





摘要
t箱转录因子4基因的罕见变异(TBX4)最近被认为是儿科肺动脉高压(PH)的一个新原因。它们的病理生理和对新生儿持续性肺动脉高压(PPHN)的贡献尚不清楚。我们试图定义与之相关的临床表现和组织病理学谱TBX4PH在新生儿和儿童中的变异。
我们评估了19例PH(包括PPHN)儿童的临床资料和肺组织TBX4下一代测序和拷贝数变异阵列鉴定的罕见变异。
变体包括6个17q23的删除,包括整个TBX4位点和邻近基因,以及12个可能的破坏性突变。10例患儿出现新生儿缺氧呼吸衰竭和PPHN,随后出院回家。PH在婴儿期或儿童期被诊断出来。3名儿童死亡,2名需要肺移植。相关异常包括动脉导管未闭、间隔缺损、足部异常和发育障碍,后者在缺失携带者中发病率较高。7例婴儿组织学显示肺远端发育异常和肺动脉高压重塑。
TBX4突变和17q23缺失是一种新的发育性肺部疾病的基础,在出生时和/或婴幼儿后期表现为严重的双相PH,通常与骨骼异常、心脏缺陷、神经发育障碍和其他异常相关。
摘要
TBX4突变和缺失与远端肺发育异常、新生儿持续肺动脉高压、儿童肺动脉高压、多重先天性异常和发育障碍有关http://bit.ly/2UXDrl3
简介
肺动脉高压(PAH)在婴儿和儿童中是一种罕见的疾病,患病率为每百万人4.8至8.1例[1- - - - - -3.],导致进行性右心衰和高死亡率,尽管最近在诊断和治疗方面取得了进展[4].多环芳烃是一种毛细血管前病变,是肺动脉高压(PH)的一种亚型。尽管多环芳烃具有异质性,但与肺循环相关的遗传缺陷是大多数家族性多环芳烃病例和特发性多环芳烃病例的基础。骨形态发生蛋白(BMP)受体2型基因突变(BMPR2)和其他bmp相关基因在约70%的家族性多环芳烃病例和20%的成人和儿童特发性多环芳烃病例中被发现[5- - - - - -8].最近的研究显示,T-box转录因子4 (TBX4),与小髌骨综合征(SPS)相关的基因[9],在儿科多环芳烃[8,10- - - - - -12].
在围产期,新生儿可能出现一种被称为新生儿持续性肺动脉高压(PPHN)的PH值,这是一种具有不同潜在病因的情况,导致肺血管阻力持续升高,并无法从胎儿循环模式过渡到产后循环模式。PPHN比儿科PAH更常见,发生率为0.18%,其中20%似乎是特发性的[13].尽管PPHN大多是可逆的,死亡率<10%,但一小部分典型的对治疗无反应的病例患有发展性肺部疾病[14].最近,TBX4在3例发育性肺病引起的新生儿缺氧呼吸衰竭中描述了罕见的变异[15,16,扩大了与这些基因缺陷相关的症状的范围。
考虑到潜在的重要性TBX4肺发育过程中的表达与TBX4和儿科PH值,我们收集了19例确诊的儿科患者的数据TBX4变异和寻求更精确地确定在婴儿和儿童的表现谱。
方法
本系列病例选取自2014年1月至2017年12月,来自各临床中心(补充表S1)以7例右心导管检查(RHC)或12例超声心动图(表1)在婴儿期或儿童期和aTBX4发现罕见变种通过临床或研究测试。小核苷酸变异(SNVs)是通过经认证的临床实验室或定制研究小组的下一代测序或通过Sanger测序(补充表S1).对于错义变体,通过polyfen -2 (http://genetics.bwh.harvard.edu/pph2)和Combined Annotation Dependent Depletion v1.3 (CADD)评估其对蛋白质结构的功能影响[17].次要等位基因频率(<0.05)通过搜索外显子组聚合联盟数据库[18].与ClinVar [19和ClinGen [20.)数据库。拷贝数变异(CNVs)由染色体阵列测定。根据美国医学遗传学学会cnv指南确定变异显著性[21]和snv [22].未识别的患者数据,包括生物特征、家族史和新生儿史、最初和随后的诊断研究以及功能数据、随访和结果,从登记处或医疗记录(补充表S1).临床获得的肺组织,如有,由单一病理学家(CG)重新分析。这项研究是遵照当地机构审查委员会的要求进行的。
结果
基因型描述
19例患者(包括2名兄弟姐妹)共检出18个不同杂合变异,其中6个涉及17q23.2位点的CNVs和12个TBX4SNVs (表2).cnv包括两个大小约2.2 Mb和约3.6-3.7 MbTBX4编码序列加上其他几个基因(图1一个).2.2 Mb CNV(病例1、3、4和5)是由于之前描述过的片段重复导致的17q23.1q23.2重复删除[23并在ClinGen上进行了报道。较大的CNV(病例2和6)在ClinVar仅报道过一次。snv是新颖的。其中,10个是可能的基因干扰变异,包括移码indel(案例7-11)、过早停止增益(案例12-14)和规范剪接位点(案例15和16)变异,3个是影响t盒DNA结合域共识的错义变异(案例17-19)。图1 b).在没有实验数据证明其基因破坏作用的情况下,位于T-box结构域下游的两个无意义和两个移码变异体被归类为可能致病。基于polyphen2和/或CADD得分分别为0.85-1和≥10-20,在所有脊椎动物物种中氨基酸位置的保守,以及13个人类TBX蛋白的T-box结构域的完全(p.Gly106和p.Leu186)或中度(p.Val218)保守,这三个错义突变被认为可能是致病的。在可确定遗传的8种变异中,有3种(37%)是可确定遗传的新创5例(62%)是家族性的,携带患者的兄弟姐妹感染了SPS(病例18)或确诊之前患有PAH(病例13和14),携带患者的母亲感染了SPS(病例12和15)、PAH(病例15)或无症状(病例16)。尽管被测试基因的数量和性质因中心而异(补充表S1),没有BMPR2、叉车箱F1 (FOXF1)或其他与ph相关的致病基因变异可在任何接受测试的患者中发现。

17q23删除位置和TBX4突变。a)在17q23.2区域发现基因组缺失。最小的缺失重叠区包含以下编码基因:MED13(大型Mediator复合体的亚单元,无处不在),INTS2(Integrator复合物亚基,肺表达低),BRIP1的成员DEAH解旋酶家族的贡献乳腺癌易感基因1活动,无处不在),BCAS3(与血管生成和细胞粘附、细胞外基质组织、肽酶活性和转化生长因子-β信号等相关过程有关,在肺中普遍存在高表达[49]),TBX4(转录因子参与后肢、肺和尿囊等几个发育过程),TBX2(包括心脏、大脑和四肢在内的多个器官的转录因子[42]),PPM1D(调节DNA损伤反应途径的蛋白磷酸酶,主要表达于大脑,与人类发育障碍有关[50),APPBP2(无处不在,与微管相互作用,与运输和/或处理有关)和USP32(一种泛素特异性蛋白酶)。在这些基因中,只有TBX4,TBX2而且PPM1D在人类疾病中有罕见的变异。加州大学圣克鲁斯分校(UCSC)基因组浏览器(GRCH37/Hg19) [51].b) TBX4蛋白突变位置。TBX4蛋白用灰色表示,T-box区域用粉色表示。三个误义变体(黄色)位于t框域。其中三个移码变体(紫色)位于上游或t框域内,而两个位于下游。两个无意义突变(绿色)位于下游。鱼:荧光原位杂交;CCDS:共识编码序列项目;tRNA:转移RNA。
临床表型和结果
所有患者均为足月或晚期早产(中位40.0周,四分位差(IQR) 38.0周),男女比例为2.16:1,CNVs和SNVs相似(表1而且3.).中位胎龄出生体重正常(中位胎龄3075 g, IQR 2450 g),尽管3个新生儿胎龄较小(出生体重z评分<−1.28)[24].11例新生儿需要有创呼吸支持。19例新生儿中10例(53%)最常见的表现为PPHN,其中8例严重(氧合指数>25或需要体外膜氧合(ECMO))。4例新生儿(10%)出现无PH的短暂性呼吸窘迫;其余5例新生儿的过程都很平静。所有患者在新生儿重症监护病房期间存活下来,并在中位年龄37天(范围7-180天)出院回家,其中6人使用家庭氧气,2人使用西地那非。在两名有PPHN病史的患者中,尽管接受了治疗,但在新生儿期后右心室收缩压(RVSP)仍然升高,他们都在婴儿期死亡,分别为5个月和8个月。在其余8例PPHN患者中,PH在出生后的头几个月内出现改善或消退。在童年后期,17名幸存的患者接受了心脏病学评估,通常是新发的低氧血症或心肺症状(表1).这些患者被诊断为PH(中位年龄1.5岁,IQR 0.17岁)。在本报告发表时,患者的随访时间不同,本研究是回顾性研究(中位数10.0年,IQR 4.7年)。尽管接受了多种血管扩张剂治疗,仍有3名患者发展为终末期肺病:1名患者在29岁时死亡,2名患者接受了心肺移植,1名患者11岁,1名患者18岁。11例患者在最后一次评估时仍然存在慢性PH值,尽管7例患者使用了多种PH值靶向治疗,1例患者使用了单一PH值靶向治疗,3例患者仅使用补充氧治疗。其余3例患者(病例14、15和16:3个月至10年)的PH在最后一次随访时已消退,其中1例未使用药物,而2例仍在使用单一血管扩张剂治疗。13例可检索信息的患者中有10例(77%)存在骨骼异常,包括具有典型足部异常的SPS [9].其他神经和发育障碍包括自闭症、小头畸形症、神经感觉障碍和肌肉张力异常。
心脏成像和血流动力学分析
其中7例患者仅通过超声心动图进行评估,12例患者在治疗过程中至少发生一次RHC (表4).10例(53%)患者在新生儿期出现全身性或系统上性RVSP。8例动脉导管未闭(PDA),其中4例持续超过新生儿期。其中2例患者在最初诊断PH值时出现左向右分流:1例在4岁时手术闭合(病例4),1例血流动力学不显著(病例5)。2例患者(病例5和11)在诊断RHC处持续出现右向左分流。8例(42%)患者存在房间隔缺损。在12例行心导管置管的患者中(中位年龄1.5岁,IQR 0.17岁),显示出较高的平均肺动脉压(mPAP) (60.0 mmHg, IQR 57.5mm Hg)和肺血管阻力指数(中位16.6 Wood单位,IQR 10.7 Wood单位)。然而,12例患者中只有6例符合美国胸科学会指南中严格诊断多环芳烃的所有标准[25].在6例连续RHCs患者中,随访时mPAP和肺血管阻力指数均升高或升高(数据未显示)。在10例接受急性血管反应性测试的患者中,8例(80%)未表现出mPAP下降至少10 mmHg至<40 mmHg [25].6例肺-全身血流量比降低,表明明显的右向左分流。肺血管造影显示弥漫性解剖和血管异常,包括曲折的肺小动脉,异常的毛细血管红肿,小肺静脉和小静脉,肺静脉阻塞1例(未显示)。
表型特征和变异类型
表3比较6例CNV和13例SNV携带者的临床、功能特征。尽管我们观察到CNV携带者的相关心脏和足部异常发生率更高,但只有发育障碍达到了统计学意义(100%)与33%, p=0.029),与其他研究结果一致[10].我们还观察到SNV携带者RHC功能严重程度更大的趋势,尽管这可能反映了该组首次插管时年龄较大。补充表S3,比较已发布的17q23删除,包括和不包括TBX2 / TBX4包括我们系列在内的基因座显示,先天性心脏缺陷的患病率更高(57%)与0%, p=0.02), PH的存在也有类似的趋势(57%与17%, p=0.16)TBX2 / TBX4包容的删除。
成像研究
胸部图像只能在部分病例中收集(图2,补充表S2).新生儿胸片示肺发育不全(n=5)图2一个)、漏气及/或毛玻璃浊度;婴幼儿胸片(n=4)显示鼻中隔增厚伴多灶区呼吸困难、支气管增厚和毛玻璃影(图2 b).1 - 18岁(n=5)的CT扫描显示了一系列的发现,包括多灶性磨玻璃样影、蜂窝状和交替灶性囊变、提示小叶和大叶纤维化的浓缩区域和结节(图2 c- f)。
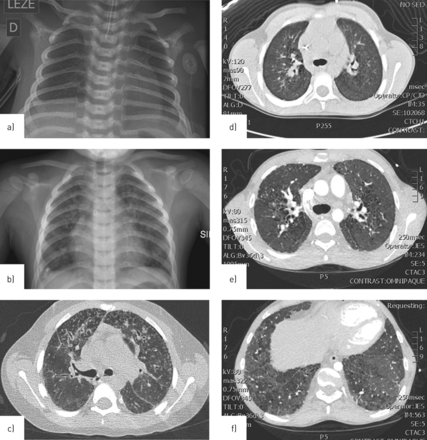
肺部成像。a)病例1:第1天胸部x线片显示肺超通透、发育不良,血管标记差。b, c)病例19:3岁胸片示磨玻璃影和支气管增厚(b);病例10:8岁时胸部CT显示弥漫性磨玻璃影、间隔增厚、肺动脉肿大、散在肺气肿;e, f) 18岁,间质纤维化明显增加,上叶和下叶中央花叶密度区,小囊性区域,部分支气管扩张,肺部分区域受累较少,舌区出现实质大泡。
肺组织病理学
7例患者获得病理材料,并对肺发育和血管重建的组织学特征进行半定量分析(表5).所有样本均表现为弥漫性肺泡生长异常和不同程度的肺动脉壁重塑,伴或不伴纤维内膜增生。未见丛状病变或血管坏死。在早期出现严重症状并在新生儿期进行活检的患者中(病例1、7、12和13;图3一i),组织学显示远端肺发育严重中断,其特征是小叶生长延迟,远端空气间隙扩张,未成熟的肺泡没有继发间隔,通常排列着反应性的立方上皮细胞。远端气腔增大,肺泡简化。所有病例均有间质增厚征象;3例显示,在肺间质糖原病中观察到苍白和未成熟间充质细胞的存在[263例间质纤维化呈斑块状。所有患者均有肺动脉高压重塑的迹象。在两个病例中可以看到背靠背的细支气管剖面,其中一个显示支气管血管招募,包括肺内支气管-肺吻合术。总的来说,这些结构变化指向远端肺发育的所有间室的严重破坏,反映了在管状或早期囊状阶段的生长停滞。儿童活检患者的肺组织学(病例10a, 18和19,10b-外植体;图3 j-r)显示支气管血管系统招募的证据,包括原支气管和扩张的支气管静脉和毛细血管,除了肺泡简化和肺动脉重构。气道重塑和功能损害的特征是气道壁增厚,肺泡内巨噬细胞和多核巨细胞的胆固醇晶体数量增加(未显示)。
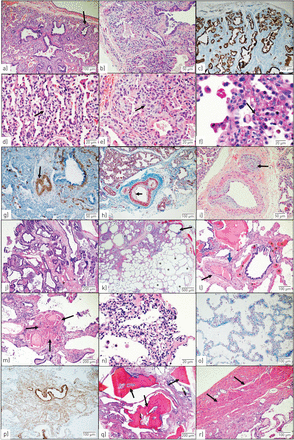
代表性组织病理学图像。新生儿期的早期活检显示明显发育不全的呼吸小叶(a, b;苏木精、伊红(H+E)染色),小而圆的未成熟肺泡,有的呈管状,有的呈囊状(c;细胞角蛋白染色)。细支气管末端延伸至鼻中隔(A上的箭头),表明肺泡发育明显迟缓。间质因未成熟的、苍白的间充质细胞的存在而增宽(d、e上的箭头;H + E染色);这些细胞呈PAS阳性(f上的箭头;PAS染色),类似于肺间质糖原病细胞的特征。肺动脉高压重塑伴肌肉肥厚(g上的箭头; smooth muscle actin stain) along with fibrointimal proliferation (arrow on h; trichrome stain) (i; H+E stain) evolving to complete pulmonary arterial obstruction (arrow on i) is identified. Samples from childhood (j–r) showed lack of alveolar development evidenced by multiple foci of back-to-back bronchiolar profile (j; H+E stain), and a terminal bronchiole extending to the pleura surface (arrow on k; H+E stain). There were diffuse alveolar growth abnormalities characterised by large, dilated and simplified alveoli (examples are labelled with asterisks on k). Marked vascular changes included pulmonary arterial hypertensive remodelling (black arrow on l; H+E stain); recruitment of bronchial vasculature including intrapulmonary bronchopulmonary anastomosis (blue arrow on l), bronchial veins and microvessels (asterisks on l); and marked muscular hypertrophy of lymphatic vessels (arrows on m; H+E stain). Pathologic remodelling of the interstitium is seen with the presence of proliferating capillaries (n; H+E stain), fibrosis (o; trichrome stain), focal smooth muscle proliferation (p; smooth muscle actin stain) and multiple areas of bone formation (arrows on q; H+E stain). In one case (10b) marked pleural smooth muscle muscularisation is noted (arrows on r; H+E stain).
病例10可进行纵向组织学分析(2岁活检,18岁移植)。最显著的组织学发现包括气道/肺泡生长障碍的进展,其特征是多灶性、明显发育不全和曲折的背对背细支气管结构,类似于先天性肺气道畸形[27或腺泡发育不良[15].此外,肺动脉、淋巴管、气道和胸膜血管显示明显的内侧壁增厚,在随后的移植体中可见骨形成区域,提示间充质发育不良。间质增厚伴有纤维化,IBA招募和间质毛细血管增生发展。
讨论
TBX4变异携带者有发生远端肺发育异常、PPHN、儿科起病性PH、包括先天性心脏缺陷和典型足畸形在内的多种先天性异常和发育障碍的风险。我们的大多数患者(63%)表现为双期临床过程,包括PPHN和新生儿呼吸衰竭,在1个月大左右明显缓解,随后是婴儿期或儿童早期的慢性PH。值得注意的是,这种形式的PH符合毛细管前表型;然而,考虑到并发肺畸形的程度,这些人在技术上不符合世界卫生组织分类第1组PH (PAH)的传统标准,很可能被归类为第3组(慢性肺部疾病和/或缺氧引起的PH) [6].我们对发展性肺部疾病患者的描述TBX4除了特发性PH外,相关PH值可能还有一些因果关系,包括慢性呼吸系统疾病和缺氧。鉴于在我们的回顾性系列中难以定义这些病因,我们使用术语PH (与多环芳烃)TBX4-相关血管疾病。
弥漫性发育性肺疾病是一种罕见的疾病,与肺气道和血管发育的主要机制异常有关,包括AD、先天性肺泡发育不良和肺泡毛细血管发育不良伴肺静脉错位(ACDMPV)等诊断,ACDMPV是一种致命的新生儿疾病FOXF1变异(28].新出现的证据表明,发育性肺疾病在表型上是异质性的。ACDMPV最近在年龄较大的婴儿中有报道,似乎毛细血管PH值前,这表明FOXF1 -相关疾病的临床范围比最初认为的更广[29,30.].TBX4据报道,在出现致命AD的新生儿中有变异[15], 1例为致死性先天性牙槽发育不良,1例为不明确的牙槽生长异常,存活时间超过8个月[16].我们的病理发现,具有更广泛的年龄和临床表现,阐明了PH的发病机制TBX4突变体,尽管我们不能排除选择偏差因为活检是在临床基础上获得的,没有统一的标准。本研究证实了各种发育异常影响的肺泡,间质和血管结构的基础TBX4这些特征表明肺内胚层生长受阻,严重影响气道/肺泡发育,间充质发育不良,表现为肺动脉高血压重塑、显著的IBA和间质疾病的其他表现。我们的纵向观察表明,这些病理胎儿过程在出生后继续,在某些病例中随着年龄的增长而进展,伴随着血管和淋巴的逐渐重塑和侧枝循环的发展,导致儿童或青年期的进行性PH和终末期肺病。
影像学研究表明,新生儿表现伴有肺发育不良和肺泡功能障碍,在某些病例中,随着时间的推移出现进行性支气管和间质改变。在一组患者(病例2、12、13、18和19)中,突出的间质性肺疾病和PH值结合表明,与肺疾病相关的机制,可能包括慢性缺氧,可能是一个促成因素。然而,RHC数据显示,在大多数情况下,无论是否有实质疾病,严重的肺血管疾病,与参考儿科PAH队列(REVEAL队列[31])。对血管反应性测试缺乏反应也被描述在患有BMPR2与无基因突变相比[32],表明PH的遗传形式具有独特的血管病理生理。
Kerstjens -Frederikse等.[10首先描述的TBX4在被诊断为特发性/家族性多环芳烃和SPS的儿童队列中,有30%发生了突变,而在对照组成人队列中,只有2.5%发生了突变。Z胡等.[11]计算出7.7%的患病率TBX4在一个更大的儿科多环芳烃队列中发现相关疾病。l产品等.[12也估计为7.5%TBX440例多环芳烃婴儿中有3例发生突变。Eyry等.[8[词汇发现TBX4法国儿童和成人患多环芳烃的患病率分别为1 / 36(2.8%)和4 / 168 (2.4%)BMPR2变异分别为19.4%和14.3%。频率较低的TBX4在成人发病型多环芳烃中检测到的变异比在儿童发病型多环芳烃中检测到的变异多,总体突变频率估计为1.5%(1633例中有25例)[8,10,11,33,34].总的来说,在这个儿科系列的中心中缺乏标准化的纳入和诊断标准,排除了任何关于患病率的推断或比较骨形态发生蛋白相关和其他多环芳烃基因
TBX4变异与多种异常相关,与肺以外关键发育过程的中断一致。并不是所有的表型特征都具有相同的表现性。SPS的外显率较高,PH的外显率较低[10].这种选择性外显可能取决于变异本身及其对蛋白质剂量和功能的影响。然而,具有共同变异的亲戚之间的表型异质性,有些只具有SPS,而另一些具有SPS和PH的组合,表明TBX4与PH相关的PH不是纯单基因的,多种先天和环境因素可能起作用,与在其他遗传形式的PH中观察到的情况类似[6].在BMPR2多环芳烃外显率仅为20%,次要因素调节表达和疾病进展[6].我们观察到2:1的女性患病率,可能归因于男性胎儿的选择性浪费或原始性别比例异常。在鉴定致病基因之前,已在成人PH中观察到这种差异[35],并在大型登记册内确认[36].性别依赖的激素因子的作用[37和修饰基因[38,随后在BMPR2与多环芳烃相关,女性居多。假设类似的机制也存在于TBX4.
PH值的严重程度不一定与蛋白质表达的预测水平相关,正如在FOXF1相关的ACDMPV [39].有些CNV病例具有完全性TBX4在我们和其他系列中,单倍不足的严重PH值低于具有较少基因破坏snv的其他基因[10],提示显性负性影响或与遗传或环境因素相互作用,这使得基于遗传的预后具有挑战性。
在我们的CNV患者中(1-6例)发育障碍的发生率高于snv患者(7-19例),这表明邻近基因的作用,尽管在CNV组中不能排除PPHN或ECMO等产后因素。将我们的案例与先前发表的17q22-q23.2删除进行比较(补充表S3),涉及连续的删除TBX2而且TBX4与保留这两个基因的人相比,这两个基因更经常导致PH和先天性心脏缺陷,这表明这两个基因在该综合征的心血管成分中起着主要作用,而发育障碍的患病率均相同TBX2/4参与,再次表明了其他邻近基因的作用。
TBX2有助于气道生长和分支[40]和心内膜垫的形成,在室间隔缺损的发病机制中起关键作用[41].TBX2在心脏间隔缺损、发育迟缓和骨骼异常的个体中发现了错义突变,但没有PH [42].我们可以推测,在先天性心脏缺陷复杂的发病机制中,TBX4而且TBX2作为致病基因或修饰基因发挥着重要而独特的作用TBX4导致了该疾病组的PH发病和严重程度。相反,发育迟缓、听力损失和骨骼缺陷在独立研究中同样具有代表性TBX2 / TBX4参与,提示多重基因相互作用在发病机制的非心血管异常。
虽然TBX4最初被认为是后肢发育的关键因素[43],在发育中的肺间质中高度表达[44,具有高度保守性TBX4调控其时空表达的增强子序列[45].纯合子的TBX4突变小鼠胚胎在胚胎第10.5天死于尿囊形成缺陷和胎盘功能不全[46和条件肺间质TBX4减氧导致肺发育受损[44].TBX4与成纤维细胞生长因子10相互作用(FGF10),是肢体和肺芽生长和气道分支的重要调节因子[47],这可能解释下肢/肺联合表型TBX4相关的疾病。的FGF10途径也调节甲状腺转录因子1 (TTF1)的上皮表达NKX2.1基因),是肺泡发育和表面活性剂合成的关键因素[48],这可能导致新生儿呼吸道症状TBX4突变体。
本研究的局限性包括招募偏向于儿科和新生儿形式的TBX4与PH值;缺乏标准化的纳入标准,无法估计的流行程度TBX4婴儿PPHN、婴儿/儿童PH和先天性心脏缺陷的变异;随访时间的变化排除了结果比较。
总之,这项研究证实了这一点TBX4变异是各种复杂的发育性肺疾病的基础,导致一系列的临床表现,包括PPHN、新生儿缺氧呼吸衰竭、间质性肺病和慢性/进行性儿科PH,通常与多系统异常相关。表型的可变性和复杂性及其与其他ph相关基因缺陷的潜在重叠要求进行彻底的分子遗传检测,包括TBX4-包括诊断面板结合CNV微阵列,以捕获小的和大的变异。我们所描述的双相发展,以出生时缺氧呼吸衰竭和后发PH为特征,提示患有严重PPHN的婴儿,特别是特发性PPHN的婴儿,应在婴儿期和幼儿期进行适当的超声心动图随访,并应进行检查TBX4阳性和/或存在提示性特征如先天性心脏缺陷、足部异常和SPS的变异。为了更好地描述基因型-表型的相关性并确定未来的诊断和治疗策略,需要进行更大规模的队列和人群基础研究。
补充材料
脚注
本文的补充资料可从www.qdcxjkg.com
作者贡献:O. Danhaive和S.H. Abman设计了这项研究并监督了该项目;O. Danhaive, C. Galambos, E.D. Austin和M.P. Mullen共同撰写了手稿;C. Galambos与J. Johnson和R. Boldrini合作进行病理学研究;J.T. Shieh在C. Surace、D. Peca、P.B. Agrawal、M.W. Pauciulo和W.C. Nichols的贡献下进行了遗传分析;M.P. Mullen和D. Ivy对心脏病学和血流动力学分析做出了贡献;E.D. Austin、M. j . Kielt、M. Griese和S.H. Abman对肺学分析有贡献;N. Schwerk, N. Ullmann, R. Cutrera, I. Stucin-Gantar, C. Haass和M. Bansal对临床数据均有贡献。
利益冲突:C. Galambos没有什么可透露的。
利益冲突:M.P. Mullen曾在联合治疗、Actelion、伊卡利亚和GSK赞助的试验中担任现场首席研究员,并在提交的工作之外获得Actelion的旅行支持。
利益冲突,j。t。谢伊没什么可透露的。
利益冲突:N. Schwerk没有什么可透露的。
利益冲突:m·j·基尔特没有什么可透露的。
利益冲突:N.乌尔曼没有什么可透露的。
利益冲突:R. Boldrini没有什么可透露的。
利益冲突:第一,斯图辛-甘塔尔没有什么可透露的。
利益冲突:C.哈斯没有什么可透露的。
利益冲突:M. Bansal没有什么可透露的。
利益冲突:P.B.阿格拉瓦尔没有什么可透露的。
利益冲突:j·约翰逊没有什么可透露的。
利益冲突:D.佩卡没有什么可透露的。
利益冲突:C. Surace没什么可透露的。
利益冲突:R.库特雷拉没有什么可透露的。
利益冲突:M.W. Pauciulo没有什么可透露的。
利益冲突:W.C.尼科尔斯没有什么可透露的。
利益冲突:M. Griese没有什么要透露的。
利益冲突:D. Ivy(通过科罗拉多大学医学院)与Actelion、拜耳、礼来和联合治疗公司签订了咨询和研究合同。
利益冲突,S.H.阿布曼公司没什么可透露的。
利益冲突,e。d。奥斯汀没有什么可透露的。
利益冲突:O. Danhaive没有什么可透露的。
支持声明:本出版物部分得到了Frederick和Margaret L. Weyerhaeuser基金会、Jayden de Luca基金会(D. Ivy, C. Galambos)、NIH赠款R01HL114753和U01HL121518 (S.H. Abman)、NIH/NCATS科罗拉多州CTSA赠款编号UL1 TR002535、NIH赠款HL105333 (W.C. Nichols, M.W. Pauciulo)的支持,以及意大利帕尔马Chiesi基金会(O. Danhaive, R. Cutrera, D. Peca)的无限制赠款。本文的资助信息已存入交叉参考基金登记处。
- 收到了2018年10月17日。
- 接受2019年4月19日。
- 版权所有©ERS 2019











![Position of 17q23 deletions and TBX4 mutations. a) Genomic deletions identified in the 17q23.2 region. The smallest deletion overlap region encompasses the following coding genes: MED13 (subunit of the large Mediator complex, ubiquitous), INTS2 (subunit of the Integrator complex, low lung expression), BRIP1 (a member of the DEAH helicase family contributing to BRCA1 activity, ubiquitous), BCAS3 (associated with angiogenesis and related processes such as cell adhesion, extracellular matrix organisation, peptidase activity and transforming growth factor-β signalling, ubiquitous with high expression in lung [49]), TBX4 (transcription factor involved in several developmental processes including hindlimb, lung and allantois), TBX2 (transcription factor involved in patterning of several organs, including heart, brain and limbs [42]), PPM1D (protein phosphatase that regulates the DNA damage response pathway, mostly expressed in brain and associated with developmental disability in human [50], APPBP2 (ubiquitous, interacts with microtubules and is associated with transport and/or processing) and USP32 (an ubiquitin-specific protease). Among these genes, only TBX4, TBX2 and PPM1D have rare variants described in human disease. University of California Santa Cruz (UCSC) Genome Browser (GRCH37/Hg19) [51]. b) Mutation positions in TBX4 protein. The TBX4 protein is represented in grey, with the T-box domain in pink. The three missense variants (yellow) are located in the T-box domain. Three of the frameshift variants (purple) are located either upstream or inside the T-box domain, while two are located downstream. The two nonsense mutations (green) are located downstream. FISH: fluorescent in situ hybridisation; CCDS: Consensus Coding Sequence project; tRNA: transfer RNA.](http://www.qdcxjkg.com/content/erj/54/2/1801965/F1.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}