





文摘
肺纤维化是许多弥漫性肺实质疾病的最后阶段。它的特点是过度矩阵的形成导致破坏正常的肺架构,最后死亡。尽管一个指数增加我们对潜在的重要介质和机制的理解,界定主要通路已经被证明是难以捉摸的。
综述易感性和有害的药物,如病毒和gastro-oesophageal回流,在启动疾病将讨论可能的作用。进一步阐述了主题是候选人辅助途径,包括免疫机制、氧化和内质网压力,激活凝血级联的干细胞的潜在作用。本文将试图为读者提供一个集成的观点对当前的知识和努力为未来的研究提供一个路线图。
探索整体的健壮的模型是很重要的发病机制,协调大量临床和科学观测。我们认为,当前数据的整合成一个“大画面”的概述纤维发生发展的有效的antifibrotic策略至关重要。后者可能会包含代理的组合目标关键途径。
肺纤维化是一些弥漫性肺实质疾病的结束阶段(DPLDs),矩阵的特点是过度沉积和破坏肺的架构,最后导致呼吸衰竭。最常见的肺纤维化,特发性肺纤维化(IPF)是一种进行性疾病,5年生存率仅为20%,反映出缺乏有效的治疗方法。>在英国,每年有3000人死于与IPF和发病率每年都在增长11% (1]。IPF的病因学仍然知之甚少,虽然提出了一些风险因素和诱发因素,包括吸烟、病毒感染和表面活性剂蛋白质多态性。
肺组织学检查IPF交替地区正常肺实质、间质炎症、纤维化和蜂窝。IPF的病理生理基础一直是有争议的问题在过去的几十年里。现在有越来越多的证据表明,IPF可能代表一个单独的疾病中,纤维发生的结果,至少在某种程度上,从multi-focal上皮36例。反复损伤肺泡上皮细胞(AEC)导致细胞凋亡,这可能会导致无序epithelial-fibroblastic交互和异常的修复过程,最终导致纤维化(2]。
在本文中,我们关注的因素使个人容易进行性纤维化的过程,可能是特工参与反复损伤和重要元素导致异常修复和肺纤维化。此外,免疫等新兴新发现将讨论过程,氧化应激,内质网(ER)压力,激活凝血级联,并可能改变的淋巴管,最后干细胞的作用。
纤维化的起始:倾向和因果代理
代理负责纤维化的起始过程定义仍然不佳。人们普遍认为重复受伤是由于遗传素质和有害环境代理之间的交互。
遗传素质
尽管大多数DPLDs零星的疾病,家族发生存在的例子。家族聚类的成人特发性间质性肺炎(IIP)表明,遗传因素可能在疾病发展发挥重要作用。有趣的新发现包括ELMOD2基因的描述和表面活性剂的作用蛋白(SP) - c。有趣的是,基因改变还在零星的IPF指出。ELMOD2 mRNA表达显著减少IPF肺与健康对照组相比。ELMOD2已被证明是与interferon-related抗病毒反应的规定(3]。
SP-C突变在患有严重特发性肺炎的儿童经常确认(4]。有趣的是,这些已确定在50%的这些孩子另外50%有零星的疾病。在儿童和成人,SP-C突变与非特异性间质性肺炎的临床实体相关联(NSIP),脱皮的间质性肺炎(DIP)和肺肺泡蛋白质沉积症。在最近的一篇论文,V盎ydF4y2Ba米oorselet al。(5)表明,5个20无关的家族性肺纤维化患者SP-C的突变,与这个特定的突变没有找到零星的疾病。
突变的表达,比如SP-A SP-C,肺泡上皮和突变的识别影响细胞稳定性,如端粒酶(hTERT和ht)突变,导致肺泡上皮的异常的体内平衡。SP-C和SP-A突变导致肺泡ⅱ型细胞(ATII)功能障碍;可能由于错误折叠的蛋白质,ER保留和激活的蛋白质反应(6]。最近的一项研究也表明,基因异常涉及航空公司可能促进肺纤维化,作为MUC5最近描述(7]。假定的启动子的常见变异MUC5B已被确认,与家族性间质性肺炎和IPF的发展。主题与IPF MUC5B在肺部的表达明显高于对照组和MUC5B表达IPF的组织学病变。然而,之间的确切联系MUC5B分泌过多的航空公司和干扰在肺纤维化肺泡上皮内稳态的存在还不清楚。
有害的药物
目前认为,IPF遗传易感性的关联结果异常上皮细胞监管环境诱因,导致制定的“多重打击假说”。纤维化疾病的病因学,纤维化是由持续伤害,可以由免疫复合物,吸入剂如石棉或直接毒性损伤,如辐射或药物。在国际信息局,提出了许多潜在的病原体;最著名的环境因素是病毒和gastro-oesophageal回流(工资)。
病毒
病毒感染被假定作为纤维化但极其难以研究的发起者由于呼吸道样本变量敏感性的文化。随着PCR等分子检测方法的发展,知识在这个领域产生了戏剧性的增长。病毒与IPF的发病机制包括eb病毒(EBV),人类疱疹病毒7和8、巨细胞病毒、丙型肝炎病毒、单纯疱疹病毒、细小病毒B19病毒和扭矩塔诺河。另一个问题是是否存在EBV和其他病毒可能仅仅反映肺泡上皮损伤增加易受感染,尽管有越来越多的证据表明因果作用的病毒在肝纤维化的起始。
EBV感染一直是最广泛的识别在IPF提供重复的细胞损伤的潜在来源,目前发病的假说的核心组件(8]。T盎et al。(9]报道EBV的存在,在五(62.5%)检测到真正的time-PCR的八个家族病例和16 (64%)25 IPF的散在病例。尽管通常EBV感染上呼吸道,它也被证明的下呼吸道感染和复制的(10]。EBV蛋白质和DNA表达已确定在IPF患者的肺组织(11,12]。在IPF活检样本,EBV gp340/220和病毒蛋白,表示在裂解阶段EBV感染,被局部的原子能委员会(11]。EBV的假定的角色发展的IPF支持其他观测。了IPF患者的不良预后与EBV潜伏膜蛋白1的表达在原子能委员会(13]EBV感染和复制的阶段。临床稳定两IPF患者口服抗病毒治疗后一直报道(9]。转化生长因子(TGF) -β1 pro-fibrotic关键细胞因子与IPF,是一个强有力的生长抑制剂在大多数细胞上皮来源的14]。EBV-infected细胞暴露于TGF-β1显示病毒裂解阶段激活和抗细胞生长抑制(15]。它也表明,EBV-AEC交互可能参与生产纤维疤痕,IPF的病理特点。此外,生物活性EBV可能诱导上皮间充质转变(EMT)。
遗传因素和宿主的相互作用可能为ELMOD2证明基因有关。ELMOD2由肺上皮细胞和肺泡巨噬细胞表达。它调节interferon-related抗病毒反应及其在响应表达式是减少病毒感染(3]。有趣的是上皮细胞和肺泡巨噬细胞是基本的细胞类型感染呼吸道病毒。这些发现强化相关性强病毒-宿主相互作用的证据和遗传易感性因素。
Gastro-oesophageal回流
从1970年代起,气油比和食管裂孔疝,DPLD之间的关联已经认可(16]。在IPF,倾心于估计患病率高达90% (17]。新兴的数据点的潜在作用慢性micro-aspiration由于小水滴的亚临床的愿望,气油比的一个风险因素。气油比和沉默micro-aspiration与一些肺部疾病相关,包括post-transplantation拒绝[18]。另一个有趣的研究Tcherakianet al。(19]提供了证据作用的气油比和IPF发病的急性发作。这些研究者研究了一组不对称IPF患者。他们发现,气油比出现在不对称的IPF患者20例(62.5%)更对称情况下这只是出现在31.3%的病人。不对称的IPF患者也报道有更多急性发作(46.9%)比对称情况下(17.2%)。作者得出结论,不对称可能突出病因相关因素如气油比的作用或特定区域的条件发展和/或肺纤维化的进展和急性恶化。
其他最近的研究已经报道了IPF急性加重,气油比之间的关系。急性发作的特点是弥漫性肺泡损伤的组织病理学模式叠加在底层的模式通常的间质性肺炎(摘要)。胃蛋白酶被认为发挥重要作用在急性发作被发现在支气管肺泡灌洗(BAL)流体的急性发作患者(20.]。甚至发现高架BAL胃蛋白酶是预测急性恶化状态,主要由子群的存在情况下与胃蛋白酶水平明显升高(33%)。
尽管令人信服的数据支持气油比与IPF之间的相关性,这可能或者被解释的气油比更经常发生在肺纤维化,甚至纤维化IPF的变化的结果,尽管这似乎不大可能。
纤维化的进展
关键的细胞类型
目前,肺纤维化疾病被认为是一种重复造成的亚临床损伤导致上皮损伤和后续alveolar-capillary基底膜的破坏。这个过程开始纤维细胞的浸润和(myo)成纤维细胞的激活。在肺纤维化炎症和间充质细胞的正常决议通过细胞凋亡和吞噬作用是特异表达。这导致正常的肺结构的破坏和损失的功能。在IPF这个过程导致死亡诊断后平均3年的时间。归纳了这些过程图1。
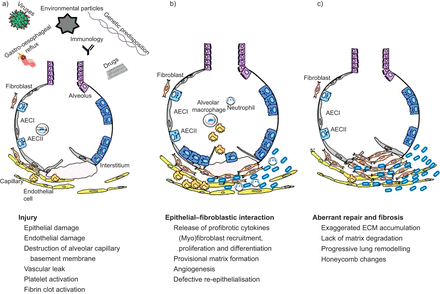
所涉及的主要元素感应和纤维化的进展。a)纤维化的发病特点是受伤和易感性进行性纤维化的形成。许多不同的有害代理已确定,导致上皮和内皮损伤,血管渗漏和纤维蛋白凝块的形成。b)这是紧随其后的是异常修复过程的特点是异常re-epithelialisation,大量的myofibroblasts和胶原蛋白的形成矩阵。c)继续过度矩阵形成过程导致建筑变形,最后死亡。ECM:细胞外基质;原子能委员会:肺泡上皮细胞。
肺泡上皮细胞
目前的体外和在活的有机体内证据表明,原子能委员会受伤在IPF的发病机制是一个关键过程。的重要观察组织的IPF患者,首次报道了Katzensteinet al。(8],增生ATII和剥蚀的肺泡上皮内成纤维细胞的焦点(21]。此外,正如前面所讨论的,突变的基因,影响再生能力或造成伤害/ ATII细胞凋亡已确定在家庭形式的肺纤维化。(22]。在这个过程中受伤的ATII是至关重要的,因为它已被证明在IPF患者的肺的70 - 80% ATII染色阳性标记细胞凋亡的23]。如图所示最近,有针对性的枯竭ATII肺纤维化小鼠模型的具体链接ATII损伤肺纤维化的发展(24,25]。增强纤维母细胞分化和胶原蛋白生产已被证明在上皮细胞/纤维母细胞共培养的上皮细胞损伤组件(24]。此外间充质细胞生存的增强是由于响应性生长因子和抗细胞凋亡增加。
经典描述,ATII受伤导致无效的大批的正常上皮细胞诱导和驱动纤维化的居民间质成纤维细胞分化成myofibroblasts [25,26]。这些是肺纤维化的关键效应细胞的特点是新创α-smooth肌肉肌动蛋白的表达,这是组织功能应力纤维和授予收缩性质(27]。他们本土化纤维化病灶纤维化和其他网站的活跃,并且负责细胞外基质(ECM)的合成和沉积和合成结构改造,导致肺泡功能的丧失。当前舆论表明myofibroblasts至少有三种可能的起源,虽然每种途径的相对贡献IPF目前未知。最简单的建议是居民直属肺成纤维细胞分化形成myofibroblasts profibrotic微环境的影响。第二个可能性是上皮细胞经过分化转化成纤维细胞/ myofibroblasts EMT形式。上皮细胞失去特征标记如钙粘蛋白和带occludens-1并获得间充质标记,如fibroblast-specific蛋白1和α-smooth肌肉肌动蛋白(28]。第三个假设表明myofibroblasts可能来自循环纤维细胞(胶原蛋白I + / CD34 + / CD45RO +)或其他来源于骨祖细胞(29日]。衰减的纤维细胞直接贩卖小鼠模型与肺纤维化的减少。循环纤维细胞的数量最近显示出与疾病活动和发展30.]。增强myofibroblast转化和扩散阻滞细胞凋亡的间充质细胞纤维化持续进展的主要元素(31日]。此外,持续恶化的矩阵沉积或缺乏矩阵进一步导致毁灭和替换正常的肺泡组织密集纤维化病变。
关键细胞因子
招聘的成纤维细胞,成纤维细胞增殖和ECM的生产过剩是由一个复杂的炎性细胞因子网络,趋化因子和细胞类型。关键细胞因子和趋化因子,诱导profibrotic环境包括肿瘤坏死因子(TNF) -αTGF-β,单核细胞趋化蛋白(MCP) 1 / CCL2,巨噬细胞抑制蛋白(MIP) 1α/ CCL3和辅助(Th) 2-chemokines如CCL17 CCL18和CCL2232,33]。TNF-α在刺激炎症反应中起着重要的作用,信息粘连和trans-endothelial迁移,以及早期的事件导致细胞因子和趋化因子的生产级联(34]。TNF-α-transgenic小鼠t细胞介导肺泡炎和随后的纤维化发展,而TNF-α基因敲除小鼠未能发展与博来霉素(纤维化治疗后35]。尽管这些令人鼓舞的结果IPF患者用依那西普进行进行的一项研究显示,疗效无显著差异端点治疗48周后(36]。
TGF-β是迄今为止最有效的profibrotic中介的特征和调节肺纤维化通过招聘和活化的单核细胞和成纤维细胞,和ECM生产的感应。肺成纤维细胞增殖是TGF-β1的间接影响通过纤维母细胞生长因子2的感应和顺向活化增殖作用的蛋白激酶(MAPK)途径37]。此外,TGF-β诱导成纤维细胞分化成myofibroblasts。TGF-β促进ECM生产通过促进ECM基因转录,包括胶原蛋白、纤连蛋白和蛋白聚糖,通过抑制基质金属蛋白酶的活性,纤溶酶原激活物物和弹性蛋白酶,这导致胶原蛋白降解的抑制38]。
MCP1 / CCL2和MIP1α/ CCL3促炎症趋化因子单核细胞负责招聘。CCL2 CCL3和显著调节BAL流体从IPF患者和肺纤维化小鼠接受博来霉素(39]。MCP1受体CCR2-deficient老鼠免受bleomycin-induced肺纤维化是由于招募巨噬细胞的损伤和功能(40),纤维发生的细胞因子表达和纤维母细胞响应TGF-β(41]。
基因组学是另一个领域,极大地促进了我们对近年来纤维发生的知识。基因表达研究利用患者的肺组织间质性肺炎有显示分子表型的IPF /摘要本文不同于NSIP,更像是后者过敏性肺炎(42]。许多基因显著增加在肺纤维化的编码与ECM的形成和相关的蛋白质降解和蛋白质表达的平滑肌细胞,包括matrilysin,基质金属蛋白酶(MMP) 743),这是一个金属蛋白酶肺纤维化的中介。它变得明显,基质金属蛋白酶发挥多效性的影响,包括蛋白水解降解ECM和处理的趋化因子,细胞因子和生长因子(44]。它甚至有可能增强的MMP的活动,因此,机械的司机IPF的进行性纤维化。分子表达特征区分进步IPF MAPK-early IPF包括成员相对稳定的增长响应1热休克蛋白70通路调节烟雾诱发炎症。然而,分子和细胞功能,如细胞增殖、迁移、入侵和细胞形态、IPF占了大多数,IPF(急性加重患者的组织45),与基因表达分析确定上皮损伤和扩散的关键分子事件(46]。总的来说,这些方法表明,在IPF肺,与肺癌相关基因和机制发展(包括Wnt /β-catenin通路,上皮细胞可塑性和EMT)被激活,与古典炎症的作用有限。
新兴的新发现
免疫机制
炎症和纤维化之间的相互关系可能被视为从各种角度。IPF的历史数据表明,淋巴细胞百分比的增加在矿山与更大的响应性皮质类固醇疗法和更好的生存47]。BAL淋巴球增多被发现与严重肺泡隔炎症和相对缺乏的蜂窝48]。后来,随着NSIP的出现作为一种独特的实体(49),它被怀疑,许多患者BAL淋巴球增多,以前与IPF诊断,事实上有NSIP。这是支持的事实,三大东亚研究显示NSIP真正引人注目的淋巴细胞水平(50- - - - - -52]。建议从高分辨率计算机断层扫描观察,这些患者可能组织肺炎/ NSIP重叠。
现在清楚的是,BAL淋巴球增多的存在不可靠的区分NSIP和摘要在个别病人53),但有数据显示,淋巴细胞计数升高落下帷幕可以帮助区分IPF和其他DPLD。在这方面提到的研究是很重要的hshimoet al。(54)报道,截止的30%,淋巴细胞水平落下帷幕了IPF的诊断的有利的辨别力。6(8%)的74 IPF患者根据临床资料和成像显示,BAL淋巴球增多≥30%。他们的最终诊断特发性NSIP (n = 3)和过敏性肺炎(n = 3)。
IPF以外的间质性肺炎
几项研究的重新分类的病人以前与IPF诊断,使用当前的标准,已经证明了组织病理学的影响模式在IIP患者长期生存。特发性NSIP,倾斜和不明原因引起的组织肺炎(COP)比IPF(有一个更好的结果55,56]。作为警察响应显著口服皮质类固醇和5年生存在NSIP∼80% (57]。它可以得出结论,炎症,当警察的指示性,显然是与更好的可逆性和生存有关。警察不是视为一种纤维化疾病,尽管目前分类作为国际信息局。
在特发性疾病,最常见的组织学模式摘要和NSIP经常遇到在结缔组织疾病有关的间质性肺病(CTD-ILD)。两个关键CTD-ILD近年来取得了进步。首先,它变得明显,生存在CTD-ILD比在IPF (58),除了肺部疾病风湿性关节炎。其次,许多更频繁地观察组织病理学特性比IIP CTD,包括NSIP的模式和淋巴聚集和生发中心摘要(以及更多的浆细胞),与边际与生存的关系(59]。密集的血管周围胶原沉积也报告了CTD。
然而,目前尚不清楚IIP的临床诊断,患者在他auto-antibodies被检测到,从那些没有抗体有不同的预后。年代昂et al。(59)最近表明,IPF患者的生存/摘要没有改变auto-antibodies的存在。早期的研究表明,抗核抗体可能会发现在10 - 20%的所谓的IPF患者(60),随后重新归类为NSIP [55,56]。抗核抗体,较少anti-neutrophil细胞质抗体(61年IPF患者)可能会发现,尽管他们的意义是不确定的。最常见的病态模式CTD-ILD NSIP,除了风湿性关节炎的特点是一个更高频率的摘要(62年]。此外,很明显,风湿性arthritis-associated摘要代表一个可能的例外改善生存在CTD-UIP IPF /摘要[相比63年]。P约柜et al。(58)报道,风湿性arthritis-associated摘要倾向于有一个生存比non-rheumatoid关节炎CTD-associated NSIP或摘要。在一个较大的研究82例风湿性arthritis-ILD更糟糕的生存患者的报告摘要高分辨率计算机断层扫描模式。此外,他们发现,估计在风湿性arthritis-UIP生存与IPF(没有显著差异64年]。
特发性肺纤维化
多的信息经肺纤维化动物模型;然而,重要的是要记住,这些模型的特点是急性损伤和炎症反应并不真正复制所有功能IPF发病机制的核心。转移到啮齿动物的肺上皮细胞各种炎性细胞因子和趋化因子使用replication-deficient运载体导致显著增加BAL炎症细胞和组织肺炎但没有残余肺改造和纤维化65年的例外),TGF-β1和细胞因子如白介素(IL) 1βTGF-β1导致过度活跃。然而,尽管不代表人类的肺纤维化动物研究都证明了一个核心作用TGF-β1作为关键profibrotic中介,也表明诱导肺纤维化并不总是依赖程度的炎症反应65年]。
虽然没有明显的炎症在IPF患者的肺部炎性细胞因子和趋化因子(凝血级联一起和类二十烷酸脂质介质)之间的相互作用导致肺上皮细胞和成纤维细胞,分化向2型辅助t细胞表型和淋巴再生(66年,67年),以及血管生成和血管生成通过一个失衡angiostatic科学家趋化因子(66年),这将在本文进一步讨论。M1和M2巨噬细胞的表型最近支持Th1、Th2反应的描述,分别。
Th2反应特征的肺部疾病,其中许多的组织改造和纤维化。转向Th2免疫反应在IPF似乎占主导地位,促进纤维化为主通过profibrotic细胞因子的分泌受伤的上皮细胞。在肺部疾病的小鼠模型,动物组织损伤的主要Th2反应类型,更倾向于肺纤维化肺损伤后比那些主要Th1反应(67年,68年]。
已经表明,CCL18 CC-chemokine受Th2细胞因子,与肺纤维化(33]。CCL17 CCR4和CCL22及其受体被发现是提升区域的纤维化肺组织与正常肺实质(38,69年,70年]。特别是CCR4被发现主要由巨噬细胞表达出现在纤维领域。而且它已经表明,中和CCL17和CCL22导致显著减少肺损伤的71年]。Th2免疫反应导致的失败re-endothelialisation re-epithelialisation,并导致profibrotic生长因子的释放到受伤的地区。这些profibrotic细胞因子启动成纤维细胞迁移到损伤的部位,促进其增殖和分化成myofibroblasts。炎症和组织改造与病理纤维化Th2反应的常见后果是肺癌和其他器官。IL-13和TGF-β1经常发生在这些反应和被认为Th2-induced病态的发病机制中扮演很重要的角色。这个Th1 / Th2假设主导我们理解免疫调节,免疫发病机理和宿主防御几十年来,尽管存在缺陷,无法解释某些数据对t细胞介导的组织损伤。
一个令人兴奋的发现是血清CCL18浓度在IPF(有预测价值72年]。CCL18 CC-chemokine由人类骨髓细胞,被描述为一个标记替代激活的巨噬细胞。巨噬细胞激活的Th2细胞因子诱导一个特殊的表型,所谓的“另类激活”。另外,激活巨噬细胞在组织修复过程中发挥作用,如伤口愈合和纤维化。P麝香猫et al。(72年]显示患者死亡率明显高于血清CCL18浓度> 150 ng·毫升−1。这些发现是有前途的,应该在设计中实现新的临床试验。
最近,两个小说描述了CD4 t细胞亚群,已经彻底改变了我们对免疫功能的理解:Th17子集,发展通过不同细胞因子信号的Th1、Th2血统,和t调节细胞亚群)。IL-17 Th17子集的特点是生产和参与自身免疫组织损伤的发病机制,包括类风湿性关节炎和有浓度过敏原特异性反应。中央介质产生效应是TGF-βCD4 + Th17子集,IL-23 IL-17。虽然在粘膜宿主防御Th17细胞是重要的,他们可以调解的免疫病理过程的事件。IL-17受体是调节在过敏性肺炎患者的肺73年]。肺纤维化的关键细胞因子profibrotic TGF-β,被发现为天真的CD4细胞的分化是必要的+t淋巴球Th17细胞在小鼠74年,75年),这可能是另一种方式TGF-β促进纤维化。在纤维化机制的全面审查,Wynn和Ramalingam(76年)建议IL-1β-IL-17-TGF-β轴的作用。
亚(CD4 + CD25 + foxp3 +)代表的第一个定义良好的扩张CD4 + t细胞功能的范围。亚群抑制免疫系统的激活和有助于维持免疫内稳态和宽容自体抗原(77年]。亚群的短缺,Th1、Th2反应的潜在振幅增加导致过度的t细胞免疫与自身免疫性疾病相关,哮喘和过敏。丰富的亚群,然而,将减少的潜在振幅Th1、Th2反应,并可能防止足够的免疫力肿瘤和传染病(77年,78年]。Treg抑制功能的一个重要障碍是明显在外周血和BAL IPF患者的液体。相同的研究结果来自肺组织活检。最后,几乎亚全球障碍的线性相关性,在功能和数值,与疾病严重程度的参数包括肺功能测试,证明,表明亚群功能障碍疾病可以作为一个可靠的预言者先进性和治疗反应,可能为临床医生提供一个新颖的工具IPF患者的危险分层79年]。可以猜测,低数字和IPF患者全身和局部treg障碍发现可能导致低效的控制既存的Th2反应或导致Th2斜(74年]。
氧化应激
氧化应激被定义为不平衡的一代活性氧(ROS)超过抵消他们的能力。生产过剩的活性氧可能导致氧化应激导致组织损伤。氧化应激可能在再生微环境,有利于促进组织纤维化。这可能扮演一个角色在发展成纤维细胞的细胞凋亡抵抗IPF (77年]。它已经表明,肺myofibroblasts分泌过氧化氢,这可能调解纤维发生的影响和诱导上皮细胞凋亡78年]。此外中性粒细胞基质降解酶的主要来源,包括中性粒细胞弹性蛋白酶,导致氧化应激。调节氧化应激可能是一个有趣的方式,防止进一步损伤,试图阻止肺纤维化的过程。有趣的新数据显示,sivestat,中性粒细胞弹性蛋白酶抑制剂,能够缓解bleomycin-induced肺纤维化(79年]。此外,缺乏抗氧化剂谷胱甘肽(GSH)在上皮衬里流体被认为扮演一个角色在肝纤维化的发生和发展80年]。氧化应激之间建立联系和IPF临床试验提供了一个强有力的理由,包括评估N乙酰半胱氨酸(NAC),一种抗氧化剂,充当一个谷胱甘肽合成的前体通过的主要代谢物半胱氨酸。IFIGENIA(特发性肺纤维化国际集团探索南京我年度)试验研究了高剂量的影响NAC除了糖皮质激素和咪唑硫嘌呤。在本试验结果表明,添加NAC能减缓用力肺活量的恶化和扩散能力下降的肺一氧化碳IPF (81年]。然而,最近的豹(强的松、咪唑硫嘌呤和南汽:一项研究评估响应在IPF)试验显示,南汽的组合与糖皮质激素用药增加死亡的风险和住院治疗与安慰剂相比82年]。这已经被认为是新的证据对这种联合治疗的起始IPF患者。这里的一个问题是IPF患者的个人特征在这个实验中比较人口(过去的研究83年),死亡的速度而安慰剂豹试验仅为2%,低于其他IPF的安慰剂对照试验。这些结果当然值得进一步的详细分析。此外,很明显,持续的豹IPF试验的结果关注NAC单一疗法与安慰剂是热切期待。
内质网应激
ER压力定义为条件造成的干扰蛋白质的加工和折叠,导致错误折叠蛋白质的积累ER和激活的所谓展开的蛋白质反应(UPR) [84年]。在过去的几年里,提供了大量的证据表明在IPF (ER应激的作用23,76年,85年]。最初的观察确定了原子能委员会从家庭SP-C突变,但他们也被观察到具有非SP-C-related家族IPF甚至零星IPF (4,23]。分泌蛋白最初送到急诊室的多肽链。此后他们正确折叠功能的三维构象,组装和糖化。强调细胞时,激活的UPR可以发生,失败的UPR应答,ER应激或太严重,可能导致原子能委员会通过细胞凋亡(细胞死亡85年]。其他研究表明,ER应激可以导致纤维化原子能委员会表型通过急诊医疗机制,这个过程可能参与调节细胞表型(86年]。此外,病毒感染开始生产大量的病毒蛋白,可能激活ER应激。有趣的是,它已经表明,疱疹病毒蛋白质表达的原子能委员会衬里地区IPF的纤维化co-localised与ER应激标记(85年]。
很明显,异常蛋白处理未来的治疗是一个潜在的目标,但在这个有趣的领域需要更多的研究。特别是,它是不清楚环境的侮辱和ER应激之间的链接,或者细胞类型除了原子能委员会也可能参与了ER stress-UPR轴。此外,表面活性剂之间可能有联系蛋白质和ER应激。Hermansky-Pudlak综合症患者,这是一种罕见的常染色体隐性障碍与oculocutaneous白化病和出血素质,有时出现间质性肺炎。它最近表明,ER应激反应中看到这种疾病可能是由于越来越多的积累SP-B / c。这些额外的ER应激原子能委员会进一步导致细胞凋亡(87年]。
血管组件及凝血级联
异常的血管重塑IPF的开发和发展是至关重要的。大量的数据显示改变平衡的重要性pro - (CXCL8, CXCL5和CXCL12)和反(CXCL9, CXCL10和CXCL11)血管生成科学家趋化因子在促进循环纤维细胞的异常neoangiogenesis和肺部招聘TGF-β的贡献。
激活凝血级联的组织损伤后最早的事件开始。凝血级联的主要功能是填补受损血管,防止失血。然而,人们越来越认识到凝血级联的功能超越止血,这级联中起着重要的作用影响炎症和组织修复项目。凝血级联的细胞反应主要是由几个执行凝固蛋白酶,表演通过特定的细胞表面受体,proteinase-activated受体(PARs)。这个家庭有四个成员(标准1为标准4),但目前的证据显示的一个主要角色的高亲和性凝血酶受体标准1的上下文中,影响细胞反应肺损伤。激活的票面价值1无数的肺细胞类型,包括肺上皮细胞、成纤维细胞和巨噬细胞,导致有效的促炎的释放和激活和profibrotic介质(88年- - - - - -90年]。此外,票面1Xa信号对凝血酶或因素对成纤维细胞合成myofibroblasts也促进其增殖和分化成矩阵(91年- - - - - -93年]。
凝血级联的证据来自可能的病理生理意义在活的有机体内bleomycin-induced肺纤维化模型。针对这些模型中的凝血级联(使用不同的策略,包括直接凝血酶抑制、组织因子途径抑制,气管内的活化蛋白C和aerosolised肝素)管理导致减少肺胶原蛋白积累和纤维化病变的发展(94年]。细胞介导的核心作用的凝血级联反应模型提供的证据表明,标准1缺乏的老鼠不受实验肺部水肿、炎症细胞招聘和纤维化(88年,95年]。
当前教条假定凝固酵素原来自循环和局部激活伤害通过组织因子(外在)凝血途径。然而,它最近表明,凝血因子X是IPF患者局部调节bleomycin-induced肺损伤的小鼠模型,代表突出与支气管和肺泡上皮细胞来源的凝血酶原(96年]。
这些发现预示着一个范式转变对我们理解组织过度pro-coagulant起源在肺部疾病的信号,表明上皮是一个主要站点的起始反应。与不同的抗凝策略的有前途的临床前研究,最近的临床试验表明,系统使用华法林抗凝治疗没有任何进步的IPF患者受益。相反,用华法林治疗与风险增加有关的死亡率IPF人口缺乏其他适应症抗凝(97年]。目前尚不清楚是否华法林是完全介导的影响通过其抗凝作用或其他维生素K-dependent酶。也不知道华法林是有效地阻止intra-alveolar隔间内的凝固。然而,这个试验提出了凝血级联的可能性在肺纤维化可能也起到保护作用。系统性的抗凝血剂,因此,不能证明有用的和未来的抗凝策略可能需要有选择地目标有害凝固在IPF intra-alveolar舱信号响应。小说一样1拮抗剂目前III期试验的心血管疾病和可能在IPF(值得进一步调查98年]。
淋巴管和肺纤维化
在正常肺组织,淋巴管存在接近航空公司和主要血管。由于薄壁结构,缺乏可靠的标记的知识在正常的人类肺淋巴管是稀疏的。Pusztaszeriet al。(99年)发现,D2-40淋巴内皮细胞的标记,遵循bronchovascular分布和一个研究报道缺乏淋巴管在肺泡空间(One hundred.]。最近,年代ozioet al。(101年)表明,小叶内淋巴管扩展超出呼吸细支气管。此外,他们提供的证据异质性小叶内的淋巴管,相比与其他肺淋巴管区(101年]。在猪肺淋巴管在薄壁组织(未找到102年]。一个有趣的发现已经发表在缺氧诱导肺泡损伤鼠模型(103年]。作者发现perialveolar淋巴管,在14天内解决。
在IPF lymphangiogenesis的数据是稀疏的。众所周知,纵隔淋巴结病存在于大量的IPF患者(104年,105年]。研究El -C丙烯酸-et al。(106年),在IPF肺淋巴管接近肺泡空间,甚至在这些地区保存完好的建筑。此外淋巴管在场在靠近主要的纤维组织和bronchovascular树。在成纤维细胞的焦点,没有观察到淋巴管,淋巴管周围可以看到。弥漫性肺泡损伤,intra-alveolar lymphangiogenesis是纤维化的过程的一个关键因素One hundred.]。
不过,也有数据表明胸膜下和小叶间淋巴管在IPF肺严重fibroconnective病变明显减少,与罕见lymphangiogenesis [107年]。此外,胸膜下淋巴管的破坏和细胞凋亡的淋巴内皮细胞中观察到IPF患者。胸膜下的消失和IPF肺小叶间淋巴管,连同lymphangiogenesis差,可能大大损害肺泡间隙,延长接触有害介质间质,提高对纤维发生的影响。急性加重,这是一个特别有吸引力的模型的凝血级联和淋巴中断可能会把最具破坏性的方式。
干细胞在肺纤维化
广泛的细胞已被证明具有分化成肺细胞的能力。这些范围从肺部祖细胞,循环细胞,间充质干细胞(msc),诱导多能干细胞,胎盘干细胞和人类胚胎干细胞(色调)。治疗肺纤维化疾病的常见策略来取代受损上皮和内皮细胞,恢复正常的修复。msc和胎盘干细胞表达的标记所示肺上皮细胞注入后肺部疾病的动物模型108年),肺上皮细胞和胚胎干细胞的特性在体外(109年]。
最近,一项研究pitalieriet al。(110年)表明,HUES-3可以区分在体外在ATII细胞。此外,HUES-3-ATII移植到硅受损的老鼠明显减少炎症和纤维化的标志。此外,肺移植msc后立即bleomycin-induced受伤已被证明有有利影响与胶原蛋白沉积减少,纤维化和MMP的水平(111年]。
目前几乎没有临床试验调查这种高度肺纤维化的干细胞具有挑战性的问题。很明显,这是一个令人激动的新的潜在治疗,迫切需要进一步调查。
结论
经过多年的相对停滞在肺纤维化治疗仅限于消炎药、知识成倍地增加了在过去的十年。新概念有了重大变化的理解这个有趣的疾病。在当前范例,肺纤维化被认为是多因素疾病过程的结果。一般来说,人们认为基因易感宿主,暴露在有害的药物可能会导致结构细胞的表型变化。这将导致异常的细胞相互作用,最终纤维化。尽管几个核心纤维化机制的瓦解,新兴数据生成新的感兴趣的领域。
最具挑战性的领域之一是遗传学领域。有一个更好的理解所涉及的因素对肺纤维化发展。有趣的新发现ELMOD2 SP-C突变,ht和hTERT MUC5B,虽然之间的确切联系MUC5B分泌过多的航空公司和纤维化的过程还不清楚。也主要关心的,是进一步探索基因异常之间的联系和纤维化的模式和严重性在个别病人。此外,有新兴的证据作用的有害的代理。有前途的新数据已经公布在病毒的作用在纤维化的起始和传播过程中,尤其是对EBV。EBV IPF肺中确实发现了,虽然有EBV诱导纤维化的机制,许多问题仍悬而未决。气油比似乎是另一个有害的代理,虽然直到现在治疗倾心于对纤维化的形成的影响还不清楚。non-acid回流是否参与,和扩展这个可以治疗,在很大程度上仍然是未知数。另外,很明显,有害的代理和遗传易感性之间的关系需要更多的澄清。
纤维化的基本机制变得更加清晰,例如ATII细胞和myofibroblasts的重要作用,以及细胞因子网络的许多元素得到进一步发掘。其他新兴区域免疫的作用机制的启动和调制纤维化的过程。免疫学最近受到了新发现的推动。其中一个最令人兴奋的新发现是血清CCL18,替代激活巨噬细胞的标记,在IPF有预后价值。这指出可能的IPF巨噬细胞的作用。直到现在这些细胞在IPF相对被忽视。虽然CD4 + t细胞功能的开发已经由Th1-Th2范式,Th17通路的发现及其与亚群之间的关系(图1)打开一个新的和有趣的时代我们对适应性免疫调节的理解。这肯定会导致小说和更有效的治疗方法在IPF的自身免疫和炎症性疾病。
氧化应激的作用仍然是一个具有挑战性的领域氧化药剂治疗IPF仍未阐明。ER应激多领域的更新和许多有趣的发现已经发表在过去的几年里。ER压力之间的关系和其他免疫细胞活性和结构,特别是ATII,需要进一步调查。血管的角色间的知识相当差,但将变得越来越重要的角色不同的凝血通路现在得到很好的证实。下一步将试图达成整合视觉通路的作用和纤维化的过程和如何开发新疗法基于这些新发现。
一个完整的新领域是肺淋巴管。这一领域的进一步发展可能会导致新途径的发现纤维化的过程。最后但并非最不重要,来自不同来源的干细胞的发展无疑是与广泛的新的可能性。在体外,有许多有前途的观察,但有用的不同类型的干细胞停止无情的纤维化和恢复适当的修复过程仍然需要探索。
虽然看似的主要步骤和关键球员在纤维化的形成已确定,仍不过总体发病机制的重要探索健壮的模型,协调大量临床和科学观测。我们认为,当前数据的整合成一个“大画面”的概述纤维发生发展的有效的anti-fibrotic策略至关重要。
确认
作者的从属关系如下。维姆·a·Wuyts:肺病学实验室,呼吸医学部门,Katholieke项目鲁汶鲁汶和呼吸医学部门,鲁汶,比利时鲁汶大学医院。卡洛Agostini:部门的临床和实验医学、临床免疫学、帕多瓦大学、意大利的帕多瓦。(Katerina m·安东尼奥由于:胸医学部门,伊拉克里翁,希腊克里特岛大学。德摩斯梯尼Bouros:部门Pneumonology,医学院,德谟克利特色雷斯大学Alexandroupolis,希腊。雷切尔·c·钱伯斯:炎症和组织修复中心,伦敦大学学院,伦敦,英国。友谊医院文森特Cottin:济贫院土木里昂,路易斯·Pradel服务de Pneumologie中心de参考des疾病Pulmonaires罕见,我大学里昂,法国里昂。吉姆·j·伊根:板牙Misericordiae大学医院和圣文森大学医院,爱尔兰都柏林大学学院。巴特n Lambrecht:分子生物医学研究部门,VIB,绅士,比利时。里克吸蜜类鹦鹉:骨骼实验室开发和关节疾病,风湿病,肌肉骨骼科学部门,Katholieke项目,比利时鲁汶。 Helen Parfrey: Respiratory Medicine Division, Dept of Medicine, University of Cambridge and Papworth Hospital, Cambridge, UK. Antje Prasse: Dept of Pneumology, University Medical Center Freiburg, Freiburg, Germany. Carlos Robalo-Cordeiro: Dept of Pulmonology, University Hospital of Coimbra, Coimbra, Portugal. Eric Verbeken: Dept of Pathology, UZ Leuven, Leuven, Belgium. Johny A. Verschakelen: Dept of Radiology, UZ Leuven, Leuven, Belgium. Athol U. Wells: Interstitial Lung Disease Unit, Royal Brompton Hospital, London, UK. Geert M. Verleden: Lung Transplantation Unit, University Hospital Gasthuisberg, Leuven, Belgium.
脚注
感兴趣的语句
利益冲突的信息可以发现与本文的在线版本www.www.qdcxjkg.com
- 收到了2012年5月8日。
- 接受2012年8月24日。
- ©2013人队











{kind=link}
{kind=link}