





摘要
哮喘患者气道上皮差异基因表达的研究与几项研究报告了对照。然而,对于哪些基因在哮喘中可重复受到影响尚无共识。我们试图使用荟萃分析方法确定差异表达基因(DEGs)的共识列表。
我们确定了8项研究,其数据符合定义的纳入标准。这些研究包括355例病例和193例对照,包括支气管或鼻上皮取样。我们进行了研究水平的分析,然后进行了荟萃分析。同样,我们将荟萃分析框架应用于研究水平通路富集的结果。
我们确定了1273个deg,其中431个在以前的研究中未被确定。450个deg表现出很大的效应量,并且在研究年龄、性别、种族/民族、药物使用、吸烟状况和病情恶化等人群差异方面具有稳健性。这450个基因的差异表达量在支气管和鼻气道上皮中高度相似。通路富集的荟萃分析揭示了一些持续失调的生物学通路,包括假定的转录和转录后调控。
总的来说,我们确定了一组在哮喘中持续失调的基因,这些基因与已知和新的生物学途径有关,并将为哮喘亚型鉴定提供信息。
摘要
超过1200个基因在哮喘患者的气道上皮中持续受到影响http://ow.ly/vxUt30k0tjf
介绍
多项证据表明,气道上皮在哮喘发病中起重要作用。气道上皮分泌的模式识别受体、细胞因子和警报器在过敏性炎症的发生中起关键作用[1]。全基因组关联研究(GWAS)的结果也涉及气道上皮表达的基因,例如IL33和TSLP[2].含有蛋白酶的过敏原(如。屋尘螨)破坏上皮屏障的完整性,有证据表明哮喘患者的屏障功能下降[3.]。最后,哮喘的标志性表型粘液分泌过多是由气道和粘膜下腺分泌细胞的功能和活性变化引起的[4]。
气道上皮的转录组学研究提供了疾病发作后该组织状态的快照。Woodruff等.[5]首次报道了22个基因在哮喘病例和对照组之间的差异表达。这个小组后来发现,只有三个基因的表达(POSTN,CLCA1和SERPINB1)区分“t辅助细胞2 (Th2)高”和“Th2低”哮喘亚型,并与吸入皮质类固醇的反应有关[6]。其他研究表明,基因表达的变化可以通过上皮甲基组的变化来反映[7,8不同的基因组与哮喘恶化有关[9,10]。因此,气道上皮不仅可能在疾病发病机制中起关键作用,而且还可以提供疾病状态的信息(即。恶化)和治疗反应。
自从对哮喘上皮细胞中基因表达的初步研究以来,更多的研究随之而来,包括更大样本量和不同人口统计学的研究。虽然这些最近的研究有助于我们理解哮喘中的上皮反应,但除了少数例外,对于哪些基因和途径在哮喘中可重复受到影响尚无共识。因此,我们开始利用现有的基因表达数据集来确定哪些基因在哮喘病例的气道上皮中持续失调与对照采用元分析方法。一些研究使用鼻上皮代替支气管上皮取样,使我们能够评估鼻上皮和支气管上皮结果的一致性。我们还在类似的荟萃分析框架中使用哮喘病例对照基因表达数据来评估生物学途径的富集。
方法
有关方法的更多信息可在补充材料.
研究鉴定
病例对照研究在PubMed、国家生物技术信息中心的基因表达综合数据库(GEO)和欧洲分子生物学实验室中得到确认-欧洲生物信息学研究所的ArrayExpress使用搜索词包括“哮喘”、“气道上皮”、“支气管上皮”和“鼻上皮”(或其组合)。我们总共确定了18项研究(补充表S1).一些出版物包含重叠的研究人群;因此,我们从每组中选择一项研究以避免重复(补充材料详情)。
在回顾研究之前,我们指定了一套研究排除标准。这些都是1)缺乏案例定义;2)任何研究组(病例或对照组)的样本量少于5人,这将限制功效;或3)基因表达或协变量(年龄、性别和吸烟)数据不可用。协变量数据是通过数据存储库或通过与研究调查人员接触获得的。应用上述排除标准排除了6项研究(列于补充表S1).在这些排除(并去除重复)之后,我们的荟萃分析包括8项研究,包括548名受试者(355例和193例对照)。所有八项研究都使用毛刷取样气道上皮:两项研究取样鼻上皮,其余研究取样支气管。除了一项研究外,所有研究都使用了微阵列平台。每项研究的描述载于表1.
数据处理、规范化和注释
数据采集、规范化、研究水平分析和元分析的总体工作流程见补充图S1.数据来自GEO或直接来自研究研究者。Affymetrix阵列数据使用鲁棒多阵列平均进行归一化[11]和Agilent阵列数据进行分位数归一化,然后进行log2变换(补充表S2).来自单个rna测序数据集(GSE85568)的规范化读取计数也进行了log2转换。在大多数情况下,我们使用GEO提供的与所使用的阵列平台相对应的阵列探针-基因注释(补充表S2).
学习水平分析
为了识别每个研究中的异常样本,我们使用了Bolstad的相对对数表达法[12]。然后,我们使用LIMMA分析识别差异表达基因(deg) [13],至少根据年龄、性别和种族进行调整,但在某些情况下也会根据吸烟状况和阵列杂交批次(表1).对于每项研究,使用错误发现率(FDR)校正方法调整p值[14],我们记录了t统计量、beta值、p值、对数折叠变化、Cohen's D和Hedges' s调整后的g用于元分析。
荟萃分析
我们使用R倡导的逆方差模型估计每个基因的总效应大小amasamy等.[15]。具体来说,我们使用Hedges调整后的g来计算合并效应大小,并使用随机效应模型来解释研究间的异质性。该分析是使用R包meta [16]。如果荟萃分析经fdr校正的p值(q值)<0.05,且该基因在8项研究中至少有2项被评估,则我们宣布基因为差异表达(DE)。
结果
学习水平分析
我们从8项研究中获得了355例病例和193例对照的基因表达和协变量数据(表1).不同研究的人口学变量差异很大。例如,两项研究只包括儿童,而其余的研究主要包括年龄范围很广的成年人(20-74岁)。在所有研究中,女性受试者的百分比从40%到70%不等。虽然大多数研究对象是白种人,但也有一些研究涉及非洲裔美国人、西班牙裔美国人、亚洲人或其他种族/族裔类别。除了两项研究外,大多数研究只涉及不吸烟的受试者。
对于每项研究,我们在排除异常值(补充图S2).由于不同的研究使用不同的阵列平台(或rna测序),测试的基因数量不同,我们通过绘制鉴定为DE的基因比例(补充图S3,补充表S2).这些值的范围为0.01% ~ 27%,样本量与检测到的DEGs分数之间没有明显的相关性。
荟萃分析
然后,我们采用了荟萃分析方法,使用随机效应模型来考虑研究间效应大小的异质性。我们鉴定出1273个DE基因,其q值<0.05 (图1,补充表S3),其中大多数(64%)表达上调。值得注意的是,超过三分之一的deg (n= 431,34%)在任何一项研究中都没有被确定为DE(在FDR q<0.05时),这突出了该方法在揭示感兴趣基因方面的实用性。就效应大小而言,CST1(调节)和APELA(下调)是最极端的基因;在统计显著性方面,CEACAM5(调节)和C3(下调)是受影响最大的基因。
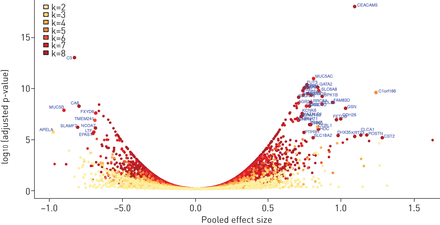
通过荟萃分析鉴定差异表达基因。基因以研究的数量(k)和调整后p值<1×10进行颜色编码−5和合并效应大小>0.7标记。
不出所料,三个Th2生物标记基因(POSTN,CLCA1和SERPINB1)一直被上调,尽管不同研究的效应大小存在一些异质性(图2).同样,与黏液产生呈正相关的基因(如使用IL13,FOXA3和MUC5AC),而MUC5B和FOXA2表达下调。内质网应激相关基因(例如AGR2和ERN1),这与黏液蛋白分泌过多有关[22,23],但也被上调了XBP1不是(补充图4).
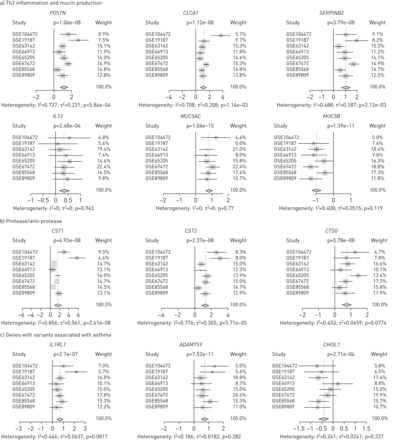
标志基因的差异基因表达研究。森林图a)与th2介导炎症相关的基因(POSTN,CLCA1和SERPINB1)和粘蛋白的产生(使用IL13,MUC5AC和MUC5B);B)蛋白酶-抗蛋白酶平衡(CST1,CST2和CTSG);c)与哮喘相关的基因变异(IL1RL1,ADAMTS9和CHI3L1).白胡须(用于CST1)表示置信区间完全包含在表示点估计的方框内。提供了额外的森林地块补充图4.
我们注意到参与蛋白酶/抗蛋白酶途径的基因明显富集,包括丝氨酸蛋白酶抑制剂(SERPINB2,SERPINB4,SERPINB8和SERPINB10)、组织蛋白酶(CTSC和CTSG被上调了,而CTSL下调)和胱抑素(CST1,CST2,CST3,CST4,CST6和CSTA) (图2,补充图4).相比之下,参与屏障功能的基因(包括CLDN1,CLDN18,CLDN4,TJP1,TJP2,TJP3,OCLN和背景)都不是DE,除了TJP1(ZO1).6个反映募集白细胞的基因是DE,包括CCL26和CLC(嗜酸性粒细胞标记)和TPSAB1,CPA3,MS4A2和HDC(肥大细胞及/或嗜碱性细胞的标记物)(补充图4).
17个与GWAS相关的基因在deg列表中(补充表S4).但总体而言,没有过度代表监管不严格与在这个列表中下调的基因,在效应大小方面表现出最大差异的五个基因(LRRC8A,ALOX15,IL18R1,IL1RL1,ADAMTS9)都被上调了。相反,三种HLA基因(HLA-DOA,HLA-DPA1,HLA-DRA)被下调。令人惊讶的是,我们发现哮喘相关基因的表达减少了CHI3L1[24]在案件中(图2).通过定位克隆鉴定的基因包括ADAM33,DPP10,PCDH1,HLA-G和SPINK5,不是DE,也不是CDHR3[25],这是一种与哮喘恶化相关的基因。
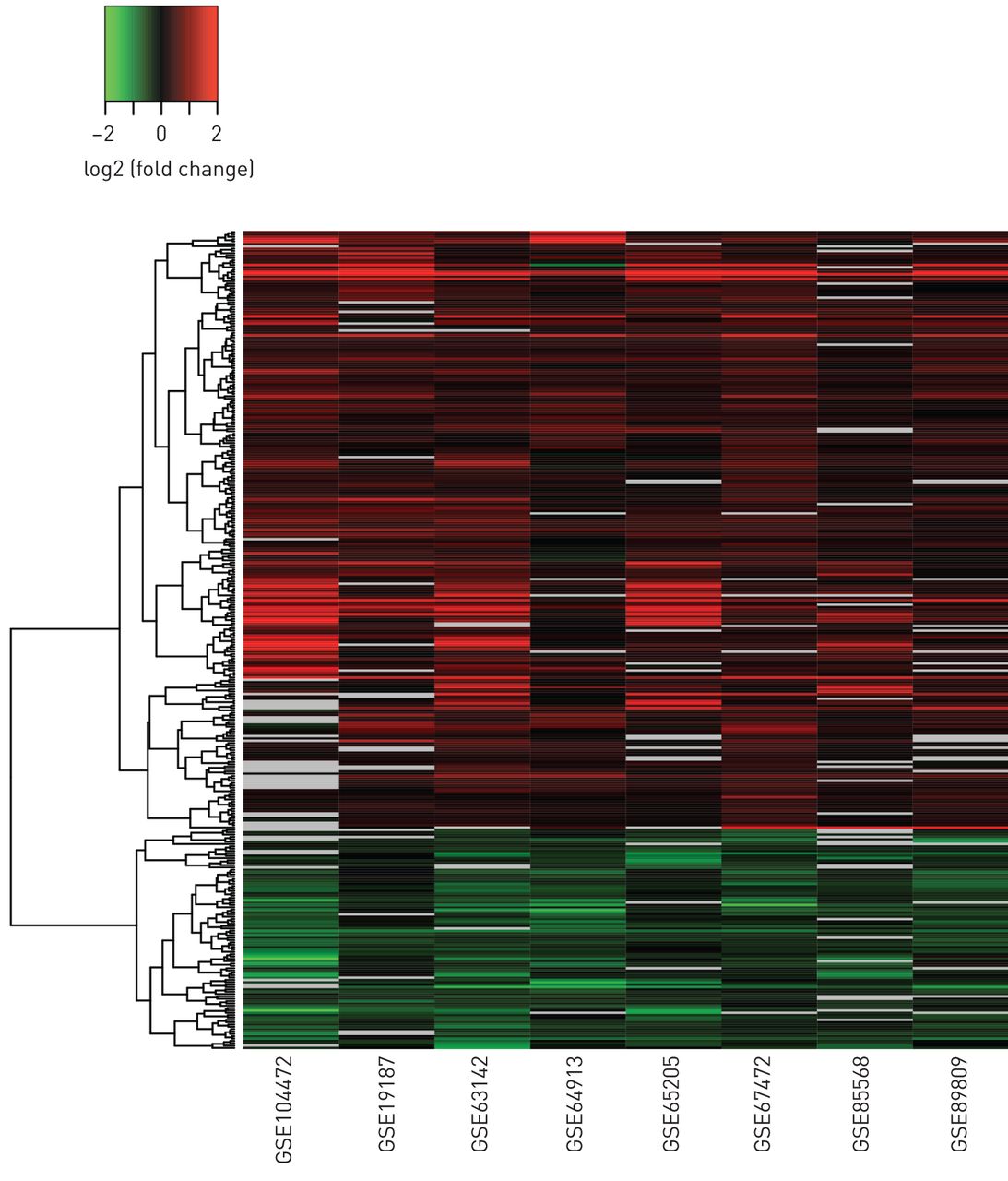
大量基因(n=450)在八项研究中的许多研究中一致被DE,并且具有较大的效应量,我们将其定义为汇集效应量(Hedges' g)值>0.5 (图3,补充表S5).这些发现表明,这些基因的差异表达对年龄、性别、吸烟状况、种族/民族、病情恶化和组织类型(支气管动脉粥样硬化)的差异具有很强的影响与鼻)。通过比较涉及支气管上皮的六项研究的合并效应大小,我们进一步研究了支气管上皮和鼻上皮之间差异基因表达的共性与两项涉及鼻上皮的研究的综合效应量(图4).该分析显示,这450个基因在不同组织类型之间的差异基因表达模式是完全一致的,并且大多数基因的差异表达幅度在鼻上皮中与支气管上皮相比仅略有下降(斜率=0.91,对于斜率=1的测试,p<0.01)CST1鼻上皮比支气管上皮更明显。
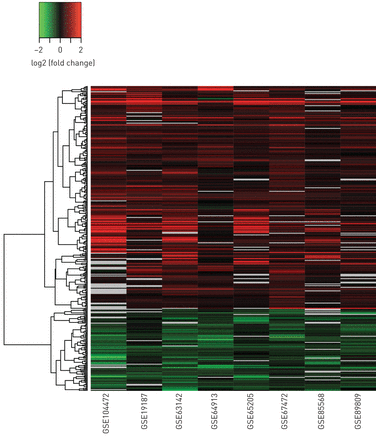
差异表达基因(DEGs)差异表达的一致性与大效应量。校正后的p值<0.05,合并效应大小为>.5。Log2 fold change值>2或<−2设置为2或−2。缺失的值以灰色显示。450个基因在不同的研究中被确定为某种程度的缺失。为了便于观察,这里显示了缺失最少的349个基因。提供了所有1273度的类似热图补充图S5.
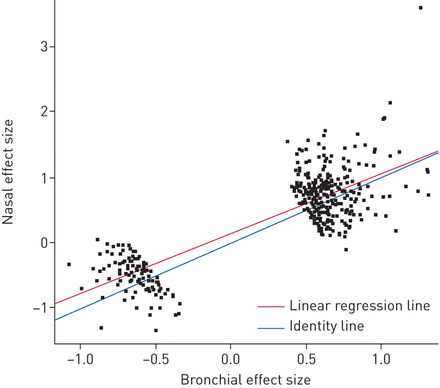
支气管组织差异表达的相似性与鼻上皮。图中显示了前450个差异表达基因的综合效应大小的比较,以及最佳拟合线性回归线(红色)和同一性线(蓝色;即。如果鼻=支气管)。右上方的异常基因是CST1.
非均质性
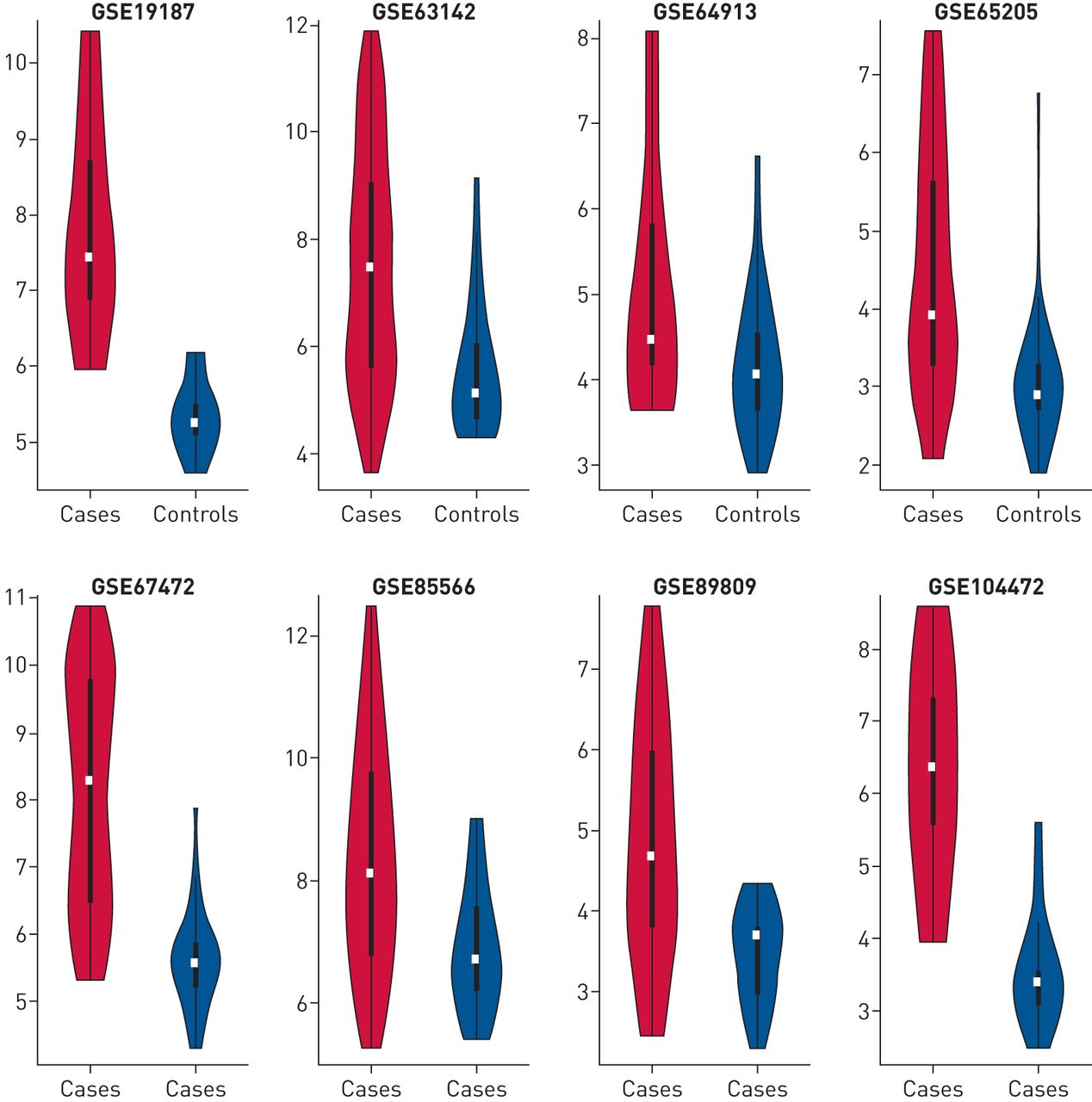
而哮喘病例差异基因表达信号的一致性与许多基因,特别是Th2标记基因的控制是明确的POSTN,CLCA1和SERPINB1,这种水平的分析模糊了潜在的异质性,可能为疾病机制和治疗提供信息[6]。为了检验这一点,我们绘制了总表达分数的分布POSTN,CLCA1和SERPINB1(图5).我们发现,虽然在平均情况下,这些基因的总体表达水平较高,但在每项研究中,分布范围很广,在某些情况下是双峰的(如。GSE67472)。此外,在一些研究中,有Th2基因表达低于对照组的病例(如。GSE63142)。
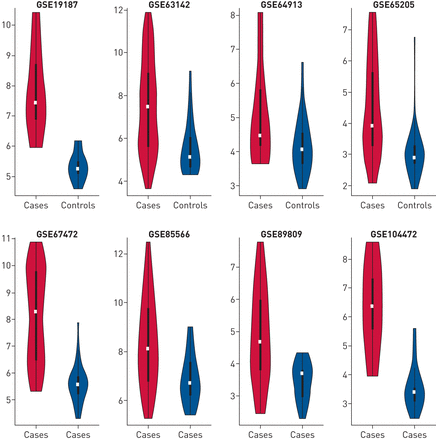
研究和样本间Th2标记基因表达的差异。基因表达的总和POSTN,CLCA1和SERPINB2(以log2为单位)绘制为每个研究中病例对照状态的函数。病例显示为红色,对照显示为蓝色。在这些小提琴图中,白点表示中位数,粗黑条对应于分布的第一到第三个四分位数,底部和顶部的须分别对应于第一个quartile-1.5×IQR和第三个四分位数+1.5×IQR,其中IQR=四分位数范围。
类固醇的作用
药物使用数据的缺乏妨碍了对吸入皮质类固醇(ICS)效果的系统分析。因此,我们将我们的deg列表与先前研究ICSs对气道上皮转录组的影响的研究进行了比较[5,26],总共鉴定出111个基因。这个ics应答基因列表包括15个哮喘基因,包括ALOX15B,POSTN,CLCA1,SERPINB1和CST1(补充表S6).
通路荟萃分析
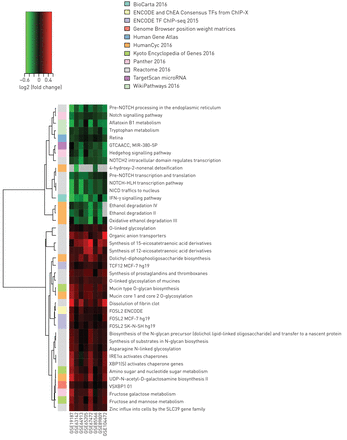
为了检验差异基因表达在生物通路水平上的影响,这可能揭示在单基因水平上不明显的其他疾病相关效应,我们对通路富集进行了荟萃分析。首先,我们使用GSVA方法进行了研究水平的通路分析[19]以检查特定基因组表达的变异(如。KEGG通路和其他预定义的基因集)在每个研究的病例和对照之间。在这项分析中,在单一研究分析或荟萃分析中未被称为DE的基因仍可能有助于途径富集。也就是说,由于GSVA的工作原理是将一组预定义的基因的表达分数结合起来,因此基因表达的适度差异(对于每个基因单独而言)不会达到统计显著性,但仍可能累积导致一组基因的差异表达。在单项研究分析之后,我们对所有8项研究的通路表达评分进行了荟萃分析,以确定持续改变的通路(图6,补充表S7,补充表S8).一些在病例中表达增加的最突出的途径与粘蛋白合成和粘蛋白的翻译后修饰有关。与前列腺素、血栓烷和二十碳四烯酸衍生物合成相关的通路也表达增加。在对照组中,干扰素γ信号、色氨酸代谢、NOTCH和Hedgehog信号是高表达的途径。此外,该分析确定TCF12、XBP1和FOSL2是上调基因的转录调节因子,miR-380-5p是对照组中表达更高的基因的转录后调节因子。我们还查询了三个蛋白质相互作用数据库,确定了335个哮喘deg (FDR q值<0.05)的推定蛋白质相互作用伙伴(补充表S9),其中大多数以前没有与哮喘联系在一起。
讨论
我们对8项研究的荟萃分析显示,1273个基因(约占人类基因组中蛋白质编码基因的5%)在哮喘患者的气道上皮中持续失调,其中超过三分之一的基因具有较大的效应量。我们的荟萃分析结果的稳健性在很大程度上是八项研究中差异基因表达相似性的函数;事实上,这八项研究使用了不同的表情测量平台,这使得结果特别有说服力。这种分析的部分用途是鉴定出在任何单一研究中都不突出的基因,这些基因占deg总数的34%。PPP1R3B就是这样一个例子。该基因在8项研究中的4项中名义上存在差异表达,但经过多次测试校正后,在任何一项研究中均未被称为DE。这个基因与脂质代谢有关[27]及阿尔茨海默病[28],但此前并未将其与哮喘联系起来。
我们的通路荟萃分析结果清楚地表明,哮喘中多种通路持续失调,尽管我们无法推断这些通路是否与疾病的发展有因果关系,还是疾病的结果。与th2介导的炎症、MUC5AC产生和粘蛋白翻译后修饰相关的基因在哮喘病例中一致且强烈上调。通路分析还涉及内质网应激(IRE1和XBP1通路),这是已知的粘蛋白产生过度的结果[22,23]。虽然XBP1被认为是哮喘deg的调节因子,XBP1基因表达在病例对照状态下没有差异,表明XBP1功能的激活可能独立于XBP1转录。与黏液蛋白基因表达升高相反,与NOTCH信号相关的基因在对照组中表达更高。总的来说,这些特殊的途径富集结果似乎表明黏液产生的改变和高脚杯的变化与气道纤毛细胞数。
在deg列表中,蛇形蛋白、胱抑素和组织蛋白酶基因家族的基因明显富集,突出了蛋白酶-抗蛋白酶失衡的作用及其后果,可能最突出的是细胞外基质重塑。先前的研究结果表明,一些蛋白酶的表达可能来自募集的白细胞,如肥大细胞胰蛋白酶和羧肽酶,以及中性粒细胞衍生的组织蛋白酶G和弹性酶。值得注意的是,这类蛋白酶的高表达也与气道黏液蛋白的生物物理特性改变有关,可能导致黏液堵塞[29]。胱抑素(蛋白酶抑制剂)的上调也可能表明暴露于含有蛋白酶的过敏原(如屋尘螨)后的代偿反应,正如嗜酸性慢性鼻窦炎的病例所表明的那样[30.]。然而,我们没有发现强有力的证据支持哮喘气道上皮存在屏障缺陷,正如人们根据蛋白酶对上皮完整性的已知影响所期望的那样[3.]。我们也没有发现类固醇对deg普遍影响的证据,但这一结果是基于对先前数据的有限分析,因此值得进行更有力的分析。
我们的主要发现之一是哮喘中的基因表达失调反映在鼻上皮中,其程度与支气管上皮高度相似。这一结果证实了早期基因表达相似性的发现[31]和DNA甲基化[8基于匹配样本的比较。由于我们的研究结果是基于跨研究的比较,我们得出结论,这种表达模式的共性可推广到多个研究人群。值得注意的是,这种差异表达的共性包括可能代表白细胞浸润的基因,例如CCL26,CLC,TPSAB1,CPA3,MS4A2和HDC).的高度表达CLC和HDC也见于变应性鼻炎[32]和嗜酸性食管炎[33](详情请参阅补充材料),提示这是跨组织嗜酸性粒细胞炎症的共同特征。
我们在哮喘患者的气道上皮中发现了17个与GWAS相关的基因。在解释这一结果时必须谨慎,因为基于物理接近的关联信号(在GWAS中)对基因的分配并不能证明因果关系是积极的。关于…IL1RL1和CHI3L1然而,其他研究有充分的证据表明,这两种基因与哮喘有因果关系。对于这两个基因,我们的荟萃分析表明,基因表达的方向性与病例状态之间存在明显差异,以及这些位点的遗传变异如何影响与疾病风险相关的基因表达。为IL1RL1,在哮喘中表达上调,但与哮喘相关的风险变异(以及血清IgE和外周血嗜酸性粒细胞计数)[34]与较低的基因表达有关[35]。这表明哮喘状态可能会导致IL1RL1气道上皮中的基因表达和/或招募的白细胞存在于哮喘气道上皮中表达IL1RL1.为CHI3L1编码YKL-40的基因,与哮喘风险增加相关的基因变异与血清YKL-40水平升高密切相关[24]和基因在全肺组织中的表达[36], YKL-40在严重哮喘患者的支气管上皮、巨噬细胞和中性粒细胞中表达[37]。然而,我们的研究结果表明,该基因在病例中的表达水平较低与整体控制。目前尚不清楚这种差异是否归因于气道细胞类型组成或疾病严重程度的变化,或者疾病发病前后气道上皮中基因表达的差异是否可能影响这种关系。最后,下游的遗传变异ALOX15都与哮喘有关[38而这个基因在哮喘患者中也显著且高度上调。该基因编码花生四烯酸15-脂氧合酶,该酶可将花生四烯酸转化为几种二十碳四烯酸(也在通路meta分析中发现),其中一些已被证明影响支气管上皮细胞的粘蛋白产生[39,40]。因此,表达和GWAS数据有助于支持该基因在哮喘中的作用的生物学上一致的论点。
虽然我们的结果清楚地表明相当数量的基因在哮喘中是DE,但这并不是说研究之间没有差异或哮喘患者之间没有异质性。的确,一些研究发现,与其他研究相比,deg的数量相对较多,这不仅仅是样本量的函数。这一结果表明,研究人群之间真正的生物学差异导致了观察到的基因表达信号的差异。似乎不同哮喘亚型的存在可能导致能量的增加或减少。例如,低th2哮喘病例的存在可以稀释病例与对照组之间th2相关基因的差异。或者,低th2病例可以提供检测非th2相关表达差异的能力。th2介导炎症的三基因标记的变异(POSTN,CLCA1和SERPINB1)的案例确实存在。至少在三项研究中,th2基因的总体表达呈双峰分布,一些哮喘患者的这些基因表达低于任何对照。确定这种哮喘亚型的遗传和临床相关性显然是当务之急。更广泛地说,利用基因表达数据来确定哮喘亚型(或内型)的生物学基础是一种很有前途的方法[6,41]。这种方法在很大程度上取决于研究对象数据(临床和/或生物标志物)的可用性,这些数据可用于评估与基因表达的相关性。哮喘患者气道中1273个持续失调基因的鉴定代表了未来这些分析的一个起点。
补充材料
补充材料
请注意:补充材料不是由编辑部编辑的,而是由作者提供的。
补充的方法erj - 01962 - 2017 - _supplement
图S1erj - 01962 - 2017 - _figures1
图S2erj - 01962 - 2017 - _figures2
图S3erj - 01962 - 2017 - _figures3
图S4erj - 01962 - 2017 - _figures4
图S5erj - 01962 - 2017 - _figures5
表S1erj - 01962 - 2017 - _table_s1
表S2erj - 01962 - 2017 - _table_s2
表S3erj - 01962 - 2017 - _table_s3
表S4erj - 01962 - 2017 - _table_s4
表S5erj - 01962 - 2017 - _table_s5
表S6erj - 01962 - 2017 - _table_s6
表S7erj - 01962 - 2017 - _table_s7
致谢
我们感谢所有将数据放入公共领域或直接向我们提供数据的研究人员,特别是Akul Singhania博士(英国伦敦弗朗西斯克里克研究所)和Timothy Hinks博士(英国牛津大学),他们尽早获得了他们的数据,以便我们能够进行这些分析。我们也感谢Lauren Donoghue (University of North Carolina, Chapel Hill, NC, USA)对手稿的批判性审查和对数据准备的协助。
脚注
本文的补充资料来自www.qdcxjkg.com
支持声明:这项工作得到了NIH拨款R01 HL122711和5P30CA01608的支持。本文的资助信息已存入交叉基金注册处.
利益冲突:没有声明。
- 收到了2017年9月26日
- 接受2018年3月30日。
- 版权所有©ERS 2018











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}