





文摘
哮喘的细胞和分子途径是高度复杂的。增加理解可以得到公正的转录组分析(RNA-Seq)。我们提出,整个人类支气管活检的转录组剖面不同哮喘患者和控制。
首先,我们调查的可行性获得RNA从支气管活检适合RNA-Seq整体。其次,我们对转录组配置文件之间的差异哮喘和控制。这个横断面研究比较四个前者过敏性哮喘患者和5个健康nonatopic控制。总RNA从四个每个主题是准备RNA-Seq活检。比较数字的读每个基因在哮喘和控制是基于泊松分布。
46个基因之间的差异表达哮喘和控制,包括pendrin periostin BCL2。10基因网络被发现参与细胞形态、运动和发展。
RNA分离从支气管活检适用于整个人类RNA-Seq,显示不同的转录组配置文件之间哮喘和控制。小说和确定的基因被发现与哮喘有关。这些结果表明,生物过程在哮喘患者的气道管理不同控制相比,这可能是相关疾病的发病机理和治疗。
文摘
转录组测序显示流程在哮喘患者的气道不同调控在转录组水平http://ow.ly/kDnln
介绍
哮喘是与航空公司包括变量的各种功能变化的气道阻塞(1),增加灵敏度和高架最大响应吸入刺激物(2和深吸气后bronchodilation减损3]。此外,结构性变化的过程称为通常在哮喘气道重构的发生。这包括增加气道平滑肌(ASM)质量,改变的细胞外基质蛋白沉积和ASM层外,杯状细胞和腺体增生和血管生成4]。因此,在哮喘气道的功能和结构特点是高度复杂的,阻碍发展的新的靶向治疗(5]。
在哮喘疾病活动似乎与气道炎症的模式和活动联系在一起。它已经表明,增加表达的辅助细胞(Th) 2-cytokines白介素(IL) 4和IL-13不仅促进嗜酸性粒细胞的炎症反应,同时也激活肥大细胞(6),microlocalisation这些ASM层中的肥大细胞主要是哮喘的病理生理学的关键因素7]。激活肥大细胞释放多种介质,从而导致增强ASM收缩和扩散6,8]。有趣的是,基因表达分析表明,Th2-cytokine表达的程度支气管活检,对于不同的哮喘病人气道重塑的相关方面和响应性吸入类固醇(9]。这表明有密切互动在哮喘炎症和重塑。
不一定是一种慢性气道重构的修复持续的炎症反应,传统上提出。最近的证据表明,可能是由气道重构的途径,在一定程度上独立的气道炎症10]。此外,气道结构组件本身可能驱动哮喘的病理生理过程11]。鉴于这种细胞和分子途径的复杂性,增加驱动机制的了解支气管哮喘最好来自客观的分析组织标本与认真的临床表现型(12]。
转录组分析航空公司的下一代(RNA-Seq)允许高通量测序和基因表达谱的详细描述在组织级别(13]。尤其是在复杂疾病如哮喘,这项技术有可能发现基因表达谱特征的疾病。据我们所知,没有直接的比较RNA-Seq概要文件从支气管哮喘和控制之间的活检。
我们提出,整个支气管活检的转录组剖面哮喘患者和健康对照组之间是不同的。因此,本研究的目的是:1)调查的可行性获得RNA从支气管活检适合RNA-Seq整个人类,和2)检查的差异转录组的整个人类之间过敏性支气管活检前者轻度哮喘患者和健康nonatopic控制。
方法
关于学习方法的附加信息包含在网上补充材料。
设计
这个横断面研究由两个访问。在访问1,受试者筛选资格参与按照纳入和排除标准。其次是肺功能测量,包括肺量测定法,用力呼气量在1 s (FEV1)可逆性和醋甲胆碱bronchoprovocation测试。在访问2,进行支气管镜检查支气管活检的集合。
主题
研究人群包括前者过敏性哮喘(n = 4)患者和健康nonatopic控制(n = 5)。哮喘患者控制疾病根据哮喘指南[全球倡议14),在6周内没有恶化之前参与。对照组没有历史或肺部疾病的症状。本研究的一部分研究在荷兰注册试验注册(在哮喘气道平滑肌层的作用;NTR1306)。
RNA-Seq和数据分析
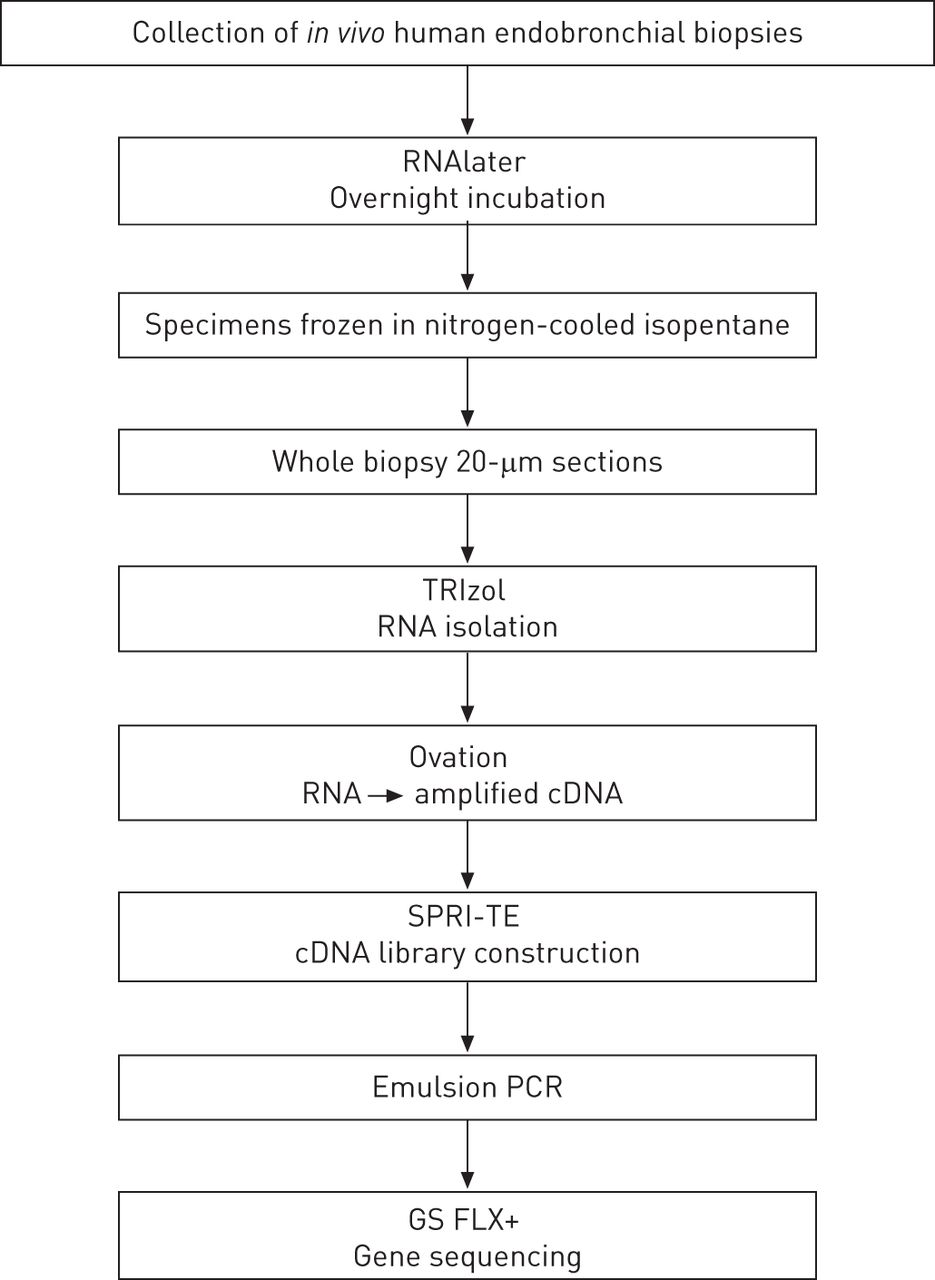
支气管活检每四个主题收集和冷冻过夜孵化后RNAlater (Venlo试剂盒,荷兰)。试剂盒(美国表达载体,卡尔斯巴德,CA)是用来隔离RNA。放大cDNA准备与喝彩RNA-Seq系统(美国NuGEN圣卡洛斯,CA),同时构建互补脱氧核糖核酸数据库使用SPRIworks片段库系统II(美国beckman coulter、沥青、钙)。Emulsion-based克隆扩增都使用了GS FLX钛emPCR工具包Lib-L(罗氏,潘茨堡,德国)准备丰富图书馆DNA测序的珠子GS FLX +系统(454;罗氏公司)。提出了一种严格的工作流程的概述图图1。

工作流的活检标本。RNAlater (Venlo试剂盒,荷兰);试剂盒(美国表达载体,卡尔斯巴德,CA);热烈欢迎RNA-Seq系统(美国NuGEN圣卡洛斯,CA);SPRI-TE核酸提取器(美国beckman coulter,沥青,CA);GS FLX +系统(罗氏,潘茨堡,德国)。
对人类基因组测序读被映射(hg19) (15)使用GS引用映射器(罗氏)。创新路径分析(异丙醇;智慧系统公司红木市,美国CA)被用来识别基因网络。异丙醇生成网络的分数,给组基因的可能性在这个网络可以解释的机会。网络得分≥2≥99%的信心,他们并不是偶然产生的。定量PCR (qPCR)进行验证测序数据。RNA-Seq数据上载到基因表达综合(GSE38994)。验证测序数据,qPCR进行选择哮喘和控制之间的差异表达基因。
统计分析
学习小组的主题特征比较使用未配对t或Mann-Whitney U-tests。哮喘之间比较读取每个基因的数量和控制是基于泊松分布修正读取的总数。科恩的影响大小计算量化读数字的大小不同个体基因之间哮喘和控制。Bonferroni调整为多个测试应用。的假定值< 0.05被认为是具有统计学意义。
结果
主题
表1节目的主题特征哮喘患者和健康对照组。肺功能特点主要是两组之间的比较。正如预期的那样,挑衅的乙酰甲胆碱的浓度导致FEV下降了20%1在哮喘病人相比,对照组低。
支气管活检标本
4∼沿支气管活检/主题1 - 2毫米3成功收集到的所有参与者。的总RNA分离从支气管活检标本每四个研究主题范围从900到9300 ng。
RNA-Seq
放大cDNA准备从50 ng孤立的RNA,和平均长度数量从6120年到9630年的250个基点。互补脱氧核糖核酸库平均长度为700个基点。互补脱氧核糖核酸分子的数量范围从0.25到1.58×109分子·μL−1。DNA珠子克隆扩增后16 - 24%浓缩的互补脱氧核糖核酸数据库使用乳液PCR用于测序。
中值读取长度与测序获得运行199个核苷酸最多867个核苷酸。读总数为349 999年239年和399年获得了哮喘和控制,分别。哮喘病人的87%可以映射到人类基因组hg19读取,这是控制为88%。
转录组映射到10 167个独特的基因控制哮喘和11 006 (图2)。中等长度的重叠群读的261个基点,在当前的研究中报道大约是24×[19]。多个测试校正后,46个基因的效应值0.5是哮喘和控制之间的差异表达(表2)。在参考基因被发现无显著差异,包括3 -磷酸甘油醛脱氢酶(GAPDH)β-actin (ACTB)β-2-microglobulin (B2M)和乳酸脱氢酶(LDHA)。
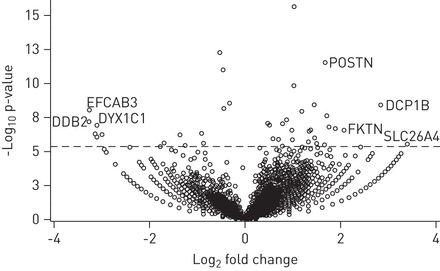
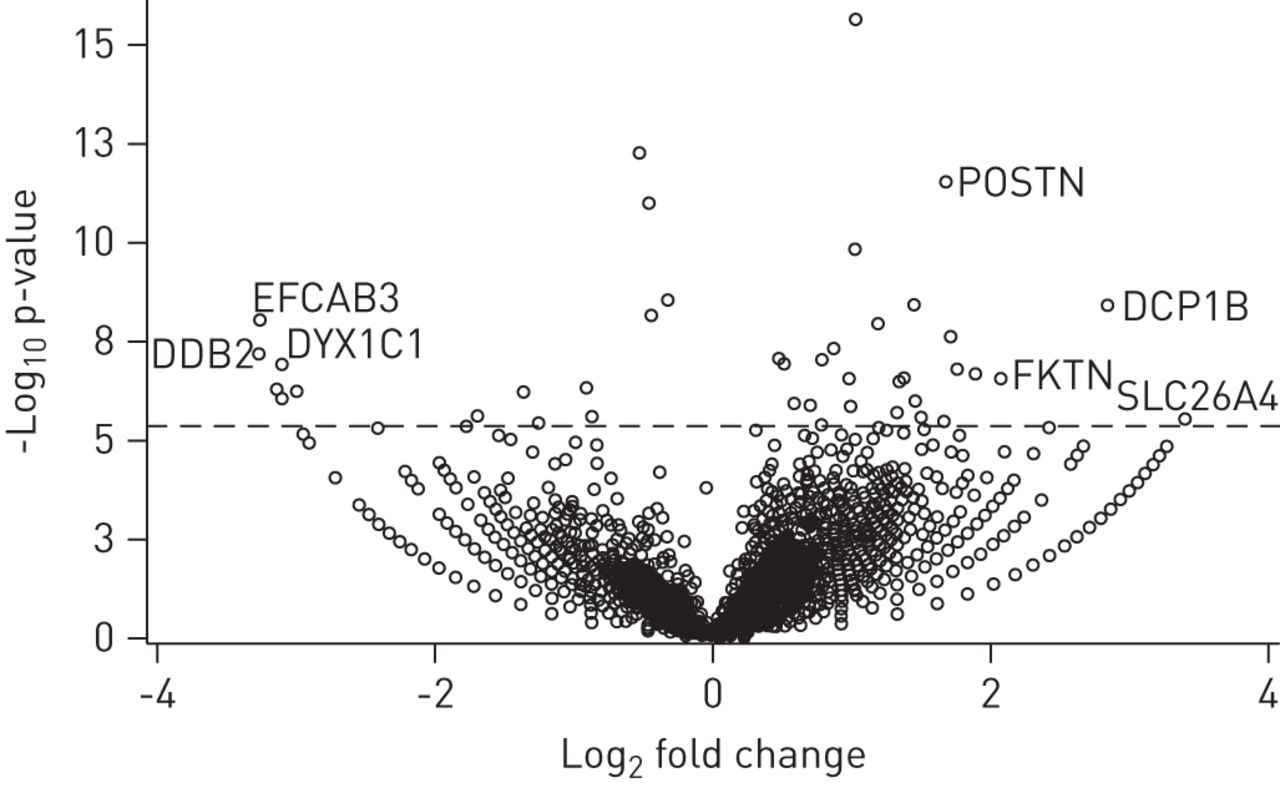
Volcano-plot哮喘病人的基因与控制。单个基因的褶皱变化的对数(x设在)策划与负以10为底的对数的假定值(y设在)。积极的日志2(褶皱变化)值代表upregulation哮喘与健康对照组相比,和消极的价值观。差别代表对这些圆上面的虚线代表的是哮喘和控制之间的差异表达基因修正多个测试后p < 0.05。DCP1B: DCP1开瓶酶同系物B (酿酒酵母);DDB2: damage-specific dna结合蛋白2 (48 kDa);DYX1C1:阅读障碍易感性1候选人1;EFCAB3:钙类ef - hand绑定域3;FKTN: fukutin;POSTN: periostin;SLC26A4: pendrin。
基因网络识别
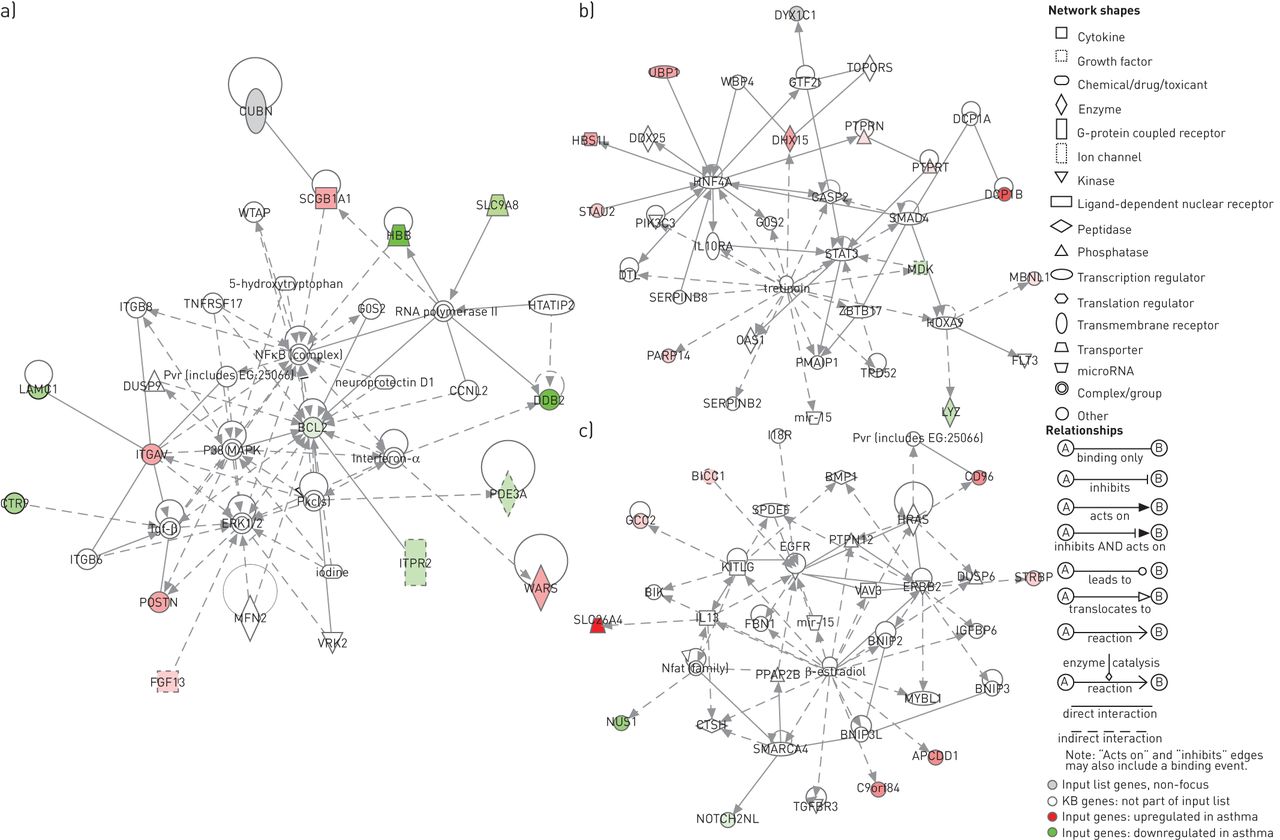
之间的差异表达基因46哮喘和控制被用来识别基因网络出来的软件应用程序。10基因网络与网络评分≥2。得分最高的网络有一个网络32分和相关网络功能细胞运动、细胞死亡和细胞形态。第二和第三网络排名,得分21日和17日与遗传性疾病和细胞运动和发展有关,分别是(图3)。基因b细胞慢性淋巴细胞白血病/淋巴瘤2 (BCL2),这是一个46哮喘和控制之间的差异表达基因,似乎是一个关键的组件在网络网络分(最高的图3)。其他关键部件在这个网络增殖蛋白激酶(MAPK) 1,也称为细胞外signal-regulated激酶(ERK) 1/2,核转录因子(NF) -κB p38 MAPK和转化生长因子(TGF) -β。14的46个输入基因可能位于这个网络。第二个信号传感器和网络排名中,激活转录(STAT) 3和staufen (STAU2) (图3 b),而第三个网络由表皮生长因子受体(EGFR)和pendrin (SLC26A4) (图3 c)。
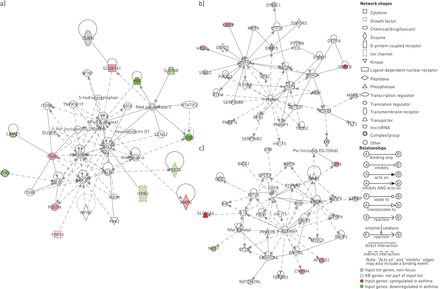
三大基因网络的创新路径分析(IPA)(智慧系统,Inc .,红木市,CA,美国)。)网络网络得分最高的32岁,包括b细胞慢性淋巴细胞白血病/淋巴瘤(BCL) 2、增殖蛋白激酶(MAPK) 1、核转录因子(NF) -κB p38 MAPK和转化生长因子(TGF) -β,与网络有关的功能细胞运动、细胞死亡和细胞形态。b)第二高得分的网络,包括信号传感器和激活转录(STAT) 3和staufen (STAU2)和c)第三网络,包括表皮生长因子受体(EGFR)和pendrin (SLC26A4),与遗传性疾病和细胞运动和发展有关,分别。红色——或者绿绿的形状表示的强度,差别的程度,或者对这些。KB基因:基因中IPA的知识库。
测序数据的验证
显著的相关性被发现之间的每个基因序列读取次数和Cq-ratio tryptophanyl-tRNA合成酶(战争)(r = -0.652;p = 0.05), Secretoglobin (SCGB) 1 a1 (r = -0.671;p = 0.05)和BCL2 (r = 0.724;p = 0.04)。这些结果表明,高测序读数量与低Cq-ratio(高数量的cDNA)反之亦然。因此,qPCR结果证实RNA-Seq发现的数据。
讨论
在这项研究中RNA从整个人类支气管活检标本分离成功测序,显示转录组配置文件不同哮喘和支气管组织的控制。因此,RNA与支气管活检适合RNA-Seq当处理根据严格的工作流程。RNA-Seq数据的分析揭示小说的表达基因尚未与哮喘和呼吸道炎症。此外,基因网络识别显示几个46哮喘和控制是关键组件之间的差异表达基因在基因网络与一个或多个细胞功能的规定,包括细胞形态、运动和发展。这些发现表明,生物过程在航空公司的规定显然是哮喘和健康对照组之间的不同,这可能是相关疾病的发病机理和未来的治疗。
据我们所知这是第一个研究应用RNA-Seq代替微阵列研究转录组的概要在活的有机体内人类支气管哮喘患者和健康对照组的活检。这种下一代高通量测序方法使基因表达的客观的方法分析,因为它并不局限于选择数量的已知基因或序列,与微阵列(一样13]。因此,下一代测序促进的发现新创成绩单和新颖的疾病相关基因的识别。
我们发现46之间的基因差异表达的支气管活检哮喘和控制。这些基因已经被证明的几个施加各种影响信使rna降解和转化过程影响细胞功能,如STAU2 [20.和战争21]。然而,大部分尚未与哮喘有关的差异表达基因。通过微先前的研究已经表明,气道上皮细胞的基因表达谱是不同的在哮喘与健康对照组相比22,23]。这包括SLC26A4(也称为pendrin,在我们的研究中也发现的一个最显著的调节基因在哮喘。它已经表明,这种基因与哮喘的病理生理学(密切相关24]。更具体地说,刺激IL4 /使用IL13受体复杂导致SLC26A4表达增加通过转录因子STAT6,导致气道表面的粘液生产和增加粘度液体(25]。此外,它已被证明之前,periostin (POSTN)氯通道调节器1 (CLCA1)和serpin肽酶抑制剂,支系B,成员2 (SERPINB2)高度诱导气道上皮细胞(9,22]。我们目前的研究扩展了这些调查结果,表明这三个基因的表达是在哮喘控制相比,尽管只有periostin达到意义(表2)。这可能是由于这样的事实,我们整个支气管活检的转录组测序,而不是只气道上皮细胞。
此外,国际音标的46个差异表达基因输入数据库产生10基因网络,在细胞水平相关的监管职能。除了这一事实的46个基因在这些网络关键职位,还应该指出的是,特定的细胞因子,趋化因子和蛋白质复合物,是众所周知的在炎症过程为主要角色,如TGF-βinterferon-α,p38 MAPK和NF-κB这些相同的网络(26,27]。除了p38 MAPK和NF-κB外,其他两个主要节点出现在IPA得分最高的基因网络BCL2和ERK1/2。有人提出抑制BCL2导致了调节免疫反应伴随着一个增强释放促炎细胞因子和增强功效坏死的细胞(28]。我们的结果显示显著减少的表达BCL2相比,哮喘控制。此外,磷酸化ERK1/2已被证明是增加了咆哮,eotaxin而抑制ERK1/2减少ASM的水平扩散引起的趋化因子(29日]。第二个和第三个最高IPA-scoring基因网络(图3 b和c)包含STAT3和表皮生长因子受体等。STAT3已被证明是相关的转录机制胸腺基质淋巴细胞生成素诱导炎性基因表达在ASM细胞(30.),而阻断EGFR可能导致减少粘液分泌过多(31日]。总的来说,这些结果表明,生物过程在航空公司的规定显然是不同的相比,哮喘控制。此外,这三个基因网络可以让我们更好的理解轴从病理学和炎症(p38 MPK、NF-κB BCL2和表皮生长因子受体)的生理(ERK1/2, STAT3)在哮喘气道。
几个方法论的问题需要解决。包含特征明显的主题似乎代表了我们的研究力量。只有前者哮喘患者疾病包括控制,从而避免了潜在的偏见的类固醇使用基因表达谱的气道壁。所有哮喘患者保持稳定和控制在研究期间以最小的临床症状。尽管他们被允许使用吸入短效β2-agonists抢救治疗,这些患者在研究期间使用任何。因此,基因表达谱可能不会影响药物的使用。
我们设计了一个严格的标准操作程序引起的支气管活检。此外,通过结合鼓掌RNA-Seq系统和GS FLX +我们可以最大化的效用可能较低的收益率,甚至部分降解RNA在活的有机体内获得人类支气管活检。首先,成功的RNA加工成放大cDNA,所需输入RNA鼓掌RNA-Seq系统≥500 pg,这确实是适合当前获得的孤立的RNA (900 - 9300 ng)。第二,放大3′端和随机启动整个转录组,从而最大化覆盖作为阅读的形式分布于整个整体转录组,以及潜在的不利影响最小化退化RNA的基因表达分析。最后,样本容量是基于详细的功率计算显示一个可接受的错误发现率为4%。
基因网络分析的音标是基于已知的途径。差异表达基因中发现当前的研究可能是小说通路的一部分,还没有包括在IPA数据库,即使它是每周更新的发现与其他数据库包括Entrez基因,RefSeq、人类和基因本体。我们使用异丙醇因为我们协会旨在阐明基因表达谱中发现哮喘患者哮喘的病理生理学。随着基因测序领域推进各种生化过程的知识,新颖的途径与哮喘相关可能在将来的研究中被发现。
然而,我们的研究也有一些局限性。首先,放大的Ovation RNA-Seq系统可以引入偏差由于代短cDNA片段。这可能导致更少的代表共识序列发现,用于映射参考基因组。因此,基因少于实际样品中可被检测到。然而,它是不可能的,这影响了我们的结果,因为我们生成的高覆盖率深度和GS FLX的使用+系统确保长序列读取比竞争技术。
其次,活检标本取自4 - 6水平的肺。随着哮喘的气道壁改造和炎症程度可以随位置的支气管树和哮喘严重程度(32),可能是基因表达谱的外围航空和更严重的哮喘患者可能是不同于本研究中观察到。后来的研究专注于这些位置的航空公司在哮喘患者更严重的疾病需要补充我们现有的数据,以提供更全面的概述哮喘气道的基因表达。此外,应该检查颞可变性,将受到道德的约束。
第三,RNA是独立于整个支气管活检。我们选择执行整个切片分析,因为单个组件的贡献气道壁的病理生理学哮喘在很大程度上仍然是未知的33]。此外,哮喘和控制之间的差异表达基因46不包括记录特定的炎性细胞,但各种细胞过程相反,表明RNA-Seq整个活检并不影响差异的炎症细胞存在于活检材料。
基于目前观察到的差异基因表达谱的整个活检,重复分析选择个人气道组件,如。上皮和ASM,激光捕获显微解剖看起来是合理的。这将允许个人气道壁组件的基因表达谱与气道功能和疾病活动。高通量下一代测序技术有可能实现这一目标。它不仅可以导致新基因或通路的发现对于哮喘的发病机制是至关重要的,但也可能成为subphenotyping病人与临床结果的工具或治疗反应。在新招募的患者足够的外部验证后,新的治疗靶点可能出现,这可能促进发展本质上是小说哮喘治疗。此外,通过实现系统生物学方法在未来哮喘研究[12),整个生化过程,从翻译的DNA, RNA,蛋白质,这些蛋白质的翻译后修饰和监管,将揭开通过组合不同的“组学”技术,包括基因组学、转录组、蛋白质组学和代谢组学。
结论
目前的研究表明,RNA分离适合RNA-Seq从支气管活检是整个人类。转录组的分析整个活检显示46哮喘和控制之间的差异表达的基因,大部分不是与哮喘有关。此外,路径分析表明,几个46个差异表达基因的基因网络的关键组件与一个或多个细胞功能的调节。这些结果表明,不同的生物过程在哮喘患者的气道管理在转录组水平与健康对照组相比。
确认
我们感谢Saheli Chowdhury(部门实验免疫学、学术医疗中心,阿姆斯特丹,荷兰)支持的设计定量pcr引物组。
脚注
可以从本文的补充材料www.www.qdcxjkg.com
支持声明:本研究被研究给予支持荷兰哮喘基金会(项目编号3.2.09.065)。泰国人Mauad由CNPQ(巴西研究委员会)(302828/2012-9)。
利益冲突:披露可以找到与本文的在线版本www.www.qdcxjkg.com
- 收到了2012年7月24日。
- 接受2012年12月17日。
- ©2013人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}