)(图5一个)。
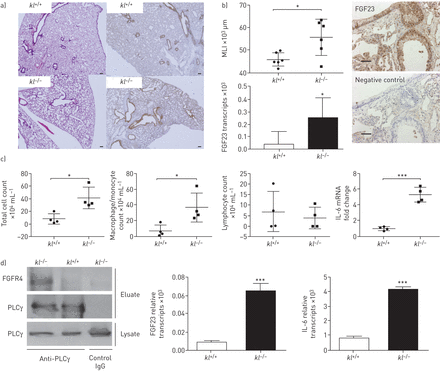
Klotho表达的缺失导致小鼠气道炎症的增加。MLI:平均线性截距;FGF(R):成纤维细胞生长因子(受体);BALF:支气管肺泡灌洗液;MTEC:小鼠气管上皮细胞;IL:白介素;PLCγ:磷脂酶Cγ。a)苏木精与野生型肺组织抗fgf23染色(KL+ / +)和亚等位基因的Klotho(KL- / -老鼠)。酒吧规模:50μm。b)左面板:MLI分析显示KL- / -全肺组织中肺标本和FGF23 mRNA水平均上调KL- / -当与比较KL+ / +老鼠。每组6只小鼠。右图:使用抗fgf23免疫组化方法对石蜡包埋的小鼠肺组织切片进行染色,结果显示支气管上皮内存在信号。酒吧规模:50μm。c) BALF中总细胞的评估KL+ / +与kl- / -老鼠。每组4只小鼠。也显示BALF巨噬细胞/单核细胞计数、BALF淋巴细胞计数和IL-6在BALF中的变化KL+ / +与kl- / -老鼠。d)通过在肺组织裂解物用抗PLCγ或抗绿色荧光蛋白(对照)从内源FGFR4的免疫共沉淀KL- / -或KL+ / +小鼠显示FGFR4转录本增加KL- / -肺。对FGFR4 mRNA进行实时荧光定量PCR检测KL- / -MTECs与野生型MTECs相比,在基底外侧介质中IL-6蛋白水平显著升高KL+ / +和KL- / -MTECs。对于小鼠实验,进行了三个不同的独立实验;在每个实验中,细胞来自n= 3-4只动物;每组4只小鼠进行形态测定和BALF分析。*:p < 0.05;***:P <0.001。
平均线性截距分析用于定量腔扩大,这是显著高于的Klotho缺陷小鼠的肺与野生型相比(KL+ / +)同窝出生的(图5 b,左上面板)。此外,FGF23 mRNA水平被上调KL- / -肺的情况相比KL+ / +(图5 b(左下)。使用抗fgf23的肺切片免疫组化分析显示在支气管上皮有强烈的信号(图5 b右面板)。与它们的野生型同窝伙伴相比,BALF来自KL- / -小鼠含有更多的细胞,特别是嗜中性粒细胞23]和巨噬细胞/单核细胞(图5 c)。有两组之间淋巴细胞的数量没有什么差别,但IL-6 mRNA水平也增加(图5 c)。为了确定FGFR4 /PLCγ的活性,我们免疫沉淀从PLCγ小鼠肺组织提取物,接着为洗脱液的FGFR4的免疫印迹。我们发现在共纯化FGFR4增加肺部的KL- / -小鼠与野生型同窝相比(图5 d左面板)。此外,FGFR4 mRNA水平在培养的增加,完全分化的鼠气管上皮细胞(MTECs)从KL- / -老鼠 (图5 d,中图)。他们还发现IL-6的显著增加基底外侧分泌(图5 d右面板)。这些发现表明,在缺乏克罗索FGF23升高的存在,FGFR4 / PLCγ信号和生产的炎性细胞因子升高鼠标肺。
可溶性Klotho可保护COPD-BECs免受FGF23和香烟烟雾的促炎作用
Klotho的胞外结构域可以以可溶性形式存在[24]。已显示可溶性Klotho的对不同类型的细胞保护作用,抗炎作用〔25,26]。Ťo test if soluble Klotho interferes with the effects of FGF23 in the lung, we co-incubated COPD-BECs with FGF23, cigarette smoke and the ectodomain of recombinant human α-Klotho at 0.1 µg·mL-1for 30 min. As shown earlier, FGF23 and cigarette smoke caused an increase in PLCγ phosphorylation without affecting total PLCγ levels. In the presence of FGF23 and cigarette smoke, treatment of COPD-BECs with soluble Klotho also led to a decrease in IL-1β secretion (图6 b)。
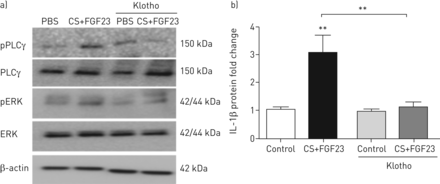
补充人类重组克罗索抑制香烟(CS)和纤维母细胞生长因子(FGF) 23-induced白介素(IL) 1β分泌。p:磷酸化;PLCγ:磷脂酶Cγ;ERK:细胞外信号调节激酶。a) Representative immunoblots show FGF23 (25 ng·mL-1for 30 min) induced phosphorylation of PLCγ. Inhibition of PLCγ phosphorylation occurs by pre-incubation with Klotho (1 μg·mL-1第3道;0.1 μg·mL-1,莱茵4)。b)可溶性克罗索抑制CS和增加FGF23-mediated IL-1β蛋白质的水平,评估ELISA基底从媒体。所有的n=3个独立的实验来自3个不同的肺。* *:p < 0.01。
过表达或在小鼠FGF23变造拦截的Klotho的/ FGFR4信令
因为长期接触香烟烟雾的老鼠[27]没有显示FGF23血清水平的上调(补充图S1b中)中,我们使用的香烟烟雾暴露急性小鼠模型。中号ice were exposed to 1 week of cigarette smoke to induce acute airway inflammation. Western blot analyses of total lung tissue showed an increase in FGF23 levels in some mice (six out of 11 cigarette smoke-exposed mice and three out of 11 control mice) (图7)。然而,有在控制和香烟烟雾暴露组之间血清FGF23水平没有显著增加(图7),并有在肺组织中的Klotho或FGFR4的mRNA水平没有差异显著(图7)。在比较野生型小鼠和Klotho过表达小鼠时[28在肺中,这是独立的香烟烟雾暴露],后一组显示增加的Klotho mRNA水平(图7 b左面板)。我们检测到血清中FGF23水平和支气管肺泡灌洗液中的总细胞数无明显差异(图7 b、中间板)。与野生型小鼠相比,烟熏诱导过表达Klotho的小鼠肺中IL-6的增加减少(图7 b右面板)。为了测试Klotho细胞缺乏症的效果,因为KL- / -老鼠病得太重,不能暴露在香烟烟雾中,两者都分离出了MTECsKL+ / +和KL- / -老鼠。在基线,KL- / -与野生型细胞相比,MTECs具有更高的IL-6和FGFR4 mRNA水平(图7 c)。
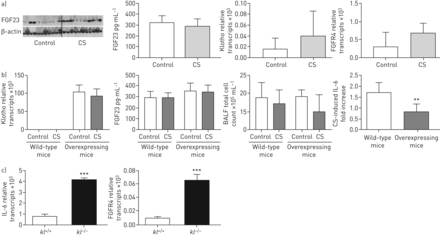
Klotho过表达和Klotho缺乏对小鼠成纤维细胞生长因子(FGF)23信号通路的影响。CS:吸烟;FGFR4: FGF受体4;BALF:支气管肺泡灌洗液;IL:白介素。a)具有代表性的免疫印迹显示,对照组小鼠和急性CS暴露小鼠的肺总裂解液中FGF23蛋白水平。条形图显示了两组小鼠的血清FGF23水平、肺总Klotho水平和FGFR4 mRNA水平。b) Klotho mRNA水平、血清FGF23水平、BALF总细胞数和CS诱导的小鼠肺IL-6总表达量在急性CS暴露后以及与未暴露对照组小鼠(野生型和Klotho过表达小鼠)相比均有增加。c)野生型小鼠气管上皮细胞IL-6和FGFR4转录水平(KL+ / +)和亚等位基因的Klotho(KL- / -老鼠)。* *:p < 0.01;* * *:p < 0.005。
为了进一步表征为直接或间接的,从MTECs FGFR4组成型活性的转基因小鼠的Klotho缺陷,FGF23增加和升高的FGFR4表达之间的联系进行了分析并没有表现出与对照相比MTECs IL-6水平的任何差异。有趣的是,也有对这些转基因FGFR4的MTECs增加的Klotho mRNA水平的趋势(补充图S2a和b)。MTECs从FGFR4- / -小鼠虽然从FGF23和香烟烟雾诱导的IL-6的增加表明保护伴有的Klotho表达的下调(补充图的S2c和d)。
总之,我们的研究结果表明,在COPD中存在吸烟导致的FGFR4表达上调和可溶性Klotho下降,从而使FGF23升高发出信号通过FGFR4 /PLCγ/ NFAT诱发气道炎症(图8)。此外,与可溶性Klotho共给药可阻断COPD-BECs中FGF23介导的信号通路,从而抑制FGF23的促炎作用。

对于香烟烟雾(CS)和成纤维细胞生长因子(FGF)FGF受体4(FGFR4)的23介导的调控可能的信令机制/磷脂酶Cγ(PLCγ)活化的T细胞(NFAT)途径的/钙调神经磷酸/核因子。示出FGF23和CS的直接影响在慢性阻塞性肺疾病(COPD)气道上皮细胞:FGFR4的CS诱导上调和降低了支气管上皮的Klotho表达。升高的FGF23血浆水平激活FGFR4 /PLCγ/钙调磷酸酶/ NFAT信令以释放白细胞介素(IL)-1β,由此诱导炎症。
讨论
在此,我们首次展示了FGF23在支气管上皮细胞中诱导的信号通路,并对FGF23及其协同受体Klotho在吸烟诱导的慢性支气管炎中的相反作用提供了新的见解。我们证明1)等离子体FGF23水平升高轻度到中度COPD患者和一个相关的炎性表型和杯状细胞增生,2)香烟烟雾暴露结合FGF23诱发IL-1β分泌人类BEC初级文化从慢性阻塞性肺病患者,3)香烟诱发增加FGFR4克罗索的表达减少,造成激活PLCγ/ NFAT信号和炎症,和4)可溶性克罗索变弱的香烟烟雾诱发IL-1β分泌。
血浆FGF23水平和吸烟之间的正相关性已经描述了以前使用的604例患者患有慢性肾病多变量回归分析[29]。然而,在该研究中,吸烟者并没有进一步明确慢性阻塞性肺病的特征。在另一份报告中,COPD患者中FGF23水平升高与低磷血症有关,但其升高的机制和循环FGF23的来源尚未确定[30.]。在这里,我们表明,FGF23水平似乎并不与疾病的严重程度本身,因为严重慢性阻塞性肺病患者不存在全身性FGF23升高。一种解释可能是,FGF23/Klotho扰动可能在晚期疾病中减少,因为随着存活的肺上皮的残余质量减少,“烧毁”。如果是这样的话,FGF23/Klotho失调可能不仅是疾病的有效标志,而且是“危险”肺组织肿块的标志。因此,我们通过评估慢性阻塞性肺病患者血浆和经支气管活检标本的炎症反应,将研究重点放在这组轻度至中度慢性阻塞性肺病患者身上。Klotho是FGF23的共同受体,在COPD患者的气道上皮细胞中下调[10]。此外,以前的报道显示的Klotho的基因沉默在BEC线诱导的IL-8的分泌是由香烟烟雾提取物进一步增强[10]。
我们的数据还显示,在COPD小鼠模型(由于Klotho细胞的缺乏)有增加支气管发炎,这是至少通过的BEC和巨噬细胞和中性粒细胞的流入部分生成的。我们进一步表征潜在新颖信号通路对具有潜在的用于将来的治疗干预香烟烟雾诱导的炎性气道疾病,即。FGFR4活化和连续PLCγ/钙调磷酸酶/ NFAT信号,这发生在Klotho的缺乏和FGF23的上调的状态。它是具有挑战性的实验表征Klotho细胞缺乏症之间的联系,我们的数据表明两者的该条例是紧密相连。先前已经表明的Klotho敲除小鼠具有升高的FGF23水平[19]。目前,还不清楚如何可溶性Klotho细胞块BEC中的FGF23影响,因为ERK磷酸化没有改变。然而,我们发现,可溶性Klotho的是不是一个循环共受体在肺中的是介导ERK信号FGF23。因此,需要进一步研究。
有趣的是,我们还观察到ERK活化在香烟烟雾诱发IL-1β表情,似乎FGF23 / FGFR4独立,因为没有改变后pre-incubation FGFR4抑制剂(图3 b)。由于香烟烟雾下调Klotho的表达,FGF23信号发生通过FGFR4,正如我们之前在肝脏和心肌中所证明的[17,18]。这最终导致增加的IL-1β分泌,病毒诱导的肺部疾病的关键细胞因子[31]。的IL-1β抗体的施用减少在流感的小鼠模型中肺部炎症[32]。此外,IL-1β升高总肺组织和慢性阻塞性肺病患者痰液(33],并与频繁COPD恶化以及一个IL-1β相关的痰蛋白质组签名相关联[34,35]。
确定新的COPD相关的信号通路重要的是要开发新的未来的治疗策略。FGFR的遍在表达,而FGFR4主要存在于肺,心脏,肾脏和肝脏。发信号通过FGFR4与炎症变化相关,我们证明了FGFR4信号通路与空气重塑和炎症之间的几个联系。此外,一种FGFR4阻断抗体和小分子抑制剂已被开发用于治疗肝细胞癌[36,37]。因此,靶向FGFR4或COPD炎性亚型中的可溶性Klotho水平的治疗可能代表新的治疗选择。
补充材料
致谢
作者感谢J.S.,D.邓肯和S.哈奇森(阿拉巴马大学伯明翰大学,伯明翰,AL,USA)的技术援助丹尼斯(迈阿密,迈阿密,佛罗里达州,美国的大学)。他们还感谢G.霍尔特和E.唐娜(迈阿密大学,迈阿密,佛罗里达州,美国)获得本研究中使用支气管镜标本协助和R.亚伯拉罕和J.班戈(U3制药,马丁斯里德,德国)的提供所述抗FGFR4阻断抗体。
脚注
这篇文章有提供补充材料www.qdcxjkg.com
利益冲突:无申报。
支持声明:该作品是由乘务员医学研究所(YFAC152003到S.克里克,YCSA113380到P.拉蒂和CIA13033到M. Salathe),囊性纤维化基金会(CFF KRICK1610到S.克里克和SALATH14G0到M.支持Salathe),美国心脏协会(A. GRABNER和C.福勒),美国糖尿病协会(C.福勒),詹姆斯和Esther王佛罗里达生物医学研究发展计划(5JK02到M. Salathe)和美国国立卫生研究院(R01HL128714到C.福勒)。本文资金的信息已交存交叉引用出资者注册。
- 收到2017年7月15日。
- 公认2018年4月27日。
- 版权所有©ERS 2018











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}