





摘要
背景肺动脉高压(PH)是COPD的常见并发症,与死亡率和发病率增加有关。有趣的是,肺血管改变被认为是肺气肿发展的驱动因素。之前,我们发现诱导型一氧化氮合酶(iNOS)是发生和逆转吸烟诱导的PH和肺气肿的必要酶,并表明iNOS在骨髓来源细胞中的表达驱动肺血管重塑,而不是实质破坏。在这项研究中,我们的目标是确定inoos表达细胞类型驱动烟雾诱导的PH,并破译相关的促增殖途径。
方法为了解决这个问题,我们使用了1)慢性烟雾暴露中的骨髓细胞特异性iNOS敲除小鼠,2)巨噬细胞和肺动脉平滑肌细胞(PASMCs)共培养来破译潜在的信号通路。
结果骨髓细胞特异性iNOS敲除可预防小鼠烟雾诱导的PH,但不能预防肺气肿。此外,骨髓细胞中iNOS的缺失改善了间质巨噬细胞中M2极化标记CD206的表达增加。重要的是,观察到的对肺巨噬细胞的影响是缺氧独立的,因为这些小鼠产生了缺氧诱导的PH值。在体外在与m2极化巨噬细胞共培养时,吸烟诱导的PASMC增殖可以通过吞噬细胞中iNOS的缺失以及PASMC中细胞外信号调节激酶的抑制来消除。关键是,免疫组化显示,cd206阳性和inos阳性巨噬细胞在COPD患者肺部重构血管附近聚集。
结论总之,我们的研究结果表明,髓系细胞中iNOS的缺失对烟雾暴露小鼠的PH值具有保护作用,并为m2样巨噬细胞和pasmc之间的iNOS依赖通信在潜在的肺血管重塑中提供了证据。
摘要
骨髓细胞中iNOS的缺失可以保护小鼠免受吸烟诱导的ph的影响。M2巨噬细胞与PASMCs的交叉对话驱动吸烟诱导的肺血管重塑,并依赖于巨噬细胞中iNOS的表达和PASMCs中的ERK活性。https://bit.ly/3itDtuV
简介
慢性阻塞性肺病是全球五大死亡原因之一。慢性阻塞性肺病的病理改变是由吸入有害物质如香烟烟雾引起的,包括慢性气道炎症和进行性肺泡破坏,导致慢性支气管炎和肺气肿[1,2]。导致COPD的潜在分子机制包括氧化应激增加、蛋白水解活性和抗蛋白水解防御之间的不平衡以及炎症细胞的涌入[3.,4]。此外,临床前模型的最新发现[5- - - - - -8]和慢性阻塞性肺病患者[9]提示了肺血管改变是COPD病理的早期现象和肺气肿的可能驱动因素的假设[10]。在暴露于烟雾中的小鼠和豚鼠中,肺血管重塑先于实质破坏[5- - - - - -8]和现有的肺动脉高压治疗方法影响肺气肿的发展[11- - - - - -13]。此外,在大多数未发展为COPD的COPD患者和吸烟者中发现了肺血管重塑的组织病理学征象[9]。此外,高达90%的COPD患者存在异常高的平均肺动脉压。根据目前世界卫生组织的分类,copd相关肺动脉高压(COPD-PH)被列入PH值的第3组(肺部疾病/缺氧引起的PH值)[14- - - - - -16]。尽管PH值与COPD患者病情加重风险增加和生存期降低有关,但目前还没有有效的药物选择[14]。COPD-PH发病机制包括内皮功能障碍、缺氧、血管修剪和毛细血管床丧失[17]。此外,炎症细胞的激活可能有助于COPD的PH发展,因为几种细胞因子如肿瘤坏死因子-α、c反应蛋白[18]和白细胞介素-6 [19]与COPD患者的PH值相关,血管周围炎症细胞数量与肺血管改变相关[20.]。这些发现更加有趣,因为炎症在肺血管重塑中的突出作用已经在其他形式的PH中得到确认[21- - - - - -23],尽管先前研究表明,COPD-PH中发生的血管基因调控与其他类型的肺血管疾病有很大不同[5]。此外,我们最近发现了诱导型一氧化氮合酶(iNOS)在烟雾诱导的PH和肺气肿的发病机制中起关键作用。在烟雾暴露小鼠中,iNOS抑制阻止并逆转实质破坏、PH值和肺血管重塑。此外,我们证明了骨髓来源细胞中iNOS的表达驱动肺血管改变,而不是肺气肿的发展[5]。然而,目前尚不清楚是哪种骨髓来源的细胞类型驱动了这一过程,以及各自的机制是什么。我们假设导致吸烟诱导PH值的病理信号是由骨髓细胞中iNOS表达升高触发的,巨噬细胞起着关键作用,并且与小鼠相似的过程可能发生在人类身上。一氧化氮合酶是一种可诱导的、不依赖钙的、高通量的一氧化氮合酶异构体,一种产生一氧化氮的酶l-精氨酸和分子氧。这种酶在免疫系统中具有突出的作用,其作用超出了其抗菌和抗真菌活性,并涉及对表达细胞的表型的影响,而且还涉及对邻近免疫细胞的功能和组成的影响[24]。
材料与方法
动物模型
成年雄性和雌性iNos LysM-Cre小鼠(Nos2tm2904.1Arteartelyz2 Tg (CAG-flpe) 2tm1 lfo (cre)/), 3-4月龄,年龄和性别匹配的野生型(WT)对照组(C57BL/6N;查尔斯河实验室,苏兹菲尔德,德国)被随机分配到烟草烟雾暴露组或不暴露组,低氧组或常氧组。所有的动物实验都得到了区域动物福利委员会的批准(Regierungspräsidium吉森,德国)。
人肺切片免疫组化染色
使用了来自捐赠者和COPD患者的人类肺样本。该研究符合《赫尔辛基宣言》,组织捐赠方案得到了德国吉森贾斯特斯-李比希大学医学院伦理委员会的批准。免疫组化染色既往报道[25有细微的修改。一抗的使用如下:CD68 (Cat#ab955, Abcam,剑桥,英国;1:200);CD206 (Cat#ab64693, Abcam,英国剑桥;1:100),抗inos (Cat# NB300-605, Novus biicals, Littleton, CO, USA;1:250), phospho p42/p44 (Cat#4370, Cell Signaling Technology, Danvers, MA, USA;1:200)。
统计分析
使用Prism 6 (GraphPad, LaJolla, CA, USA)进行统计分析。所有数据均以均数±表示扫描电镜.多组间比较采用双向方差分析和Tukey's事后不同组不匹配样本(动物实验)与Sidak的比较检验事后匹配样品的检测(细胞培养实验)。采用独立t检验比较两组间均数是否相等。p值<0.05为有统计学意义。
关于本研究中使用的所有其他方法的信息可以在补充材料.
结果
骨髓细胞中iNOS的缺失可以防止吸烟小鼠的PH值和右心室肥厚
考虑到我们之前的发现,表达iNOS的骨髓源性细胞驱动烟雾诱导PH的发育,我们通过杂交iNOS生成了骨髓细胞特异性iNOS敲除小鼠液氧/液氧用溶菌酶M启动子驱动的cre表达小鼠(以下简称LysM-Cre),来评估骨髓细胞特异性iNOS表达在烟雾诱导PH发展中的作用在活的有机体内.证实敲除小鼠成功生成在体外通过量化骨髓来源巨噬细胞中iNOS的表达(补充图S1a, b),原位,以免疫荧光染色小鼠肺切片(补充图S1c)。
慢性烟雾暴露后的血流动力学测量显示,与WT小鼠相比,暴露在烟雾中3和8个月时,骨髓细胞特异性iNOS敲除小鼠对PH的影响受到保护(图1一个)。此外,通过富尔顿指数和超声心动图(图1 b, c).此外,与未接触香烟(室内空气)的对照组相比,暴露于香烟烟雾的WT小鼠在3和8个月后肺小血管的肌肉化增加(图1 d-f)。相比之下,iNOS LysM-Cre小鼠完全免受烟雾诱导的肺血管重塑。
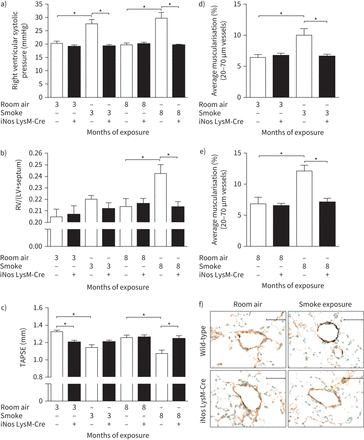
骨髓细胞类型特异性诱导型一氧化氮合酶(iNOS)缺失可防止小鼠吸烟诱导肺动脉高压的发展。小鼠分别暴露在香烟烟雾和室内空气中3个月和8个月。a)右心室收缩压(n= 9-12)。b)右心室结构改变,表现为右心室(RV)与左心室+室间隔(LV+室间隔)质量之比(n= 9-12)。c)超声心动图评价右心室收缩功能(n= 11-13),采用三尖瓣环形平面收缩偏移(TAPSE)。d - f)吸烟暴露3个月(n= 4-5)或e) 8个月(n= 5-7)后,肺小血管(20-70µm外径)的重塑表现为总血管计数的平均肌肉化;f)暴露于香烟烟雾8个月和各自房间空气对照的小鼠肺切片的代表性图像,对α-平滑肌肌动蛋白(紫色)和血管性血友病因子(棕色)进行联合染色。比例尺=50µm。数据以均数±表示扫描电镜.*: p < 0.05。双向方差分析(与Tukey的多重比较事后采用Test)进行统计分析。
骨髓细胞特异性iNOS敲除不能预防小鼠慢性烟雾暴露后肺气肿的发展
同时,我们研究了iNOS LysM-Cre小鼠肺气肿的发展。我们首先使用荧光分子断层扫描(FMT)结合微计算机断层扫描(µCT)和基于膜联蛋白v的成像探针来量化细胞凋亡在活的有机体内发现WT和iNOS LysM-Cre小鼠在烟雾暴露8个月后,与各自未暴露(室内空气)对照组相比,肺细胞凋亡均有所增加(图2一个)。使用FlexiVent系统进行功能测量(图2 b),用µCT评估功能剩余容量(图2 c)和形态测量分析,包括基于设计的体视学(图2 d而且补充图S2)证实WT和iNOS LysM-Cre小鼠在慢性烟雾暴露8个月后出现肺气肿。敲除动物肺功能变化的基线值和时间动态不同,可能是由于先天性影响。
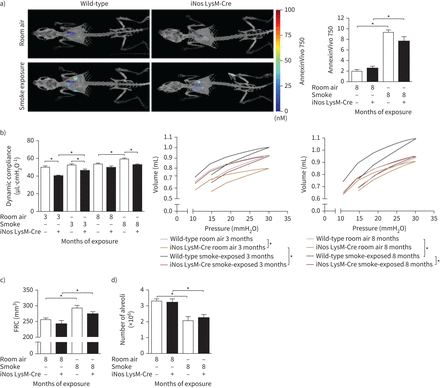
慢性烟雾暴露导致骨髓细胞特异性诱导型一氧化氮合酶敲除小鼠肺气肿的发展。小鼠分别暴露在香烟烟雾和室内空气中3个月和8个月。一)在活的有机体内肺细胞凋亡定量(n= 6-7),荧光分子断层成像系统检测,使用AnnexinVivo 750探针。b)肺功能(n= 8-11)表现为动态顺应性和呼吸压力-容积循环。c)通过微计算机断层扫描评估功能剩余容量(FRC) (n= 11-13)。d)基于设计的体视学估计的肺泡数(n= 7-9)。数据以均数±表示扫描电镜.*: p < 0.05。双向方差分析(与Tukey的多重比较事后采用Test)进行统计分析。
慢性烟雾暴露后,骨髓细胞中iNOS的缺失影响肺部炎症细胞的组成
由于肺部炎症细胞的组成和表型是其他形式PH的重要因素,我们使用流式细胞仪检测了骨髓细胞特异性iNOS缺失对肺匀浆中吸烟诱导的免疫细胞变化的影响。慢性烟雾暴露8个月后,间质巨噬细胞的部分显著增加(特征如图所示)图3一)在CD45+与未暴露于烟雾的对照组(室内空气)相比,在暴露于烟雾的WT动物的肺匀浆中发现了细胞,但在iNOS LysM-Cre小鼠中不存在细胞(图3 b)。考虑到巨噬细胞向m2样表型的转变有助于其他PH形式的肺血管重塑,我们检测了慢性烟雾暴露后肺匀浆中发现的间质巨噬细胞CD206的表达。该分析显示,在WT动物中CD206的表达显著增加,表明烟雾诱导间质巨噬细胞向m2样表型转移。重要的是,CD206在iNOS LysM-Cre小鼠肺间质巨噬细胞中的表达保持不变(图3 cd)。
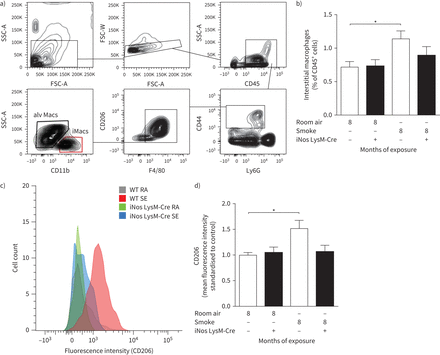
骨髓细胞特异性诱导型一氧化氮合酶(iNOS)的缺失可以防止这些细胞上烟雾诱导的间质巨噬细胞的积累和CD206(“m2样”极化的标记物)的上调。将小鼠暴露于烟雾(SE)或室内空气(RA)中8个月,用流式细胞术分析肺匀浆中的巨噬细胞。a)典型的流式细胞术门控方案,用于分离肺泡和间质巨噬细胞。b)烟雾暴露小鼠和各自未暴露(室内空气)对照组(n= 4-5)肺间质巨噬细胞含量,流式细胞术评估,以CD45百分比表示+细胞。c)显示CD206的代表性直方图+野生型(WT)和骨髓细胞特异性iNOS敲除小鼠肺中的细胞。d)间质巨噬细胞上CD206的表达(n= 4-5),流式细胞术评估,并给予标准化的平均荧光强度作为对照。数据以均数±表示扫描电镜.SSC:侧散射;FSC:向前散射。*: p < 0.05。双向方差分析(与Tukey的多重比较事后采用Test)进行统计分析。
此外,我们发现吸烟诱导CD4比例增加+CD45中的t细胞+与iNOS LysM-Cre动物相比,WT组肺匀浆中细胞数量显著增加。同样,吸烟诱发的CD4细胞和CD4细胞均有较高的升高趋势+和CD8+WT与iNOS LysM-Cre小鼠支气管肺泡灌洗液中t细胞的变化(补充图S3a, b)。
髓系细胞中iNOS缺失不影响缺氧诱导的PH值和右心室肥厚的发展
由于呼吸功能损害引起的肺泡缺氧可促进COPD血管重构[17],巨噬细胞的M2表型对于缺氧诱导PH的发展很重要[26],我们研究了骨髓细胞中iNOS的缺失是否也可以保护暴露于慢性缺氧后的小鼠免受PH的影响。然而,骨髓细胞特异性iNOS敲除小鼠出现缺氧诱导的PH水平与WT小鼠相同。从血流动力学测量(图4一)、右心室肥大及功能减退的测定(图4 b, c).慢性缺氧导致两种基因型肺小血管肌肉化程度的相似增加(图4 d)。
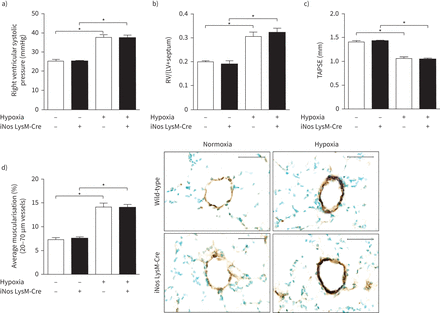
骨髓细胞特异性诱导型一氧化氮合酶(iNOS)敲除不能保护小鼠对抗缺氧诱导的肺动脉高压(PH)。小鼠分别暴露在常压常氧或缺氧环境中28天。a)右心室收缩压(n= 6-8)。b)右心室结构改变,表现为右心室(RV)与左室+室间隔(LV+室间隔)质量之比(n= 6-8)。c)超声心动图通过三尖瓣环形平面收缩偏移(TAPSE)评估右心室收缩功能(n= 6-8)。d)肺小血管(20-70 μ m外径)的重塑,表现为慢性缺氧暴露后总血管计数的平均肌肉化(n= 6-8),以及暴露于慢性缺氧和各自常氧对照的小鼠肺切片的代表性图像,抗α-平滑肌动蛋白(紫色)和血管性血友病因子(棕色)共染色。比例尺=50µm。数据以均数±表示扫描电镜.*: p < 0.05。双向方差分析(与Tukey的多重比较事后采用Test)进行统计分析。
烟草烟雾提取物治疗M2巨噬细胞可促进共培养肺动脉平滑肌细胞的增殖和迁移,并以inos依赖的方式进行
基于烟雾暴露后肺间质巨噬细胞的显著inos依赖变化和表型,我们研究了小鼠原发性肺动脉平滑肌细胞(PASMCs)与骨髓来源和香烟烟雾提取物(CSE)刺激的巨噬细胞共培养的增殖。图5一个)。对于治疗,我们选择了即使长时间暴露也不影响巨噬细胞活力/代谢活动的CSE浓度(补充图S4a)。M2预处理,M1或naïve与CSE共培养6 h和24 h后PASMCs增殖增加(图5一个)。有趣的是,M2巨噬细胞中iNOS的删除消除了cse依赖性的PASMC增殖的增加(图5一个)。此外,这种效应依赖于共培养细胞类型之间的交流,因为当pasmc未共培养,但用巨噬细胞培养条件培养基(CM)处理时,这种交流是不存在的(补充图S4b)。

诱导型一氧化氮合酶(iNOS)在巨噬细胞中的活性通过激活细胞外信号调节激酶(ERK)途径增加邻近肺动脉平滑肌细胞(PASMCs)的增殖。PASMCs与骨髓来源的naïve、M1-或m2极化巨噬细胞共同培养,用1%香烟烟雾提取物(CSE)预处理3小时。a)骨髓来源的巨噬细胞与PASMCs共培养的实验装置示意图,以及PASMCs在与巨噬细胞共培养后6小时(n=4)和24小时(n=5)标准化以控制PASMCs后的增殖情况,通过BrdU试验进行评估。数据给出了与非cse暴露对照共培养的差异。b)与CSE-和/或L-NIL处理的M2细胞共培养1小时和3小时的PASMCs ERK磷酸化的代表性照片和Western blot分析(n=5)。数据以磷酸化和总ERK之间的比例给出,并标准化以控制共培养(未暴露于CSE和未处理的L-NIL)。上图所示的pasmc染色为磷酸化ERK(绿色),α-平滑肌肌动蛋白(SMA)(红色),并用Hoechst反染色(蓝色)。比例尺=50µm。c)用ERK抑制剂SCH772984和条件培养基(CM)处理的PASMCs与CSE-和/或L-NIL处理的M2巨噬细胞共培养,通过BrdU试验评估PASMCs的增殖情况。增殖与对照(用PASMC CM治疗)标准化。 Data are given as the difference between the effects of CM from CSE-exposed and unexposed co-cultures. d) Western blot analysis of the ERK phosphorylation in pulmonary vessels captured by laser microdissection from lungs of animals exposed to smoke for 3 months and respective unexposed controls (n=3). Data are given as the ratio between phosphorylated and total ERK, standardised to control (unexposed wild-type). Data are presented as mean±扫描电镜.*: p < 0.05。双向方差分析(与Sidak的多重比较事后测试在体外和Tukey的多重比较事后测试在活的有机体内实验)进行统计分析。
此外,我们还通过单培养与共培养CM处理PASMCs,研究了巨噬细胞与PASMCs的交叉对话对PASMCs迁移和凋亡的影响。如在共同培养的情况下(图5一个)或共培养CM处理的PASMCs,共培养CM对PASMCs的促迁移作用通过CSE处理M2巨噬细胞而增强,并通过抑制这些细胞中的iNOS而消除(补充图S5a)。然而,共培养的CM降低了PASMC凋亡,这种效果不依赖于sse对M2巨噬细胞的处理,也不依赖于这些细胞中iNOS的活性(补充图S5b)。
IL-4和丝裂原活化蛋白激酶信号通路可能促进了共同培养中cse诱导的inos依赖性PASMCs增殖
接下来,我们研究了共培养的M2巨噬细胞触发PASMCs增殖的机制。这些研究发现共培养后,PASMCs早期时间点(1小时、3小时和6小时)细胞外信号调节激酶(ERK)磷酸化增加。在L-NIL处理吞噬细胞后,这种磷酸化erk的增加是不存在的(图5 b而且补充图S6a)。ERK磷酸化的模式类似于PASMC的增殖,这一事实表明ERK磷酸化在cse - m2 -巨噬细胞驱动的增殖中起着功能性作用。为了证明这一建议,我们研究了共培养CM时ERK抑制是否影响PASMC增殖。事实上,ERK1/2抑制剂SCH772984抑制了由PASMC- m2 (cse预处理)巨噬细胞共培养的CM诱导的PASMC增殖的增加(图5 c而且补充图S6c)。这种效果与iNOS抑制的效果相似(图5 c)。关键是,这些结果与我们的研究结果一致在活的有机体内数据显示,吸烟会增加肺血管中的ERK1/2磷酸化(图5 d)和肺匀浆(补充图S6c),而不是击倒动物。
此外,共培养培养基中的细胞因子分析显示,当用L-NIL处理PASMCs与M2巨噬细胞共培养时,IL-4浓度降低(图6, b).在功能上,重组IL-4在PASMCs与M2巨噬细胞共培养中的应用逆转了由l - il处理M2巨噬细胞引起的sse诱导增殖的减少(图6 c)。
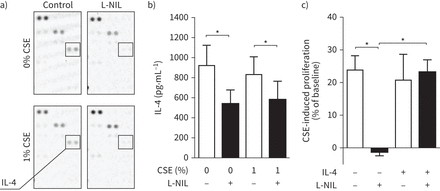
白细胞介素(IL)-4与香烟烟雾提取物(CSE)诱导的M2巨噬细胞和肺动脉平滑肌细胞(PASMCs)共培养促增殖信号有关。M2巨噬细胞与PASMCs共培养条件培养基中的IL-4,通过a)细胞因子阵列和b) ELISA法评估(n=7)。c)在与对照或l - nil处理的M2巨噬细胞共培养时,在共培养基中不存在或不存在IL-4的情况下,csc诱导PASMCs增殖(n=6)。用BrdU法评估增殖,并与对照pasmc标准化。数据给出了与非cse暴露对照共培养的差异。数据以均数±表示扫描电镜.*: p < 0.05。双向方差分析(与Sidak的多重比较事后采用Test)进行统计分析。
伊诺+和CD206+COPD患者肺中巨噬细胞聚集在重构血管附近
为了研究上述发现与人类COPD的相关性,我们对人类肺切片进行了巨噬细胞标记物CD68和iNOS的双染色,发现与供体相比,COPD患者在重构血管附近表达iNOS的巨噬细胞数量增加(图7)。此外,我们对人类COPD肺进行CD68和CD206染色,CD206是一种由交替激活的m2样巨噬细胞高度表达的蛋白质,并在COPD肺重构血管周围发现大量双阳性细胞(图7 b)。与我们在小鼠肺中的发现相似,与健康供体对照相比,免疫组化染色在COPD肺的血管基质中定位了磷酸化ERK1/2的高表达(图7 c)。Western blot分析也证实COPD患者肺中ERK1/2磷酸化水平较健康供体增加(图7 d)。这些数据支持了我们关于M2巨噬细胞在肺血管重塑中的重要作用的结论。

诱导型一氧化氮合酶(iNOS)+和CD206+COPD患者肺中巨噬细胞聚集在重构血管附近。供体和COPD患者肺切片的代表性图像。a) iNOS(绿色)和CD68(红色)共染色。箭头:CD68+伊诺+细胞。b) CD68(上红色)、CD206(下红色)连续切片染色。箭头:CD68+CD206+细胞;#: CD68+CD206−细胞。c)磷酸化细胞外信号调节激酶(ERK)染色(棕红色)。箭头:正信号。比例尺=50µm。d) Western blot检测COPD患者和供体肺匀浆中ERK磷酸化水平(每组n=10)。数据以磷酸化和总ERK之间的比率给出,并标准化以供对照(供体组)。数据以均数±表示扫描电镜.*: p < 0.05。统计学分析采用t检验。
讨论
我们的研究表明,骨髓细胞特异性的iNOS缺失1)可以阻止烟雾诱导PH的发展,但不能阻止肺气肿的发展;2)相反,它不能防止缺氧诱导的ph。骨髓细胞特异性的iNOS缺失抵消了烟雾暴露肺部间质巨噬细胞M2极化标记物(CD206)表达的增加。此外,我们还提供了以下证据:4)m2极化巨噬细胞和PASMCs之间的inos依赖的串话,在香烟烟雾诱导的PH中驱动PASMCs的增殖;5)磷酸化erk和IL-4参与下游信号传导过程(图8)。与我们的小鼠模型中类似的m2样,含inos的巨噬细胞招募发生在人类COPD肺靠近肺血管的地方。
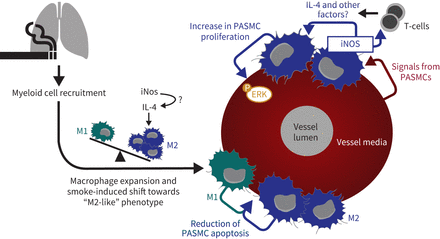
COPD烟致血管重构病理机制的研究方案。M2巨噬细胞与肺动脉平滑肌细胞(PASMC)之间的通信,导致细胞外信号调节激酶(ERK)激活和PASMC增殖,通过巨噬细胞的烟雾暴露而增强。这取决于这些细胞中诱导型一氧化氮合酶(iNOS)的表达和血管环境中白细胞介素(IL)-4的浓度。
由于肺血管重塑是COPD病理的早期标志[9,有人假设这种重塑可以促进甚至驱动肺气肿的发展。然而,在烟雾暴露后骨髓细胞特异性iNOS敲除动物中肺气肿的存在支持了我们之前的发现,PH值和实质破坏可以独立发生[5]。然而,有可能是血管系统中的分子改变(而不是重塑)本身)正在推动实质的破坏。这种情况可以解释在没有肺血管重塑但由血管分子改变(共同)驱动的情况下肺气肿的发展,因为主要在肺血管室中的iNOS上调导致实质破坏[5]。
尽管炎症细胞及其介质在其他PH形式中的关键作用已被啮齿动物模型和患者的实验证据充分证实[21- - - - - -23],只有在引言中提到的少数研究解决了炎症在COPD-PH中的作用。然而,据我们所知,我们的研究首次将巨噬细胞作为炎症细胞类型,以inos依赖的方式驱动烟雾诱导的PASMC增殖和随之而来的肺血管重塑。我们使用了一种靶向所有骨髓细胞的驱动系,其特异性超越了以前骨髓移植实验的研究,并排除了这些先前实验方法的缺点,如辐射和重建的影响[5,27- - - - - -29]。我们的实验揭示了巨噬细胞似乎是髓系细胞谱系中最重要的候选者,1)因为iNOS在这些细胞中发挥着重要作用[30.,31];2)因为它们在烟雾暴露时存在于肺血管中;3)由于它们对PASMC增殖的影响在体外实验。有趣的是,我们证明了巨噬细胞和pasmc之间存在着一种交叉对话,这种对话可以通过抑制或删除M2巨噬细胞来源的iNOS来阻止这些血管细胞的增殖。与我们的发现一致,最近的一项研究报告了M2巨噬细胞和PASMCs之间的双向通信,这种通信依赖于CCR2和CCR5,并驱动PASMCs的增殖在体外特发性肺动脉高压、动物缺氧和SU5416/缺氧PH模型肺血管重塑在活的有机体内[22]。然而,尚无第3组PH的数据[16];在第3组ph中,iNOS在这些细胞中的作用也没有得到阐述。尽管我们的数据支持M2巨噬细胞来源的信号是肺血管重塑的重要贡献者这一概念,但我们在CCR2和CCR5水平上没有发现烟雾或iNOS依赖性的变化(未发表的观察结果),这表明在COPD-PH中确实存在独特的血管分子特征,正如我们实验室之前提出的那样[5]。这一结论得到了我们的研究结果的支持,即骨髓细胞中iNOS的缺失可以防止吸烟诱导的PH,但不能防止缺氧诱导的PH(详细讨论请参阅补充材料)。
烟雾暴露时,骨髓细胞特异性iNOS缺失对肺炎症细胞的组成和表型有两个重要影响,可能会放大在活的有机体内在共培养体系中观察到iNOS抑制对M2细胞的抗增殖作用。
首先,在我们的敲除小鼠中没有观察到吸烟诱导的间质巨噬细胞CD206表达的增加,这表明对吸烟诱导的向m2样表型转移的保护作用。这是更有趣的,因为iNOS被认为是M1极化的经典标志。然而,最近和部分矛盾的报道表明,这种多效酶可以影响表达巨噬细胞的极化。V一个窝Bosscheet al。[32]报道了iNOS的表达可以阻止巨噬细胞从M1向M2表型的再极化,可能是通过抑制氧化磷酸化作用。相反,我uet al。[33]表明,骨髓细胞来源的iNOS抑制M1巨噬细胞的极化,并推测它支持这些细胞的去分化和表型可塑性,而不影响M2巨噬细胞种群。虽然iNOS是巨噬细胞基因表达和表型的调节剂的概念可以很容易地应用于我们的发现,但所描述的场景都不能完全解释在共培养中,骨髓细胞特异性iNOS敲除对吸烟诱导的PH或WT和iNOS缺陷M2巨噬细胞对PASMC增殖的差异吸烟诱导效应的保护。由于巨噬细胞表型的动态性质,很可能是微环境因素,如烟雾和来自pasmc的信号在体外,以及动物的年龄,肺上皮细胞的凋亡和其他炎症细胞类型和介质的存在在活的有机体内有助于在我们的模型中观察到的巨噬细胞在烟雾暴露时的特定行为。
骨髓细胞特异性iNOS缺失对炎症细胞浸润成分的第二个重要后果是肺免疫细胞群体中t细胞的保留比例(在补充材料)。
在潜在的分子机制方面,我们的研究结果表明iNOS在巨噬细胞极化、M2巨噬细胞对烟雾的反应以及PASMCs和M2巨噬细胞之间的促增殖通信中起调节作用。由于在M1细胞中未观察到这种效应,从机制上讲,它们可能不是由大量一氧化氮的产生和随之而来的亚硝化应激引起的,而是由蛋白质硝化作为翻译后修饰的调节作用引起的。在自然杀伤细胞和t细胞功能成熟过程中,IL-12受体下游的Janus酪氨酸激酶-2和视黄酸受体相关孤儿受体-γT的调控也分别观察到这种情况[34,35]。在我们的吸烟挑战M2巨噬细胞中观察到的inos依赖性调节的特异性,进一步得到了以下发现的支持:在共培养中,该酶的删除消除了它们对PASMCs的吸烟诱导的增殖和亲迁移信号,但不影响来自这种共培养的条件培养基对PASMCs施加的抗凋亡作用。有趣的是,从我们的在体外在实验中,iNOS在M2细胞促增殖信号传导中的调节作用在特定条件下是独特的,其中包括巨噬细胞暴露于烟雾和巨噬细胞与pasmc之间的通信。重要的是,人类肺部免疫组化染色提示COPD也存在类似的情况,COPD患者肺中聚集在重构血管附近的巨噬细胞中iNOS和CD206呈阳性。
针对inos依赖的信号事件,我们的实验显示,在与M2巨噬细胞共培养的PASMCs和烟雾暴露的WT动物的肺中,ERK信号上调。在我们的共培养模型中,在PASMCs中抑制这一途径成功地抵消了M2巨噬细胞的促增殖信号。我们证明了其与人类COPD的相关性,因为我们对磷酸化ERK的免疫组化分析显示,位于COPD患者肺部重构血管中的ERK显著上调。尽管先前在其他解剖病变的背景下研究了COPD肺中该通路的激活[36,37],据我们所知,我们的研究首次为ERK通路参与人类COPD肺血管介质重塑提供了证据。综上所述,这些结果表明ERK信号通路与其他形式的PH有关[38- - - - - -40],可能对烟雾诱导的血管重塑也很重要,并由iNOS驱动。此外,我们的结果表明,ERK通路的激活和随之而来的PASMC增殖是接触烟雾挑战M2巨噬细胞后的早期事件。
此外,我们的研究结果表明,IL-4是控制M2巨噬细胞对CSE刺激反应的因素,从而调节它们对PASMCs的促增殖信号。沿着这些线,K奥马尔et al。[41]表明IL-4与IL-13协同作用,在转化生长因子(TGF)-β-介导的过程中发挥关键的致病作用曼氏裂体吸虫-诱发肺动脉高压。根据我们的发现(数据未给出),作者没有发现总TGF-β水平升高;相反,他们提出M2巨噬细胞接收IL-4/IL-13信号激活了这种生长因子的潜在形式通过一种尚未被阐明的机制。有趣的是,TGF-β通过激活ERK通路诱导血管平滑肌细胞增殖[42,43]。但iNOS与IL-4之间的关系有待进一步研究。由于iNOS可促进糖酵解[32,44]乳酸刺激IL-4和IL-13的产生[45],可以想象,代谢变化是在cse诱导的PASMC增殖中连接这两个重要角色的缺失环节。
结论
我们的数据表明,骨髓细胞中iNOS的缺失可以保护小鼠免受吸烟诱导的PH的影响;M2巨噬细胞是最可能的骨髓细胞系候选者,在烟雾暴露后驱动肺血管重塑;M2巨噬细胞和PASMCs之间的交叉对话是这方面的一个必要过程。依赖于iNOS的ERK-和il -4信号已被确定为机械性下游信号(图8)。与小鼠模型中相似的过程也发生在人类COPD中。我们的数据进一步支持了肺血管重塑和肺气肿可以独立发生的概念,但不排除血管细胞类型的分子改变可以驱动实质破坏。
补充材料
可共享的PDF
确认
作者感谢Karin Quanz, Ewa Bieniek, Ingrid Breitenborn-Müller, Nils Schupp, Carmen Homberger, Susanne Lich, Miriam Wessendorf和Elisabeth Kappes (justus - liebig大学,吉森,德国)的技术援助。
脚注
这篇文章有补充资料可从www.qdcxjkg.com
利益冲突:M.格雷迪克没有什么可透露的。
利益冲突:C-Y。吴没有什么可透露的。
利益冲突:S. Hadzic没有什么可透露的。
利益冲突:O. Pak没有什么可透露的。
利益冲突:R. Savai没有什么可透露的。
利益冲突:B. Kojonazarov没有什么可透露的。
利益冲突:S. Doswada没有什么可透露的。
利益冲突:A. Weiss没有什么可透露的。
利益冲突:A.韦格特没有什么可透露的。
利益冲突:A. Guenther没有什么可透露的。
利益冲突:r。p。布兰德斯没有什么可透露的。
利益冲突:R.T. Schermuly没有什么可透露的。
利益冲突:f·格里明格没有什么可透露的。
利益冲突:W. Seeger报告了Actelion、拜耳公司、诺华公司、Vectura、Medspray和联合治疗公司在提交工作之外的个人费用。
利益冲突:N. Sommer报告来自Actelion的个人费用,在提交的工作之外。
利益冲突:S. Kraut没有什么可透露的。
利益冲突:N. Weissmann在研究期间报告了德国研究基金会的资助;并拥有一项用于COPD患者肺再生的L-NIL专利(EP2591777A2)。
支持声明:这项工作由德国研究基金会(DFG)资助-项目编号268555672 - SFB 1213, A07给N. Weissmann, CP02给B. Kojonazarov。本文的资助信息已存入交叉参考基金注册.
- 收到了2020年3月19日。
- 接受2021年8月3日。
- 版权所有©作者2022。
本版本根据知识共享署名非商业许可4.0的条款发布。为商业复制权利和权限联系权限在}{ersnet.org











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}