





文摘
长期口服皮质类固醇(OCS)使用严重哮喘患者与重大的不良影响。
这40周,随机,双盲试验评估的OCS-sparing潜力tralokinumab严重,患者不受控制的哮喘需要维护口服避孕药治疗+吸入型皮质类固醇激素/长效β2受体激动剂。总体而言,140名患者被随机tralokinumab 300毫克或安慰剂每组(n = 70)皮下接种每2周。主要终点是百分比变化从基线平均40周后口服剂量,同时维持哮喘控制。二级终端包括规定维护患者口服剂量的比例≤5毫克,那些规定维护口服剂量减少≥50%和哮喘恶化率。安全也得到了评估。
从基线下降40周,最后平均每日口服剂量tralokinumab和安慰剂之间没有明显不同(37.62%与29.85%;p = 0.271)。没有任何二次端点处理间差异显著。总体而言,报告的不良事件和严重不良事件是相似的tralokinumab和安慰剂组。虽然更大比例的tralokinumab-treated患者上呼吸道感染(35.7%与14.3%),没有报告病例的肺炎。
总的来说,tralokinumab并不证明OCS-sparing效应严重哮喘患者。
文摘
Tralokinumab没有展示重要OCS-sparing好处与安慰剂严重哮喘http://ow.ly/kzCt30mC43j
介绍
哮喘是一种异质性疾病的特点是慢性气道炎症和高反应性影响全世界∼3.34亿人(1]。这些,估计有5 - 10%的患者有严重的哮喘仍控制不足,尽管治疗高剂量吸入激素(ICS) /长效β2受体激动剂(腊八粥)[2]。这些病人可能规定维护口服皮质类固醇(OCS)来管理他们的症状和/或预防急性加重(2]。然而,累计使用(包括频繁和间歇使用)的口服避孕药与重大不利影响(3,4),从而减少病人的健康相关的生活质量(4]。因此,新疗法减少商务接触频繁的设置需要严重的哮喘。
白介素(IL) -13,可以诱发炎症的多效细胞因子(5,6),已被确定为一个潜在的治疗目标患者严重,不受控制的哮喘(7,8]。Tralokinumab,免疫球蛋白G4人类单克隆抗体,强有力地特别是中和IL-13,从而抑制信号通过IL-13受体(9,10]。在第二阶段试验中患者的严重,不受控制的哮喘,tralokinumab改善肺功能,但没有减少哮喘恶化年率(aa)或改善哮喘控制的措施11,12]。然而,在事后患者亚组分析分组人口的一个激活的证据IL-13轴,如高浓度血清periostin或dipeptidyl peptidase-4, tralokinumab改善aa (11]。这个观察表明,某些亚种群的严重哮喘患者可能回应tralokinumab治疗。两大三期试验,STRATOS 1和2,对流层之前报道,证实tralokinumab没有改善aa的所有人口患有严重哮喘(13]。此外,在第二阶段试验相比,periostin和dipeptidyl peptidase-4没有显示预测响应tralokinumab治疗要么STRATOS 1和2。然而,这些试验表明,在与分级分组人口的严重哮喘患者呼出一氧化氮(F伊诺)浓度≥37磅的,可能有一个增强的好处与tralokinumab [13]。
考虑到需要减少严重哮喘患者口服避孕药的要求,治疗可能允许逐渐减少口服避孕药没有丧失疾病控制是必要的。然而,这个试验开始时,没有临床试验已经在严重哮喘患者进行商务代理,减弱IL-13信号维护。因此,这一阶段的目的3对流层试验(NCT02281357)是评价的能力减少口服避孕药使用tralokinumab严重哮喘患者需要维护口服避孕药治疗结合ic /腊八。的主要目标是确定附加tralokinumab治疗与安慰剂比较,提供OCS-sparing福利。次要目标是评估tralokinumab的影响在这些患者的比例规定商务维护≤5毫克剂量的治疗期间,患者的比例≥规定OCS维护用量减少50%,与安慰剂相比。此外,aa tralokinumab是评估的影响。
方法
试验设计和病人
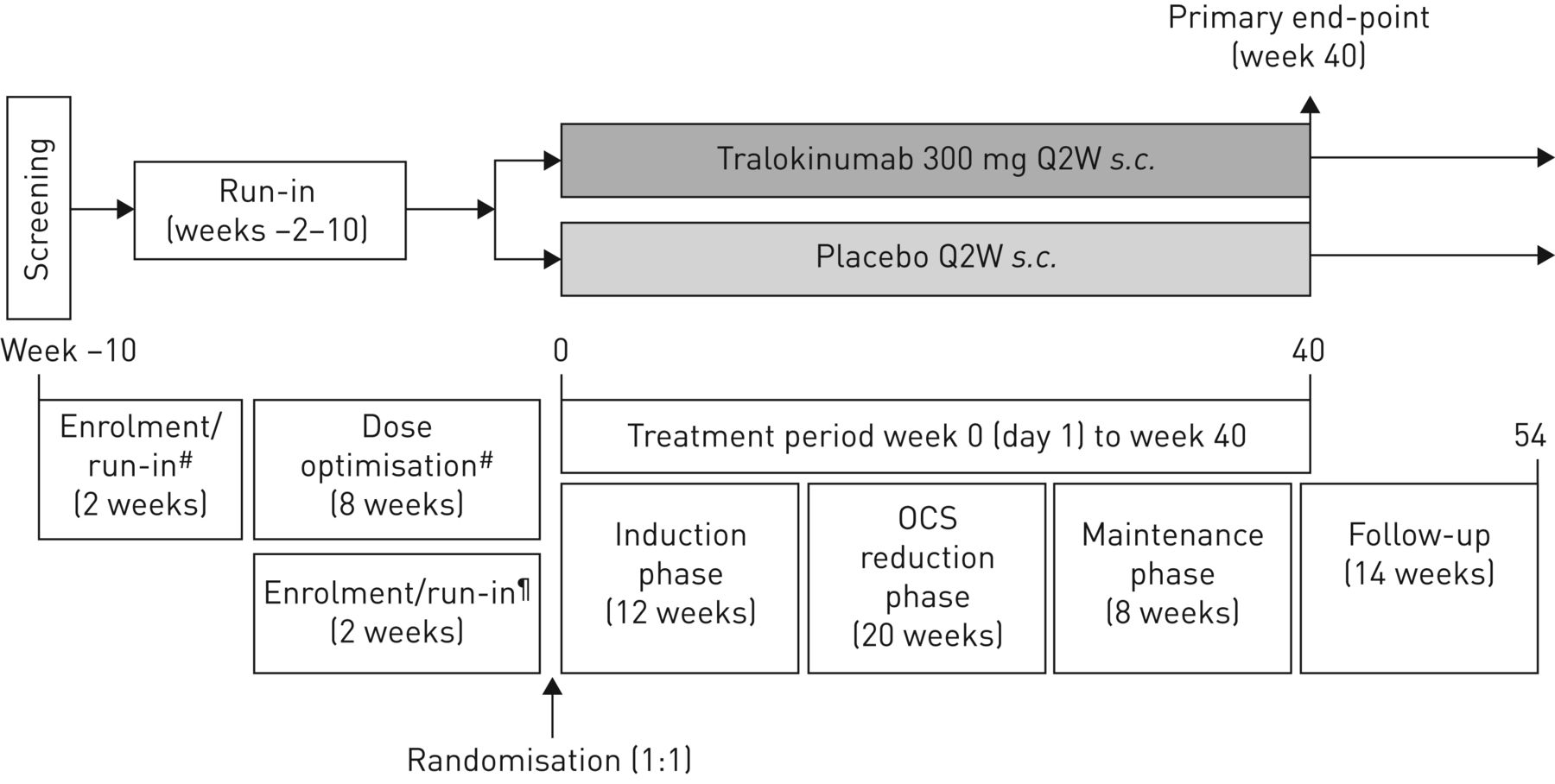
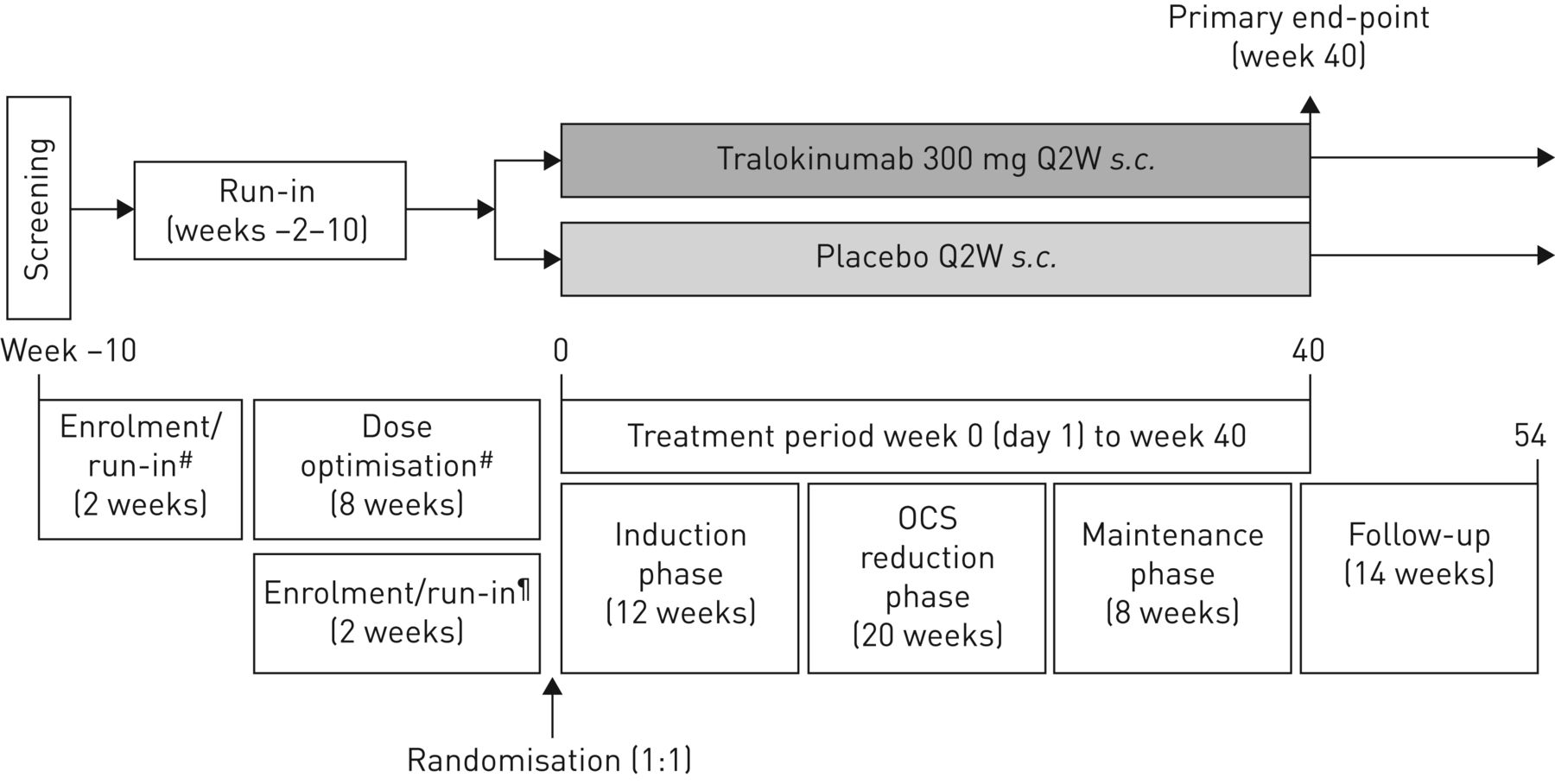
试验设计的全部细节已经公布之前(14]。简而言之,这是一个随机、双盲、安慰剂对照,与这些相应平行的组织,多中心,3期临床试验患者的严重,不受控制的哮喘需要维护ICS /腊八和口服避孕药。12 - 75岁的男性和女性都有资格,需要有以下标准包含:哮喘≥12个月,与中期的日常要求或大剂量ICS(每日总剂量≥500µg丙酸干粉或同等剂量)≥6个月12个月前报名;physician-prescribed ICS(每日总剂量≥500µg丙酸干粉配方当量)和腊八≥3个月前报名;口服避孕药治疗6个月前访问1;和商务之间的每日剂量稳定≥7.5毫克和≤30毫克(或强的松龙等效)每日或每日等效≥1个月前报名。提供了完整的纳入和排除标准的第1部分在线补充附录。
在最初报名(参观1)之后,病人进入一个试车周期2周(如果有记录失败的口服剂量减少≤6个月前访问1)或2周加上一个试车期8周优化时期(有些病人的优化周期比较短(即。那些在早些时候达到最佳剂量)建立最低有效剂量口服避孕药(由每2周剂量滴定(Q2W);图1)。
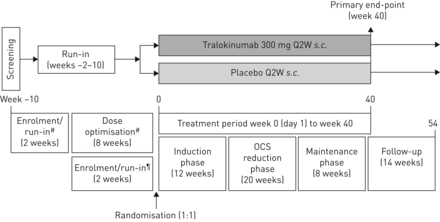
对流层试验设计。在最初报名(参观1)之后,病人进入试车周期2周或磨合过程加上一个2到8周优化时期建立规定的最小有效剂量口服皮质类固醇(OCS)。患者保持在目前规定吸入皮质类固醇/长效β2受体激动剂和任何额外的控制器药物,没有改变,从招生,整个试车/优化和治疗时间。所有患者进行了随机(1:1)和进入40周的治疗期组成的感应,口服避孕药的剂量减少和维护阶段。安全评估,尽早随访期间(后处理周44和54)。#:没有记录失败的患者在口服避孕药剂量减少≤6个月前访问1被要求完成8周口服剂量优化期间每2周(建立剂量滴定(Q2W)),在完成两磨合过程;¶:记录失败患者在口服避孕药剂量减少≤6个月前访问1被认为是最佳的口服避孕药剂量和直接进入试车周期2周。
符合条件的患者被随机1:1比例在0周(基线)接收tralokinumab 300毫克或安慰剂南卡罗来纳州。Q2W和进入一个由40周的治疗时期三个阶段:12周的诱导阶段,提供商务减量阶段和一个8周维护阶段。随机患者按年龄分层组(成年人与青少年)和基线口服剂量(成人;≤10毫克与> 10毫克或强的松龙)。两次的随访期内由周44和54安全评估。随机的细节和致盲(包括流程用于管理试验药物)和完整的试验过程提供了第二节在线补充附录。
这个审判符合赫尔辛基宣言和良好的临床实践(国际协调会议)和阿斯利康生物伦理学政策。审判独立伦理委员会批准,所有参与中心和所有病人提供书面知情同意。
端点
主要疗效终点是基线的变化百分比在最后40周后平均每日口服剂量,同时维持哮喘控制。二级终端包括规定维护患者的比例≤5毫克口服剂量,患者的比例规定维护OCS剂量减少≥50% aa 40周。一个哮喘恶化被定义为哮喘恶化,需要临时增加系统性皮质激素≥3天或导致一个急诊室或紧急护理访问造成哮喘,导致临时增加全身糖皮质激素治疗≥3天症状或因哮喘住院住院。
探索性终端包括减少患者的比例从基线在最后平均每日口服剂量,和最小二乘(LS)平均绝对百分比变化从基线prebronchodilator用力呼气量在1 s (FEV1),从基线在哮喘控制问卷(ACQ) 6和哮喘生活质量问卷(AQLQ;标准化病人≥12岁)分数和一个评估基线生物标志物之间的关系(F伊诺)和tralokinumab对商务的影响减少剂量和临床疗效。安全终端包括不良事件和严重不良事件的发生率和频率(节约)和收集血样测定的临床化学、血液学和验尿。安全方面的试验是由一个独立的监控数据和安全监测委员会。所有住院治疗上,急诊室和urgent-case互访,恶性肿瘤和心血管事件/脑血管事件被一个独立的专家委员会裁决。潜在的过敏反应事件被蒙蔽的外部评估评估者。更多细节的评估试验终端提供的第三节补充附件。
统计分析
据估计,≥55岁患者每组需要试验来检测之间的差异50%主要终点tralokinumab和安慰剂,90%的力量,使用一个双边测试在5%的显著性水平。基于STRATOS 1的分析试验的结果,确定F伊诺高(≥37磅)作为biomarker-positive人口(14),试验方案的修改包括患者F伊诺≥37磅作为主要分析人口,> 50%的试验提供人口高F伊诺水平(≥37磅)。如果< 50%的人口F伊诺≥37磅,主要分析人口包括患者F伊诺≥30磅。如果< 50%的人口F伊诺≥30磅,例如人口(即。所有患者任何级别的F伊诺)将成为主要的人口进行分析。占多重性,分层测试策略是用于中小学的结果。生物识别技术和信息科学的统计分析,阿斯利康,使用SAS(版本9.4;SAS研究所卡里、数控、美国)和额外的验证软件(在适当的地方)。更多细节的统计分析方法,分析了小学、中学和探索性的终端可以在第四节补充附件。缺失数据的影响在小学、中学和探索性端点使用敏感性分析(第五节的探索补充附件)。
结果
疾病病人的人口统计数据和基线特征
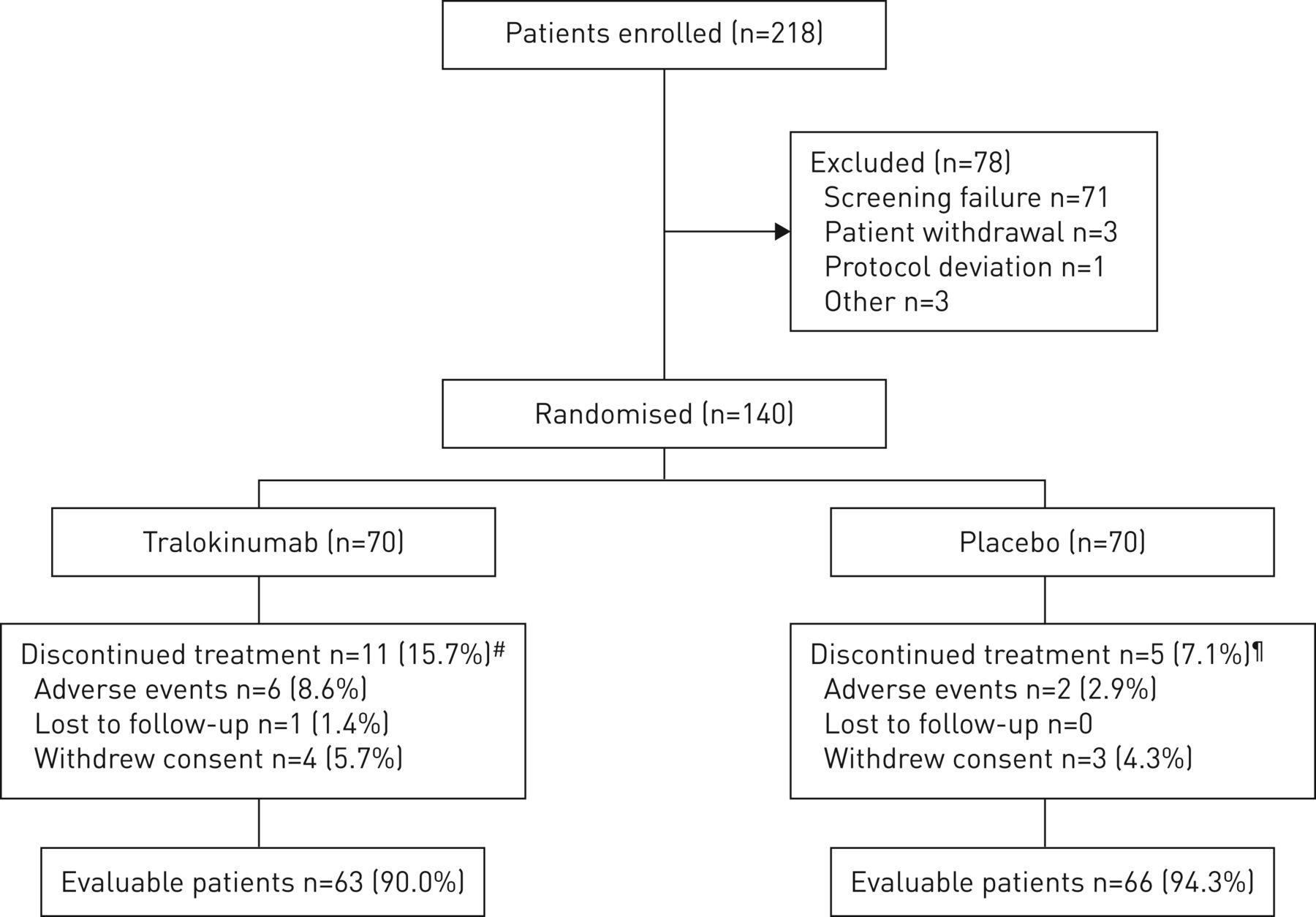
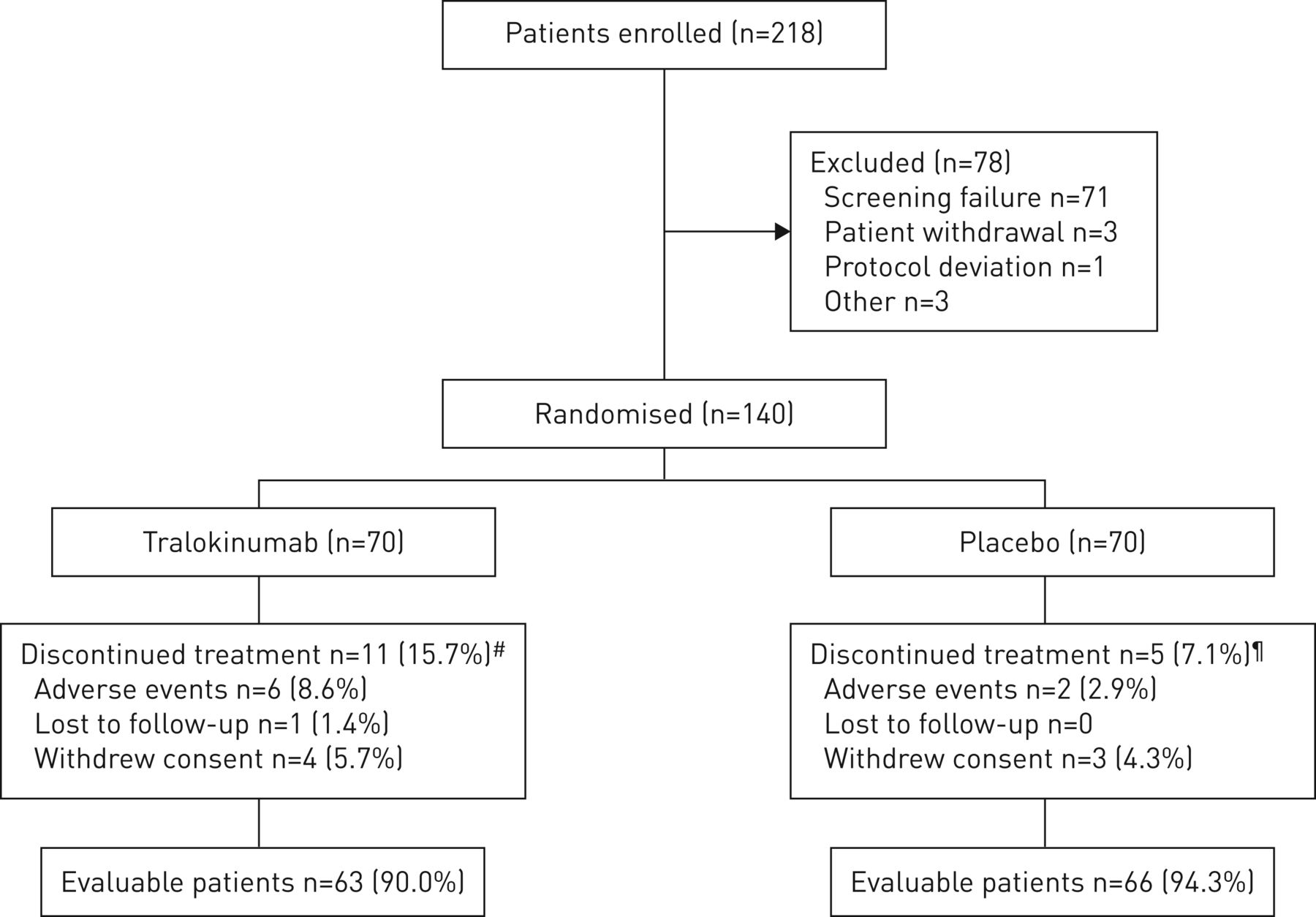
试验是2015年2月至2017年9月进行的。总共218名患者来自56个参与网站在欧洲和美国。从44个网站,140名患者被随机治疗要么tralokinumab (n = 70)或安慰剂(n = 70),其中129(92%)完成了试验和124(89%)完成了治疗(图2)。试验归纳了退出的理由补充表S1。患者基线人口统计学和临床特点是平衡整个治疗组(表1和补充表S2)。在基线,患者平均年龄为54.7岁,与哮喘诊断以来中位数是24.0年。大多数患者是女性(62.1%)。均值±sd基线F伊诺水平为35.22±29.50磅;∼在每个治疗组有51%的病人F伊诺< 30磅的水平。因此,有效性分析的主要人口是所有人口。
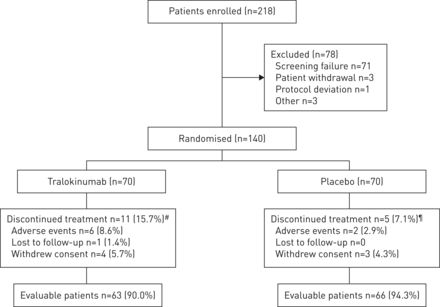
病人的入学率和性格。#:tralokinumab组、11个患者中断治疗,其中四个试验完成;¶:五个病人停止治疗的安慰剂组,一个病人完成了试验。
总的来说,在每个治疗组48例(69%)患者进入商务dose-optimisation阶段随机前8周,平均1.36毫克的剂量减少tralokinumab组和安慰剂组(0.99毫克补充表S3)。
功效
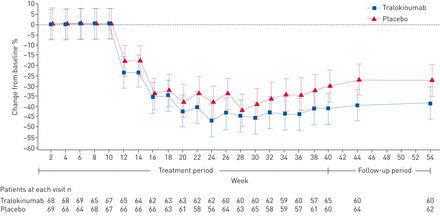
二次端点
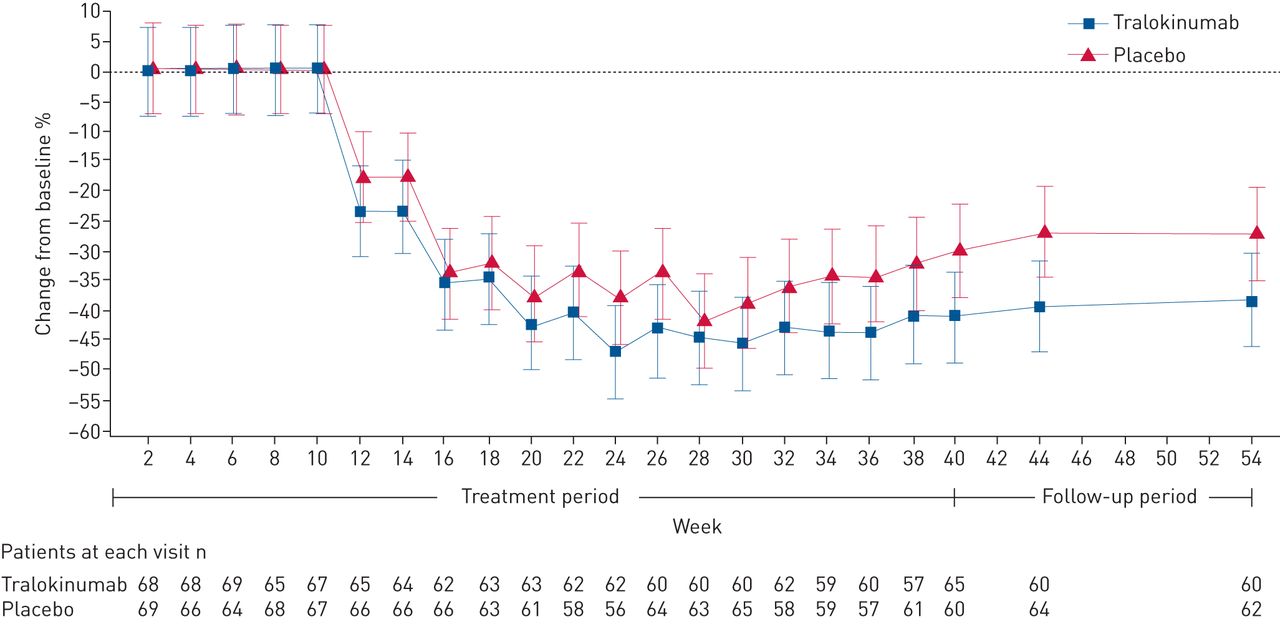
没有统计上显著的差异观察tralokinumab和安慰剂组之间任何次级终点(表2)。患者最后的比例平均每日口服剂量的≤5毫克40周tralokinumab组的45.7%与在安慰剂组(40.0%或1.33,95%可信区间0.65 - -2.73;p = 0.442)。总体而言,44.3%的患者有一个减少≥50%的基线最后平均每日口服剂量在40周tralokinumab组与安慰剂组37.1%的患者(或1.38,95%可信区间0.70 - -2.74;p = 0.356)。
约67.1%的患者tralokinumab至少经历了一次哮喘恶化在40周的治疗期间,恶化总数为925天,相比之下,75.7%的病人使用安慰剂治疗,恶化总数为1201天(补充表S4)。aa是通过一周40 tralokinumab组低20% (1.84与2.31;率比为0.80,95%可信区间0.57 - -1.12;p = 0.186)。
探索性的端点
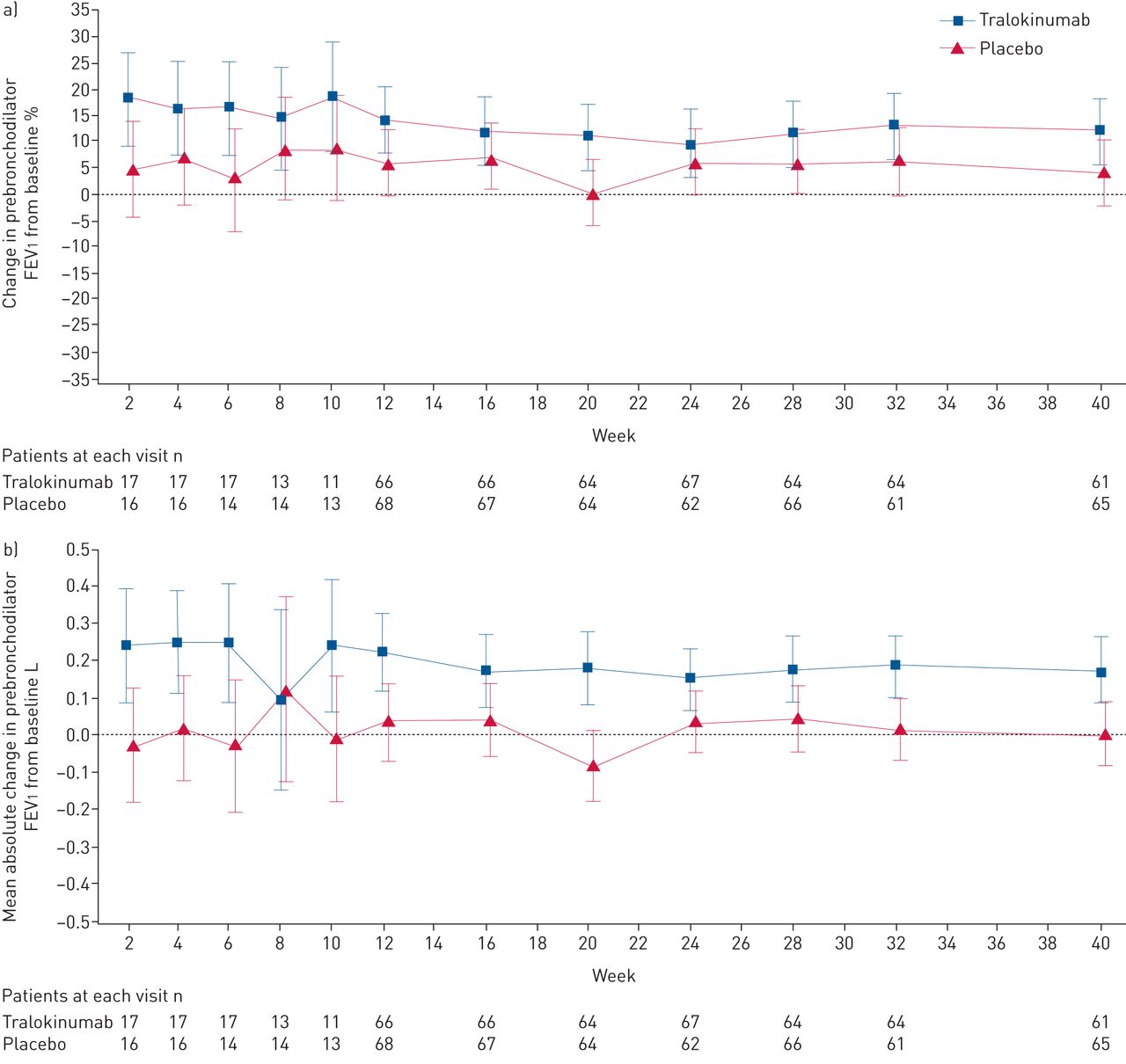
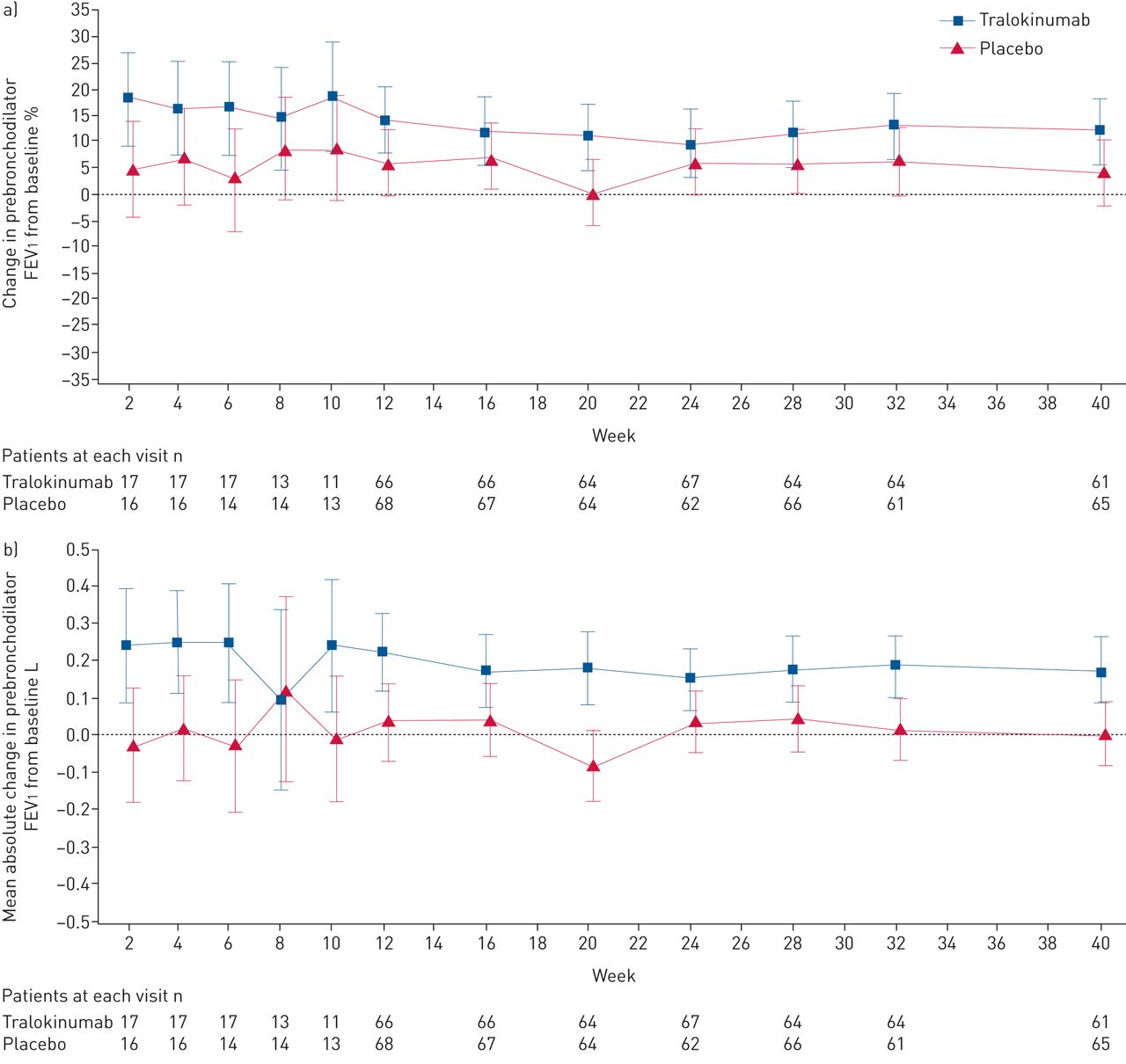
患者的比例≥75%的口服剂量减少21.4% < 90% tralokinumab组与安慰剂组的8.6% (补充表S5)。虽然没有统计上的显著差异指出tralokinumab和安慰剂组之间在LS意味着变化百分比从prebronchodilator FEV的基线1在一周40 (12.1%与4.3%;图4一),一个名义上的显著,临床有意义的改善LS平均绝对prebronchodilator FEV基线的改变1(升)指出在40周tralokinumab与安慰剂比较(LS平均差0.17,95%可信区间0.05 - -0.29;p = 0.0053;图4 b)。没有观察到显著的改进意味着变化的基线ACQ-6和AQLQ评分与安慰剂相比tralokinumab 40周(补充数据S1和S2)。
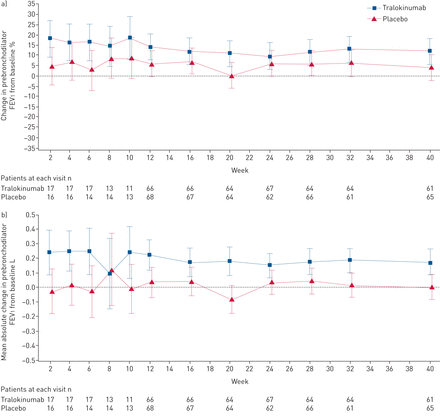
改变prebronchodilator用力呼气量在1 s (FEV1随着时间的推移)(全分析集)。prebronchodilator FEV)从基线变化百分比1;b)平均绝对prebronchodilator FEV基线的改变1。数据提出了最小二乘方法(95% CI);两组患者的数量在每个访问以下图所示。基线是最后non-missing测量记录之前随机访问(通常6)。所有患者的全分析集由随机和收到任何剂量的tralokinumab或安慰剂,不论他们的协议坚持和继续参与审判。
在患者的人口F伊诺高(≥37磅)或F伊诺低(< 37磅),没有观察到显著差异tralokinumab和安慰剂组之间在LS意味着百分比减少最后平均每日口服剂量在40周(F伊诺高37.73%与32.27%,95%置信区间-31.03 - -20.12;F伊诺低37.56%与28.62%,95%置信区间-26.15 - -8.28;补充表S5)。同样,tralokinumab和安慰剂组之间没有明显差异的任何辅助的端点F伊诺高或F伊诺低人口(补充表S6)。
安全
127名(90.7%)患者经历过至少一次不良事件在40周的治疗期(tralokinumab组的65名(92.9%)患者和62例(88.6%)患者在安慰剂组;表3)。最常报告的不良事件(> 5%),tralokinumab和安慰剂治疗组之间的不同,是病毒性上呼吸道感染(URTI;35.7%与14.3%)、支气管炎(15.7%与24.3%),哮喘(11.4%与22.9%)和背部疼痛(10.0%与2.9%)。治疗期间,14例(10.0%)患者出现注射部位反应(11例(15.7%)患者tralokinumab组和三个(4.3%)病人安慰剂组)。最常见的注射部位反应是红斑(6个(8.6%)病人tralokinumab组)和疼痛(6例(8.6%)患者tralokinumab组和两个(2.9%)病人安慰剂组)。
总的来说,25例(17.9%)患者至少有一个SAE (tralokinumab组9例(12.9%)患者和16例(22.9%)患者在安慰剂组;补充表S7)。哮喘是最常见的SAE、报告tralokinumab组5例(7.1%)患者和8例(11.4%)患者在安慰剂组。总共八个(5.7%)病人停止治疗由于不良事件(6例(8.6%)患者tralokinumab组和两个(2.9%)病人安慰剂组)。所有不良事件都是单一事件。一个病人在每个治疗组停止由于哮喘恶化和一个女病人tralokinumab组停止由于乳腺癌。不良事件导致停药治疗被认为是治疗有关的三个病人接受tralokinumab注射部位(两个事件,一个事件angio-oedema)。11例(7.9%)患者在治疗期间严重感染(6例(8.6%)患者tralokinumab组和5个(7.1%)病人安慰剂组)。没有报告病例的肺炎在审判。严重的感染提供了信息补充表S8。
在40周,意味着从243年细胞·µL嗜酸性粒细胞计数增加−1在382细胞·µL基线−1tralokinumab组;在安慰剂组,意味着从210年细胞·µL嗜酸性粒细胞计数变化−1在246细胞·µL基线−1在40周。总共8例(7个病人tralokinumab组和安慰剂组)经验丰富的血液嗜酸性粒细胞(> 1500细胞·µL−1)。在安慰剂组病人增加了不良事件的慢性嗜酸性肺炎中等强度225天邮报》率先剂量的临床实验的产品。事件是不严重,被研究者视为与临床实验的产品。病人最终从事件中恢复过来。没有主要心血管或脑血管事件或速发型过敏反应病例报告在这个审判。在治疗期间没有病人死亡。没有检测到禁毒抗体tralokinumab 300毫克或安慰剂组中观察到。
讨论
结果从这个阶段3对流层审判没有说明一个重要OCS-sparing tralokinumab效果与安慰剂比较严重哮喘患者。Tralokinumab并未导致显著减少最后平均每日口服剂量在40周与安慰剂比较(主要终点)。此外,tralokinumab和安慰剂之间并没有显著的效果观察患者的比例≤5毫克的最后平均每日口服剂量,减少患者的比例≥50%最后平均每日口服剂量从基线和aa(二级终端)。有20%的减少aa tralokinumab治疗与安慰剂相比,但这并没有统计学意义。
与对流层相比,其他几个试验检查的OCS-sparing效果单克隆抗体(benralizumab, mepolizumab和dupilumab)严重哮喘患者报道积极的结果(15- - - - - -17]。北风试验报道,两个benralizumab剂量方案(30毫克南卡罗来纳州。每4或8周)显著降低平均最终OCS剂量从基线的75% (p < 0.001)严重哮喘患者比较15]。同样,风险审判的结果报道,dupilumab商务使用治疗减少,同时减少严重恶化的速度,增加FEV1(口服剂量变化百分比−dupilumab组的70.1%与−在安慰剂组41.9%;p < 0.001) (17]。tralokinumab未能提供OCS-sparing福利或减少哮喘发作在对流层可能解释为缺乏抗炎效应和IL-13和il - 4细胞因子的功能冗余性哮喘(18]。事实上,结果最近tralokinumab第二阶段便试验,在中度到重度哮喘患者进行支气管活检之前和之后的12周的治疗tralokinumab报告不影响气道嗜酸性粒细胞炎症(19]。缺乏抗炎效应可能解释了为什么以前的试验疗法针对IL-13,如lebrikizumab,没有证明一致减少哮喘急性加重(20.,21]。
值得注意的是,和符合第二阶段试验的结果(11,12),治疗tralokinumab(探索性端点),肺功能改善显著,prebronchodilator FEV临床上有意义的改善1在40周。有趣的是,结果便第二阶段试验(19)和一个事后分析数据的一个子集患者tralokinumab 2 b阶段试验表明,tralokinumab可能提高肺功能影响气管平滑肌的语气(22]。然而,tralokinumab治疗并没有导致其他显著改善探索性端点与哮喘控制或对流层的生活质量。
F伊诺是气道炎症的生物标志物,这是诱导通过IL-13 axis-mediated通路(23,24]。在关键STRATOS 1试验分组人口严重哮喘患者F伊诺≥37磅被确认为最可能有增强反应tralokinumab [13]。出于这个原因,截断符号之前的试验结果和协商后与美国食品和药物管理局,对流层的统计分析计划修订进行主要分析F伊诺高分组人口(F伊诺≥37磅)如果有足够比例的病人。鉴于这个族群的小样本大小,应急计划是设计来确保主分析中只会被执行F伊诺高分组人口如果≥70患者包括(阈值≥37磅≥30磅)。在对流层中,超过一半的患者F伊诺水平低于30磅的阈值越低,因此,所有的主要和次要分析人口。然而,主要和次要的端点基于探索性分析F伊诺高水平并未透露F伊诺水平(≥37磅)和tralokinumab预测一个更大的治疗效果。关键的第三阶段试验STRATOS 2也未能展示有意义的减少aaF伊诺高患者[13]。
Tralokinumab演示了一个可以接受的安全性,这与先前的报道是一致的(11,12]。总的来说,不良事件的发生率相当tralokinumab和安慰剂组,患者虽然tralokinumab报道URTIs和注射部位反应发生率更高。解释利率的差距URTIs尚不清楚,因为没有报告病例的肺炎试验和严重感染患者的比例在治疗组相似。有趣的是,在风险试验类似的观察观察,最常见的不良事件是URTI (dupilumab组中9%的患者和18%的患者在安慰剂组)(17]。同样,自由哮喘试验的结果,患者接受的附加dupilumab剂量的200或300毫克或安慰剂52周,URTI是最常报道的不良事件(25]。
增加一个小血嗜酸性粒细胞计数是观察tralokinumab集团与安慰剂组,更多tralokinumab-treated患者增加嗜酸性粒细胞计数1500个细胞·µL之上−1比接受安慰剂的患者(7与一个)。然而,尽管这一发现,没有相关的不良事件报告那些tralokinumab-treated增加患者血液中嗜酸性粒细胞计数。这一发现符合风险报告的试验,在瞬态血液嗜酸性粒细胞观察病人dupilumab组比安慰剂组(14%)与1%),没有任何临床后果或相关的不良事件(17]。在自由哮喘试验中,嗜酸性粒细胞作为不良事件被报道在4.1%的患者接受了dupilumab和0.6%的患者接受了安慰剂;然而,在这种情况下,在总人口的0.2%,这些不良事件是伴有临床症状,两个事件报告为节约(hypereosinophilia恶化和慢性嗜酸性肺炎)25]。
电流的强度试验的试验设计,这在很大程度上,复制方法之前用于其他OCS-sparing试验(15,16,26,27),但有两个例外。首先,基于调查结果从tralokinumab发展计划(11,12),12周的诱导阶段纳入了对流层对FEV试验设计,以确保最大的影响1。其次,维护阶段延伸至8周,以评估长期疗效相比以前OCS-sparing试验报告(16,26- - - - - -28]。值得注意的是,减少口服避孕药使用安慰剂组中观察到在对流层ZONDA试验(类似报道15),表明试验设计是足以证明一个OCS-sparing效果。然而,这个试验有局限性,包括常见的选择性和一个相对比较小的样本大小的限制,这意味着一些分析,如嗜酸性粒细胞增加水平的影响,不能被执行。
总之,tralokinumab没有演示临床重大OCS-sparing或改善哮喘控制效果与安慰剂比较严重,患者无法控制哮喘。更高水平的F伊诺(≥37磅)没有与tralokinumab预测一个更大的治疗效果。因此,本试验发现,与之前报道STRATOS 1和2试验,结果表明,针对IL-13本身并不是一个有效的策略来管理严重哮喘。
补充材料
确认
我们感谢卫生保健提供者,研究人员,患者和医护人员参与试验。医学写作提供的支持是Khund细哔叽,穆罕默德Najeeb和弗朗西斯赌博仙人掌通信(印度孟买),由阿斯利康(英国剑桥),按照良好的出版实践(GPP3)指南(www.ismpp.org/gpp3)。作为一个抽象的第28届国际大会上欧洲呼吸学会的巴黎,法国;188bet官网地址2018年9月15 - 19日。
脚注
可以从本文的补充材料www.qdcxjkg.com
这项研究是在注册clinicaltrials.gov与标识符NCT02281357。这手稿中描述数据基础研究结果可以获得符合阿斯利康的数据共享政策描述https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure
利益冲突:W.W.会报告个人费用从阿斯利康咨询公司,在进行研究;个人费用咨询公司从3 m,勃林格殷格翰的发言,葛兰素史克、诺华Sanofi-Regeneron Teva,个人费用数据和安全监测委员会工作从波士顿科学基因泰克,爱思唯尔的个人费用(版税),个人费用从起到了教育视频,个人费用联合监督委员会工作图标临床研究有限,和个人费用从辉瑞、罗氏、阿斯利康和切尔克西亚和赠款NIH-NIAID和NIH-NHLBI外提交的工作。
利益冲突:和Brusselle报告谢礼顾问委员会/或讲座从阿斯利康,勃林格殷格翰的发言,基耶西,葛兰素史克、诺华、辉瑞、赛诺菲安万特/ Regeneron Teva, UCB Zambon,在进行这项研究的。
利益冲突:美国从Almirall Korn咨询报告和演讲费,阿斯利康,勃林格殷格翰的发言,基耶西,Teva,罗氏,从葛兰素史克和诺华和赠款和个人费用,在进行这项研究的。
利益冲突:从亚当·p·库纳报告个人费用,Allergopharma,碱,阿斯利康,拜耳Celon制药,基耶西,西班牙,哈尔过敏,Lekam, Polpharma,辉瑞,山德士和梯瓦,在进行研究;和个人费用从柏林化学,勃林格殷格翰集团和诺华,在提交工作。
利益冲突:a . Magnan报告个人费用和非金融支持葛兰素史克、诺华、勃林格殷格翰集团,阿斯利康,Stallerg nes,碱性,MundiPharma, Teva, Menarini梅达制药公司,在过去的5年。
利益冲突:d·科恩是阿斯利康的一名员工。
利益冲突:k·鲍文是一个员工,在阿斯利康的股票。
利益冲突:t Piechowiak是一个员工,在阿斯利康拥有股票期权。
利益冲突:M.M.王是一个员工,在阿斯利康拥有股票。
利益冲突:g . Colice是一个员工,在阿斯利康拥有股票和股票期权。
支持声明:本研究是由阿斯利康。资金信息,本文已沉积的Crossref资助者注册表。
- 收到了2018年5月21日。
- 接受2018年10月30日。
- 版权©2019人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}