





文摘
结节病是一种疾病的特点是肉芽肿形成。有一个未满足的需要新的治疗策略超出皮质类固醇。先天免疫细胞的NLRP3 inflammasome途径表达和感知危险信号引起炎症白介素(IL) 1β;它最近成为制药靶点。这促使我们测试的角色NLRP3 inflammasome和肉芽肿形成IL-1β通路和结节病。
19肉质的患者和19个健康志愿者招募到这个试点研究。NLRP3 inflammasome活动以支气管肺泡灌洗(BAL)细胞和肺和皮肤活检使用免疫组织化学、免疫印迹、逆转录酶PCR和ELISA。为在活的有机体内实验中我们使用了海藻糖6,6′-dimycolate-granuloma小鼠模型和评估肺肉芽肿负担mir - 223基因敲除和NLRP3基因敲除小鼠,以及MCC950和anti-IL-1β抗体疗法的治疗效果。
我们发现强upregulation NLRP3 inflammasome通路,就是表达激活NLRP3 inflammasome组件,包括裂解caspase-1 IL-1β肺肉芽肿,并增加IL-1β释放BAL细胞结节患者相比,健康志愿者(p = 0.006)。mir - 223的mRNA水平微rna表达下调NLRP3,是减少和NLRP3 mRNA相应地增加肺泡巨噬细胞从肉质的患者(p < 0.005)。NLRP3基因敲除小鼠显示下降和mir - 223基因敲除小鼠肉芽肿的形成与野生型小鼠相比增加。药理干预使用NLRP3通路抑制剂MCC950或anti-IL-1β抗体导致肉芽肿形成减少(p < 0.02)。
总之,我们的数据提供证据的调节inflammasome和IL-1β通路激活在结节病和建议都有效的治疗靶点。
文摘
这个试点研究强调了一个关键的角色NLRP3 inflammasome和mir - 223在结节病的发病机制。的有益影响NLRP3 inflammasome通路抑制剂和anti-IL-1β抗体肉芽肿形成。http://bit.ly/36StvLe
介绍
结节病是一种全身性疾病的特点是基因易感个体non-necrotising肉芽肿的形成(1- - - - - -4]。的确切病因引发因素仍未知,可能存在各种触发器(1,5]。研究结果表明,其分子模式(pamp)源自分枝杆菌或propionibacteria肉质的患者可能会触发一个夸张的免疫反应(5,6]。人们认为误导主机响应否则无害pamp放大了免疫反应在结节病3]。穆勒的团队表明,血清淀粉样蛋白A (SAA)是高度表达的肉质的肉芽肿(7]。如果需要,病人使用免疫抑制剂治疗,通常几年甚至几十年。长期使用类固醇治疗结节病往往导致增加患者负担的共病的副作用,迫切的和新的治疗方法的概念(1,2,8,9]。
在过去的三十年里已经有多项研究表明巨噬细胞结节病病人产生广泛的M1细胞因子,包括肿瘤坏死factor-αCXCL10和白介素(IL) 1β(10- - - - - -17]。一些报告表明IL-1β在结节病的致病作用11- - - - - -16,18- - - - - -20.),近30年前。然而,IL-1β家族最近恢复了巨大利益由于分子界定所谓的inflammasome通路,一个细胞内multiprotein复杂,在生产中扮演着关键角色和释放的生物活性IL-1β巨噬细胞(17,21]。是研究最多的inflammasome NLRP3 inflammasome,被发现严重参与一些疾病包括矽肺的发病机理,肺结核和阿尔茨海默病(22- - - - - -24]。其中,硅酸盐晶体和淀粉样原纤维可以激活NLRP3 inflammasome [25- - - - - -28]。NLRP3 inflammasome装配需要两个信号,启动信号,从而导致转录pro-IL-1βNLRP3,和第二个信号,促进间接激活inflammasome的因素如ATP或nigericin [17,21]。NLRP3 inflammasome oligomerisation导致caspase-1的激活和表达功能活跃的caspase-1 p10和p20子单元17]。随后激活caspase-1劈开pro-IL-1βIL-1β的生物活性形式。基于这些新见解inflammasome和IL-1β免疫生物学,一些新的治疗策略阻止NLRP3 inflammasome甚至IL-1β信号已经开发和批准某些疾病(29日]。
在这些发现的背景我们感兴趣的角色NLRP3 inflammasome结节病。我们发现这个inflammasome途径确实是活跃在人类结节病的患者的样本而健康的志愿者。此外,使用鼠标的肺肉芽肿形成模型,我们可以确定的意义IL-1β和inflammasome组件造成结节病的病理。
方法
研究人群
本研究了19例结节病,六间质性肺病患者在硬皮病和19个健康志愿者(表1)。弗莱堡大学医学中心的研究,德国和经当地伦理委员会批准。之前所有科目提供书面知情同意参与这项研究。在18 19结节病的患者,支气管镜检查中进行常规的诊断检查在早期诊断。肉质的肺疾病活动被定义为“轻度”不活跃的肺部疾病与正常肺功能和呼吸系统症状,“温和的”主动免疫抑制治疗肺部疾病没有要求根据指导方针提出的美国胸科学会/欧洲呼吸学会(188bet官网地址30.),或“严重”活跃的肺部疾病与免疫抑制治疗要求。这些病人分类与严重疾病活动需要免疫抑制治疗严重的肺实质(> 20%)参与和炎性表型(如。高浓度的可溶性IL-2R和支气管肺泡灌洗(BAL)淋巴球增多)。我们没有招募患者先进、非炎症、纤维化疾病进入本研究。所有包括归类为患有严重疾病的患者进行支气管镜检查,在常规的诊断检查在早期诊断。
落下帷幕,transbronchial活组织检查
落下帷幕,transbronchial和皮肤活检是在结节病的患者作为他们的日常的诊断检查按照标准化的协议(31日- - - - - -33]。给出进一步的细节补充材料。
人类活检组织病理学和免疫组织化学
石蜡组织切片从人类transbronchial活检或皮肤活检是苏木精和伊红染色和免疫组织化学,主要使用特定的各种标记的抗体。给出进一步的细节补充材料。
免疫印迹分析caspase-1 p20活动
BAL细胞被刺激有或没有NLRP3激活协议中详细描述补充材料和上层清液沉淀与甲醇/氯仿。裂解caspase-1 (p20)的上层清液中检测使用初级抗体兔子马伯裂解caspase-1(细胞信号技术,丹弗斯,妈,美国)。
微核糖核酸分析和逆转录酶聚合酶链反应
肺泡巨噬细胞排序从BAL细胞结节患者和健康志愿者MoFlo Astrios /贝克曼库尔特(美国在印第安纳波利斯)基于细胞特征的细胞分选仪向前/侧面的散射和典型的肺泡巨噬细胞自体荧光。总细胞微核糖核酸从106排序的肺泡巨噬细胞是孤立使用mirVana™隔离设备(热费希尔科学、沃尔瑟姆,妈,美国)。给出进一步的细节补充材料。
动物研究
NLRP3−−/老鼠(B6株背景)请提供Vishva m .迪克西特(美国基因泰克,南旧金山,CA)。mir - 223−−/老鼠(B6。Cg-Ptprca Mir223tm1Fcam/J), their respective wild-type (B6.SJL-Ptprc一个Pepcb/ BoyJ)控制老鼠和C57BL / 6 j野生型老鼠从杰克逊实验室购买(美国加利福尼亚州萨克拉门托)。进一步的细节,请参阅补充材料。
海藻糖引起的小鼠肺肉芽肿模型6,6′-dimycolate
海藻糖6.6′-dimycolate (TDM alx - 581 - 210 - m001;恩佐生命科学,Loerrach,德国)结核分枝杆菌准备在油包水乳液使用不完全弗氏佐剂(34,35)和尾静脉注入。评估肺肉芽肿形成,肺收获7天TDM注射后对所有研究。此外,野生型老鼠管理车辆(PBS)或MCC950、一个NLRP3 inflammasome通路抑制剂,在10 mg·公斤−1腹腔内天0,1,2,4,6 TDM注射后。在另一项实验中,野生型小鼠皮下接种与车辆(PBS)、同形像控制抗体anti-cyclosporine(诺华、巴塞尔、瑞士)(200µg /鼠标)或anti-IL-1β抗体(诺华公司)(200µg /鼠标)−1天,3天之后TDM注入。给出进一步的细节补充材料。
结果
研究群体
临床特点和平衡差异表1。19例结节病,11日有严重肺部疾病活动显著降低肺功能和免疫抑制治疗因为肺癌介入,要求三个有温和的和5有轻度肺部疾病活动(表1)。没有一个病人分为中度或轻度需要免疫抑制治疗,因为肺参与。所有六个硬皮病患者重要的肺部疾病和接受支气管镜检查,因为呼吸恶化和免疫抑制治疗的必要性。所有的研究参与者都不吸烟。所有受试者的白人种族除了一个非洲裔美国人。肉质的病人都是首次治疗除了一个严重疾病患者接受低剂量强的松(5毫克·天−1)。正如所料,BAL的肉质的患者中增加总细胞数量和淋巴细胞的比例要明显高于健康志愿者落下帷幕,而肺泡巨噬细胞减少的比例(表1)。所有结节患者的平均年龄明显比平均年龄的增加健康志愿者(p < 0.0001),而年龄分组的肺病患者活动没有显著差异。重要的是,年龄没有显著的影响在所有结果下面描述。
结节病肉芽肿inflammasome激活和IL-1β表达的证据
结节病肉芽肿是CD68组成+巨噬细胞(图1一个和b)。表达的活跃caspase-1描述抗体裂解/主动caspase-1(描述看到的补充图S1)所示,对应CD68领域+包含多核巨细胞肉芽肿频繁(图1 d观察和e)。这种模式在几乎所有情况下transbronchial和皮肤活检(图1一个- f,补充图S2和S3)。免疫反应性肉芽肿内观察IL-1β在所有活检(图1 g和h,补充图S2和S3)。结节病肉芽肿是主要由CD68组成+巨噬细胞,染色积极活跃caspase-1和IL-1β。IL-1β染色,盲法,由专家进行病理学家和半定量评分显示趋势高IL-1β表达式从严重的肺病患者肺肉芽肿与温和的疾病患者相比(n = 13, p = 0.1111;图1 l)。
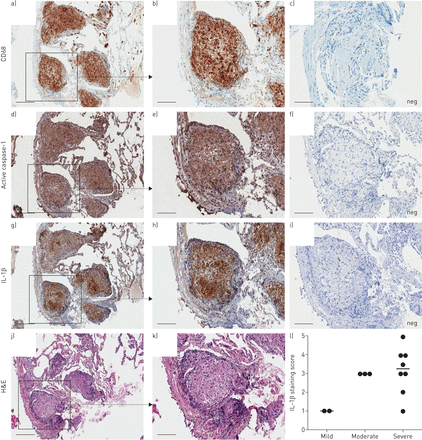
证据inflammasome和白介素(IL) 1β通路激活在肉质的肺肉芽肿。a - k)代表组织派生的连续部分的显微照片transbronchial活检(TBB)从一个肉质的严重的肺病患者活动。ⅰ)TBB组织使用巨噬细胞系的标记抗体免疫组织化学染色CD68 (a、b),激活caspase-1 (d, e)和IL-1β(g, h)。c、f i)负控制染色得到运用各自的同形像控制抗体。j, k) TBB组织苏木精和伊红染色(圆))组织病理学评估,表明non-necrotising肉质的肉芽肿。比例尺条= 100μm (a、d、g j), 200年μm (b, c, e、f、h,我,k)。l)的量化IL-1β免疫反应性肺肉芽肿是描绘与肺疾病活动。IL-1β染色是盲目的得分方法TBBs来自13个病人。
证据一个激活NRLP3 inflammasome通路在BAL细胞和单核细胞来源于肉质的病人
本构释放IL-1β肉质的BAL细胞相比显著增加的水平决定了BAL细胞健康志愿者(p = 0.0065;图2一个)或硬皮病患者(p = 0.038) (图2一个)。此外,BAL肉质的严重疾病患者的细胞显示显著增加本构IL-1β生产与健康志愿者(p = 0.0161;图2 b)。本构IL-1β版本相比,体外刺激NLRP3 inflammasome与脂多糖(LPS) / nigericin,至少有10倍增加IL-1β生产从BAL细胞来源于健康的志愿者和肉质的患者(图2一个- c)。BAL细胞从结节病的病人获得了更多IL-1β相比从健康志愿者NLRP3 inflammasome刺激(p < 0.0001;图2 c),这种能力与肺部疾病的活动(轻微增加与严重的疾病活动,p = 0.0020;图2 d)。得到了类似的结果在NLRP3 inflammasome与LPS刺激/ ATP (图2 d)。免疫印迹分析证实活跃IL-1β生产通过平衡细胞(补充图S4)。caspase-1酶的表达和激活状态,研究了BAL细胞健康志愿者和肉质的病人通过免疫印迹。我们发现增加的组成型表达激活caspase-1 p20如果BAL细胞条件培养液的肉质的患者相比,BAL细胞健康志愿者(图2 e)。水平的激活caspase-1 p20增强在有限合伙人的条件培养基/ nigericin -或有限合伙人/ ATP-stimulated BAL细胞,再次与BAL细胞来源于肉质的病人比healthy-volunteer样本含有较高水平(图2 e)。
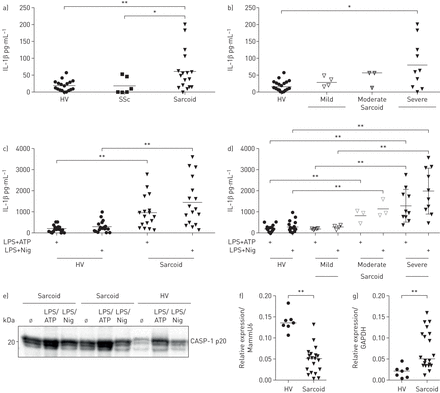
证据一个激活NLRP3 inflammasome通路在支气管肺泡灌洗(BAL)细胞结节的病人。a e) BAL细胞健康志愿者(高压),硬皮病患者间质性肺疾病(SSc)或结节病(结节)离开如果6 h (a, b)或刺激6 h和脂多糖(LPS) + nigericin(国家)或有限合伙人+ ATP激活NLRP3 inflammasome IL-1β(汉英)和水平和裂解亚基caspase-1 p20测定细胞培养上清液的ELISA和免疫印迹。a, c) IL-1β水平从健康志愿者(n = 18),肉质的病人(n = 17)和患者SSc-ILD (n = 6);b, d) IL-1β水平从肉质的患者在肺疾病活动。e)代表免疫印迹分析裂解caspase-1亚基p20,结果从caspase-1 inflammasome激活。f, g)在一个额外的研究中,肺泡巨噬细胞排序从BAL细胞来自20个严重的肉质的进行性疾病和患者需要联合免疫抑制治疗和七个健康志愿者;f)相对于MammU6 mir - 223表达和g)相对于GAPDH NLRP3表达式由逆转录酶聚合酶链反应决定。酒吧描绘中值。* *:*:p < 0.05, p < 0.01, Mann-Whitney紫外线测试。
此外,我们研究在同一队列inflammasome激活单核细胞从外周血中分离出来。注意,NLRP3 inflammasome IL-1β释放不同的激活和调节单核细胞和巨噬细胞。肉质的患者单核细胞显示增加释放在LPS刺激后IL-1β与单核细胞从健康志愿者(p = 0.0069;补充图S5)。
因为mir - 223之前的研究已经证实,表达下调NLRP3老鼠骨髓衍生细胞的转录我们组(36),我们研究了RNA的表达mir - 223和NLRP3 fluorescence-activated cell-sorted肺泡巨噬细胞通过逆转录酶聚合酶链反应。我们发现相对mir - 223表达显著下降(p = 0.0002;图2 f)和相对NLRP3表达显著增加(p = 0.0023;图2 g)排序的肺泡巨噬细胞从肉质的患者相比,健康的志愿者。因此,mir - 223水平降低可能导致增加NLRP3表达和NLRP3 inflammasome通路激活在结节病。
下游的影响IL-1β信号调节在肉质的患者样本
因为增加的IL-1β水平肉质的患者中观察到,我们成为下游IL-1β信号的影响和研究感兴趣的表达il - 1受体拮抗剂(IL-1ra)和引发,这两个被IL-1β诱导。本构IL-1ra释放BAL细胞培养的水平体外24 h高度升高对肉质的病人相比,健康志愿者(p = 0.0038;图3一)和与疾病活动增加(p = 0.0001;图3 b)。此外,中性粒细胞化学引诱物的水平引发了BAL细胞结节病的增加(p = 0.0074, p = 0.0004;图3 c和d)。我们发现IL-1ra和引发水平之间的显著相关性(r2= 0.860,p < 0.0001;补充图S6本构IL-1β和引发生产之间)和(r2= 0.559,p = 0.0006;补充图S6)。中性粒细胞组成的低分数整体BAL细胞群从肉质的患者和健康志愿者中恢复过来。然而,中性粒细胞计数当总细胞数正常/ / 100毫升BAL流体增加肉质的患者相比,健康志愿者(p = 0.0256),特别是严重相比,轻微疾病患者显著增加(p = 0.0018;表1)。此外,血液中性粒细胞计数肉质的严重肺部疾病患者活动是健康的志愿者相比显著升高(p = 0.0458;图3 e)。

本构受体拮抗剂(IL-1ra)及引发释放支气管肺泡灌洗(BAL)细胞和中性粒细胞计数增加结节病表明IL-1β途径激活。模拟)BAL细胞健康志愿者(高压)或肉质的患者培养没有进一步刺激24 h和IL-1ra水平(a, b)和引发(c, d)测量条件培养基。落下帷幕的)IL-1ra生产细胞健康志愿者(n = 17)和肉质的病人(n = 18);b) IL-1ra生产BAL细胞结节患者在肺疾病活动。c, d)引发相应的生产从BAL细胞(高压n = 13,肉质的n = 18)。e、f)血液中性粒细胞计数由fluorescence-activated cell-sorting高压(n = 17)和肉质的病人(n = 16),根据肺疾病活动。酒吧描绘中值。* *:*:p < 0.05, p < 0.01, Mann-Whitney紫外线测试。
调节SAA含量肉质的患者延续NLRP3 inflammasome活动
SAA提出了作为主要驱动力在结节病的发病机制和作为活化剂的NLRP3 inflammasome [7),我们的表达和影响分析SAA的NLRP3 inflammasome通路;提供了这些分析的细节补充图S7。
的NLRP3 inflammasome通路是活跃在肉芽肿形成的小鼠模型
glyocolipid TDM当静脉注射诱发小鼠肺肉芽肿。代表显微照片引起的肺肉芽肿TDM的野生型小鼠所示图4一- c和补充图S8和S9。总肉芽肿的左叶肺的负担和例证图4 b。我们调查的功能响应性小鼠BAL细胞NLRP3 inflammasome刺激有限合伙人/ nigericin(和有限合伙人/ ATP)。我们发现增加了TDM-treated IL-1β释放与未经处理的小鼠(p = 0.0006;图4 d),反映出增加inflammasome激活在这个模型。连续的免疫组织化学染色部分显示Iba-1的积累+巨噬细胞(图4 e和f,补充图S8和S9)和表达的IL-1β肉芽肿对应于同一地区(图4 g,补充图S9)。此外,我们可以显示TDM-induced肉芽肿(SAA染色图4 h,补充图S9)。解决的影响NLRP3 inflammasome和mir - 223肉芽肿的形成,分析了这些分子的基因消融的影响在TDM模型中。我们发现显著增加肺肉芽肿数量从mir - 223−−/小鼠(p = 0.0016),从NLRP3肺肉芽肿数量会下降−−/小鼠(p < 0.0001),而野生型小鼠(图4我- l)。此外,在mir - 223基因敲除小鼠我们观察到更大更融合比野生型或NLRP肉芽肿−−/老鼠。
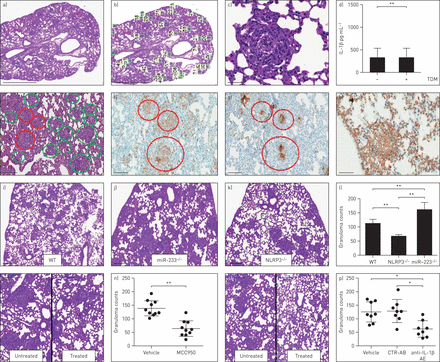
全身的海藻糖6.6′-dimycolate (TDM)小鼠的肺肉芽肿像人类的肉质的肉芽肿。参与的NLRP3 inflammasome通路,减少小鼠的肺肉芽肿负担治疗化合物针对NLRP3 inflammasome。)苏木精和伊红染色())肺收获7天从C57 / BL6野生型小鼠(WT)与TDM的挑战。b)计数策略肉芽肿的左叶肺的负担。c)高放大倍数的典型TDM-induced在小鼠肺肉芽肿。d)白介素(IL) 1β细胞释放BAL刺激体外脂多糖/ nigericin(6小时)在上层的决心。e) H&E-stained部分肺肉芽肿;红色和绿色圆圈表示肉芽肿形成。更高的放大图像f和g)所示。f-h)免疫组织化学TDM-induced肉芽肿使用f)老鼠巨噬细胞谱系标记抗体Iba-1, g) IL-1β或h)血清淀粉样蛋白A (SAA)。i (k))污渍我)WT小鼠的肺组织,mir - 223 j)−−/老鼠和k) NLRP3−−/老鼠TDM注射后7天(每n = 6)。l)肉芽肿计数WT小鼠的肺组织,mir - 223−−/老鼠和NLRP3−−/老鼠。m))污渍的肺TDM-treated C57 / BL6 WT老鼠处理车辆或NLRP3 inflammasome通路抑制剂MCC950(每个n = 2)。n)降低肉芽肿起的肺组织MCC950-treated相比vehicle-treated老鼠。o))污渍的肺TDM-treated C57 / BL6 WT老鼠同形像对待控制抗体或anti-IL-1β抗体。p)显著降低肉芽肿计数从anti-IL-1β-treated老鼠相比,小鼠肺组织同形像对待控制抗体或车辆(每个n = 8)。数据提出了个人价值观和意味着±sd。规模的酒吧,b) 1000μm, c) 10μm 500μm eg, i (k)。* *:*:p < 0.05, p < 0.01;l d n)未配对t检验,p)未配对的方差分析。
评估影响的药理干预NLRP3 inflammasome通路,在NLRP3 inflammasome通路抑制剂MCC950管理7天到TDM-treated野生型小鼠,导致肺肉芽肿形成的显著减少(p = 0.002;图4米和n)。同样,anti-IL-1β抗体治疗显著降低肉芽肿数相比,车辆控制(p = 0.0184;图4 o和p)和同形像控制抗体(p = 0.021;图4 p)。
讨论
结节病是一种肉芽肿疾病高未满足的医疗需要,迫切需要新的治疗策略。在本文中,我们演示的重要性inflammasome结节病发展的关键因素和疾病的严重程度和识别这个途径为结节病一个潜在的治疗目标。我们表明,inflammasome组件和活跃IL-1β表达在肉质的巨噬细胞和多核巨细胞肉芽肿。另外,在健康的志愿者相比,NLRP3 inflammasome由肺泡巨噬细胞的激活观察结节病和水平的患者NLRP3 inflammasome激活与疾病相关的活动。这些结果清楚地表明,NLRP3 inflammasome激活发生在肉芽肿和似乎是由存在于病变的信号。此外,我们提供支持的临床前实验中的数据模型的结节病肉芽肿的形成是由分枝杆菌诱导的醣脂类TDM,演示的放大作用NLRP3 inflammasome激活肺肉芽肿形成。
免疫组织化学显示活跃caspase-1和IL-1β蛋白质都是肉质的内高度表达的多核巨细胞肉芽肿以及肉质的组织巨噬细胞,但不是在正常的肺和皮肤。BAL细胞结节病的患者自发释放IL-1β和活跃的亚基caspase-1相比细胞来源于健康的志愿者或硬皮病患者。此外,刺激协议导致NLRP3 inflammasome激活了NLRP3 inflammasome hyperactivation肉质的BAL细胞而健康的细胞。最近发现突出inflammasomes的至关重要的作用作为病原体和危险信号的传感器(14- - - - - -16];身体需要区分无害的和有害的pamp,有关分子模式,和inflammasomes帮助直接先天免疫反应和维持肺组织恒定性。因此,在正常情况下,inflammasome激活和IL-1β生产监管严格,因为他们的潜在有害影响17,21]。然而,在肉质的病人群体我们发现特异表达NLRP3 inflammasome激活和连续IL-1β释放高度增加患者的严重疾病,并需要免疫抑制治疗。近30年前研究者指出BAL流体IL-1β水平相关性的疾病活动(13,14),增加IL-1β结节病肺泡巨噬细胞的基因表达水平和增加IL-1ra [20.,37,38),我们现在属性增强inflammasome活动在这个研究。值得注意的是,在家族性结节病,布劳综合症,Nod2基因的突变导致NF-κB激活通过另一种NLR通路是描述(39,40]。此外,在我们的肉质的队列,增加IL-1β释放与下游IL-1R1信号事件如IL-1ra和引发水平升高和随之而来的中性粒细胞涌入。高BAL中性粒细胞计数报告更严重影响患者免疫抑制治疗的必要性。(41,42]。此外,IL-1β生产增加了抗原递呈细胞如巨噬细胞和增加IL-1R信号驱动一代TH17.1细胞,最近被发现在结节病增加,可以作为一个关键致病的元素(43- - - - - -45]。因此,我们的研究结果表明,本构NLRP3 inflammasome激活在结节病的病理生理和临床意义。
最近immunohistopathological分析显示了急性期蛋白的积累SAA肉质的的中心在靠近多核巨细胞肉芽肿7,46]。事实上,其分布与CD3和靠近巨噬细胞相关+细胞涌入,作者声称,SAA坐标先天和适应性免疫反应来帮助结节病肉芽肿性炎症的发展和潜在的其他肉芽肿性疾病。此外,作者使用结节病的小鼠实验模型来证明他们的理论,SAA导致肉芽肿的发展。其他人建议NLRP3 inflammasome SAA在人类和小鼠巨噬细胞活化在体外在人类的银屑病角质细胞体外和功能型小鼠哮喘模型(25- - - - - -27]。我们注意到,与此前公布的数据,血清SAA含量增加在我们的肉质的患者(47,48]。与SAA刺激后,BAL细胞结节病IL-1β发布与细胞来源于健康的志愿者。此外,SAA-induced IL-1β生产外周血单核细胞有效地阻止了caspase-1抑制剂vx - 740和NLRP3 inflammasome抑制剂MCC950。根据以前的出版物(7),我们的数据表明,南非航空公司可能是一个潜在的内生有关分子模式能够触发NLRP3 inflammasome激活在结节病。为了证实这一点,我们还发现积累SAA肉芽肿病变小鼠模型,在mycobacteria-derived TDM唤起肺肉芽肿。此外,TDM-induced肉芽肿形成在NLRP3明显减弱−−/小鼠与野生型的动物。
一个额外的机制NLRP3 inflammasome激活规范是通过mir - 223,直接目标NLRP3表达水平(36,49,50]。从健康的志愿者相比,肺泡巨噬细胞,mir - 223结节病肺泡巨噬细胞的表达水平显著降低,而NLRP3表达显著调节。自从NLRP3表达水平与NLRP3密切相关inflammasome活动,我们的研究表明,减少mir - 223的表达肉质的巨噬细胞可能导致的本构激活NLRP3 inflammasome。自mir - 223 -缺乏小鼠显示增加NLRP3 inflammasome活动和肉芽肿形成相应的野生型相比,在结节病动物模型在活的有机体内,这种假设是钢筋。因此,我们的数据显示一个关键的角色NLRP3 inflammasome和mir - 223损耗放大肉芽肿形成。
我们使用相同的TDM肉芽肿模型测试治疗策略干扰NLRP3 inflammasome激活和IL-1β信号。MCC950是最近开发的小分子抑制NLRP3 inflammasome组装和激活(29日]。老鼠MCC950显示处理显著降低肺肉芽肿负担后TDM刺激vehicle-treated老鼠相比,表明这种新的治疗策略对肉芽肿的形成有着直接的影响。此外,老鼠接受anti-IL-1β抗体显示显著降低肉芽肿后负担TDM刺激小鼠相比处理车辆或同形像控制抗体。综上所述,这些数据显示,NLRP3 inflammasome通路是活跃在结节病的小鼠模型。
我们的发现是有限的这一事实TDM-induced小鼠肉芽肿形成后发生急性和瞬态自发的决议,10天。此外,巨噬细胞的超微结构分析显示积累而不是epitheloid细胞肉芽肿(发展中心51]。然而,目前我们缺乏任何接受为结节病小鼠模型。在我们的手TDM模型优于其他模型,我们测试了。类似于人类肉芽肿结节病,TDM-induced肉芽肿由多核表达IL-1β和SAA巨头。此外,与结节病,TDM-induced肉芽肿被认为涉及分枝杆菌pamp可能触发在最初的肉芽肿的形成。Mycobacterial-derived pamp已确定在肉质的肉芽肿和Kveim试剂(6,52基于这些考虑,我们认为TDM-induced肉芽肿模型是目前最好的模型在结节病肉芽肿形成。然而,未来在活的有机体内研究需要分析的影响NLRP3 inflammasome封锁已经建立了肺肉芽肿。我们人类研究是有限的,我们不能通过免疫组织化学方法检测结节病肉芽肿NLRP3表达由于缺乏特定的抗体在formalin-fixed组织给予可靠的染色。虽然我们的逆转录酶聚合酶链反应试验和功能实验表明NLRP3 inflammasome激活调节巨噬细胞的患者严重的结节病和免疫抑制治疗的需要,我们不能排除,另外其他inflammasome亚型可能被激活。此外,由于我们没有执行正电子发射断层扫描/计算机断层扫描在两组肉质的我们不能提供的信息是否NLRP3 inflammasome BAL细胞激活和NLRP3表达增加的患者晚期纤维化疾病和炎症。
总之,我们的研究强调的角色NLRP3 inflammasome结节病。我们的数据显示一个关键的角色NLRP3 inflammasome激活肉芽肿形成和演示的有益影响NLRP3 inflammasome通路抑制剂在肉芽肿形成。这些数据需要未来的研究解决NLRP3 inflammasome抑制患者的结节病。在结节病的临床相关性inflammasome激活病理学是目前正在进行探索试验患者的治疗IL-1β封锁的活跃的肺结节病(www.clinicaltrials.govNCT02888080)。
补充材料
可共享的PDF
确认
我们要感谢维多利亚Wirtz(弗劳恩霍夫项目,汉诺威,德国)和Marija Curcic Antje莱和流行油萜(瑞士诺华,巴塞尔)专家的技术援助。
脚注
可以从本文的补充材料www.qdcxjkg.com
作者的贡献:概念和设计:c . Huppertz贼鸥,g . Wieczorek中华民国奥利弗,做减法。Bauernfeind,诉霍农Prasse。与人类材料和实验分析:c . Huppertz贼鸥,g . Wieczorek中华民国奥利弗,做减法。Bauernfeind和a . Prasse。实验小鼠和小鼠材料和分析:c . Huppertz p .恩格尔哈德,贼鸥,g . Wieczorek做减法。Bauernfeind和a . Prasse。手稿准备:c . Huppertz贼鸥,g . Wieczorek中华民国奥利弗·恩格尔哈德a . Littlewood-Evans做减法。Bauernfeind,诉霍农Prasse。
利益冲突:c . Huppertz的员工,拥有诺华制药公司的股票。
利益冲突:Jager没有披露。
利益冲突:g . Wieczorek的员工,拥有诺华制药公司的股票。
利益冲突:p .恩格尔哈德没有披露。
利益冲突:中华民国奥利弗是一个员工,拥有诺华制药公司的股票。
利益冲突:做减法。Bauernfeind没有披露。
利益冲突:a . Littlewood-Evans的员工,拥有诺华制药公司的股票。
利益冲突:t . Welte报告从诺华赠款,在进行研究;外的个人费用咨询委员会在诺华工作,提交的工作。
利益冲突:诉霍农没有披露。
利益冲突:a . Prasse报告从诺华赠款,在进行研究;赠款和个人费用从勃林格殷格翰集团和阿斯利康讲座,个人费用从罗氏讲座,诺华和赛诺菲-安万特外提交的工作。
支持声明:这部分工作是由一个研究资助从诺弗莱堡大学医学中心(PI: a . Prasse)。提供进一步融资DZL呼吸。资金信息,本文已沉积的Crossref资助者注册表。
- 收到了2019年1月17日。
- 接受2019年12月18日。
- 版权©2020人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}