





文摘
我们从第一个每日一次的三期临床试验结果tiotropium附加吸入激素(ICS) +一个或多个控制器疗法在青少年患有严重哮喘症状。
在这个双盲、平行对照试验中(NCT0127752312 - 17岁),392名患者被随机接收每日一次tiotropium 5µg或2.5µg,或安慰剂,作为一个附加ICS +其他控制器治疗12周。的主要和关键二级终端从基线变化(反应)的峰值在1 s (FEV用力呼气量13 h post-dosing (FEV内)1 (0-3h))和槽FEV1治疗12周后,分别。
Tiotropium 5µg提供数字峰值FEV的改善1 (0-3h)反应,与安慰剂比较(90毫升;p = 0.104),并显著改善观察tiotropium 2.5µg(111毫升;p = 0.046)。数值槽FEV的改善1响应和哮喘控制观察tiotropium剂量,与安慰剂相比。的安全性和耐受性tiotropium可比与安慰剂。
每天换一次tiotropium Respimat附加ICS +一个或多个控制器在青少年患有严重哮喘症状治疗耐受良好。功效的主要终点并不满足,虽然积极趋势改善肺功能和哮喘控制观察。
文摘
Tiotropium附加治疗青少年中的数值结果的改善提供了哮喘http://ow.ly/eL8g304a9XV
介绍
哮喘是儿童最常见的慢性疾病,与11个孩子在英国大约一(112 - 18岁)和10%的青少年在美国(2患有这种疾病。
≥40%的哮喘患者据报道仍有症状尽管治疗根据建议与吸入激素(ICS)单药治疗之后,逐步添加其它控制器疗法(3- - - - - -6),> 50%的比例增加青少年患者(2]。一小部分的控制不佳的哮喘患者可能遭受频繁的哮喘症状或加重尽管坚持高剂量ICS加上其他控制器疗法,从而证明严重哮喘的诊断。这个群体特别是导致高发病率,死亡率和治疗费用7)和权证寻找新颖的治疗选项来提高控制和减少未来恶化的风险。
Tiotropium,长效抗胆碱能支气管扩张剂通过Respimat软雾吸入器(勃林格殷格翰集团、殷格翰集团莱茵、德国),已经证明是一个有效的和耐受每日一次附加至少ICS维持治疗成人轻度(8),中等(9- - - - - -12和严重的13哮喘症状。同样,最初的第二阶段研究青少年(14和孩子们15)与中度哮喘症状表明tiotropium Respimat改善肺功能和哮喘控制,安全性和耐受性与安慰剂相比。这些研究结果随后被确认第III期试验tiotropium Respimat作为附加中等剂量ICS在青少年与中度哮喘症状(16]。
第一第二和第三阶段试验后青少年与中度哮喘症状(14- - - - - -16),我们目前的疗效和安全性每日一次的第III期试验结果tiotropium Respimat 5µg 2.5µg附加ICS +一个或多个控制器在12周治疗12 - 17岁的青少年患有严重哮喘症状。
方法
研究设计
第三阶段,这是一个为期12周的随机、双盲、安慰剂对照、平行对照试验中(NCT01277523在12 - 17岁的青少年患有严重哮喘症状。后的结果PrimoTinA-asthma成年人患有严重哮喘临床试验(13),来决定是否执行这个试验结果之间的可比性成人和青少年患者人群,按照规定对儿科病人对药物开发。基于第二阶段完成概念验证试验(17按照规定,为儿科病人,药物开发在12周的治疗期在这个病人被认为是适当的。审判是在14个国家的68个站点(在线补充材料)。
研究符合赫尔辛基宣言的原则,国际会议协调好临床实践指南。在试验开始之前,试验协议和患者和家长/监护人信息表和综述了同意书和独立的伦理委员会批准和/或每个参与机构的机构审查委员会。参与审判之前,书面知情同意收到每个病人和病人的家长或监护人。
研究人群
符合条件的患者12 - 17岁和≥3个月症状哮喘史,定义为seven-question哮喘控制问卷(ACQ-7)平均得分≥1.5在筛选和随机。≥4星期前检查,所有患者被要求一直在维持治疗高剂量ICS +一个或多个控制器疗法(如。一个长效β2受体激动剂或白三烯受体拮抗剂)或中等剂量ICS +两个或两个以上的控制器疗法(如。一个长效β2受体激动剂和/或白三烯受体拮抗剂和/或缓释茶碱)。高剂量ICS被定义为> 400µg布地奈德或同等12 - 14岁的病人和800 - 1600年µg布地奈德在15岁到17岁的病人或同等。中等剂量ICS被定义为200 - 400µg布地奈德或同等12 - 14岁的病人和400 - 800年µg布地奈德或同等在15岁到17岁的病人6]。病人被要求证明prebronchodilator用力呼气量在1 s (FEV1在筛选预测)60 - 90%;FEV1可逆性≥12%,≥200毫升400年后15 - 30分钟µg舒喘灵(沙丁胺醇;如果12 - 14岁的患者表现出非常小的肺总量,积极的可逆性测试可能是完全基于FEV1可逆性≥12%);和绝对FEV的可变性1从筛选到随机值在±30%。患者还需要不吸烟者或已经停止吸烟≥1年之前报名;父母吸烟是记录。
关键的排除标准包括哮喘以外的任何重大疾病的诊断。
治疗
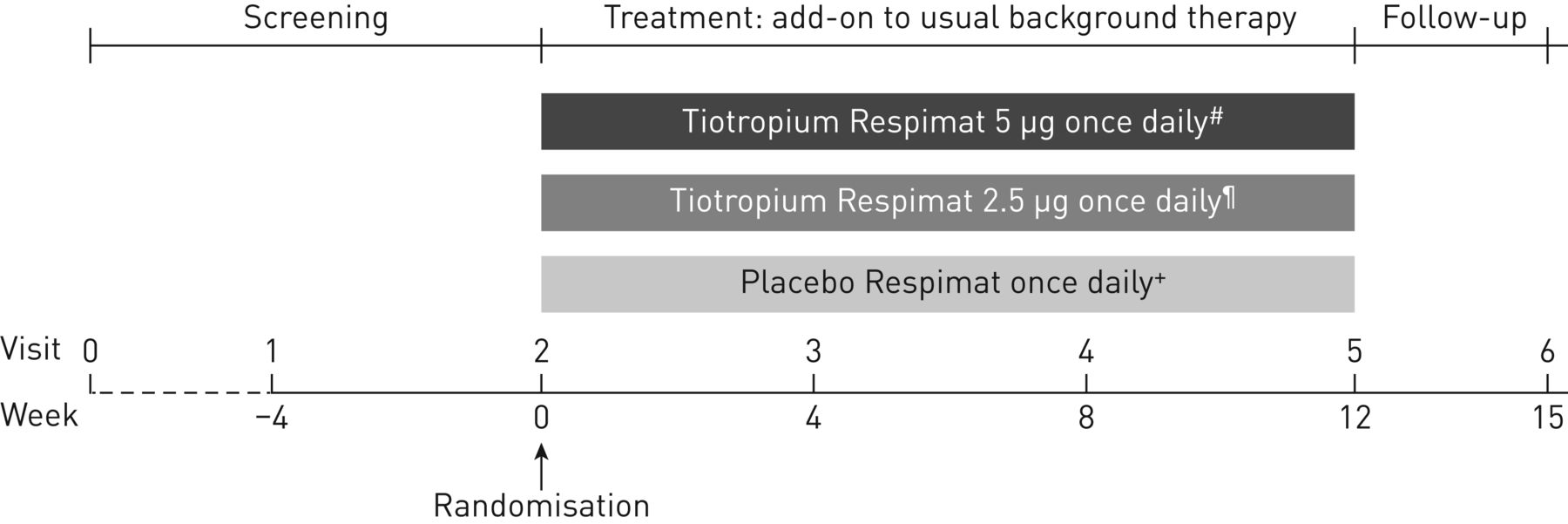
比的比例患者随机接受tiotropium 5µg(两个泡芙2.5µg)或2.5µg(两个泡芙1.25µg)或安慰剂(两个泡芙),每个交付通过Respimat软雾吸入器附加pre-enrolment背景与ICS治疗+一个或多个控制器疗法。病人进入四周筛选阶段,其次是12周的治疗进一步和3周随访期间(图1)。
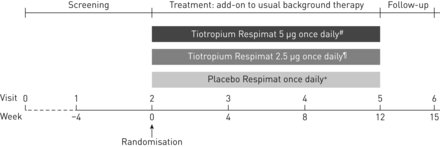
研究设计。通常的背景治疗被定义为大剂量吸入激素(ICS) +一个或多个控制器疗法(如。一个长效β2受体激动剂或白三烯受体拮抗剂)或中等剂量ICS +两个或两个以上的控制器疗法(如。一个长效β2受体激动剂和/或白三烯受体拮抗剂和/或缓释茶碱)。高剂量ICS被定义为> 400µg布地奈德或同等12 - 14岁的病人和800 - 1600年µg布地奈德在15岁到17岁的病人或同等。中等剂量ICS被定义为200 - 400µg布地奈德或同等12 - 14岁的病人和400 - 800年µg布地奈德或同等在15岁到17岁的病人6]。#:两个泡芙2.5μg;¶:两个泡芙1.25μg;+:两个泡芙。
执行随机双盲使用伪随机数生成器和验证提供的种子数量,确保分配既可再生的和nonpredictable,六块中执行,根据国家分层。生成的随机列表是勃林格殷格翰集团(Biberach der里斯期,德国)。病人自行服药每天一次在晚上17点之间7:00 h和h,在下列顺序:ICS治疗,那么其他控制器疗法,紧随其后的是试验药物。非盲舒喘灵hydrofluoroalkane metered-dose吸入器提供了用于救援药物试验过程中。允许伴随药物治疗急性哮喘加重包括临时增加ICS剂量;临时增加的剂量,或增加,系统性皮质类固醇或短效茶碱制剂;临时增加系统性β2受体激动剂或吸入短效抗胆碱能类;和抗生素。
分析
在第12周的所有功效端点进行分析。在第12周的肺活量的肺功能测量进行10分钟pre-dose 30分钟,1 h, 2 h和3 h post-dose。在每个时间点,至少三次演习执行最多八努力,以确定最高测量后三个可接受的演习。
主要疗效终点是FEV峰值1在3 h post-dosing (FEV1 (0-3h)),测量作为响应被定义为从基线(预处理值测量10分钟之前第一次剂量的试验药物管理局)。次要疗效端点是槽FEV的关键1反应,测量结束时剂量间隔,10分钟前下剂量的试验药物的管理。事后分析FEV波峰和波谷1在第12周进行12 - 14年,15 - 17岁的病人肺功能是否反应后tiotropium附加疗法不同病人之间的上下极限青春期。
其他次要疗效端点包括FEV1曲线下的面积(AUC)在3 h post-dosing (FEV1AUC(0-3h)),峰值用力肺活量(FVC) 3 h post-dosing和FVC AUC之内(0-3h)所有测量的响应。救援药物的使用在白天、夜间和完整的24小时内被记录在家里使用AM3设备(结合电子流量计和e-diary峰值;eResearch技术、业务、德国)。哮喘控制使用自我估测rq评估ACQ (ACQ-6)和ACQ-7分数和应答率在12周后,患者分为反应如果最小临床重要差异减少≥0.5 ACQ分数实现(18]。时间第一次严重哮喘恶化和哮喘恶化的第一集进行评估在12周的治疗期。严重哮喘恶化被定义为一个哮喘恶化,需要连续三个或更多天全身糖皮质激素治疗或至少一倍的系统性皮质类固醇剂量连续三个或更多天。哮喘恶化的一集被定义为一个逐步增加的一个或多个哮喘症状外,患者通常的范围和持续连续两个或两个以上的天,和/或减少病人的最好早上最大呼气流量(PEF)≥30%意味着清晨PEF连续两个或两个以上的天。
进一步疗效端点包括槽FVC,意味着用力呼气流量的25 - 75% FVC (FEF25 - 75%)在单个时间点和每周意味着pre-dose早晚使用AM3脉动电场测量设备。
不良事件监测在治疗期和后30天的最后剂量试验药物评估安全性和耐受性。
统计分析
安全分析治疗组,定义为所有患者随机收到至少一个剂量的药物研究。功效分析进行完整的分析,这是与治疗组相同。
一套零假设测试逐步的方式来控制第一类错误的概率(片面;α= 0.025):tiotropium的功效5µg然后µg tiotropium 2.5与安慰剂的主要终点,其次是tiotropium的功效5µg然后µg tiotropium 2.5与安慰剂的次要终点的关键。每一步都被认为是确认只有前面的步骤都是成功;如果前面的步骤并不成功,进一步分析被认为是描述性的。
所有疗效端点,救援药物的使用和ACQ分数反应进行分析使用限制最大基于可能性mixed-effects用重复测量模型。模型包括固定,直言“治疗”的影响,“联合国家”、“访问”和“treatment-by-visit互动”,以及“基本价值”的协变量和“基线value-by-visit互动”。“病人”作为随机效应。患者反应者的数量(至少最小的ACQ评分改善临床重要差异0.5分(18]),那些ACQ改变< 0.5分(没有变化)和那些哮喘恶化(那些与ACQ恶化≥0.5分)通过Wilcoxon排名和比较测试。时间先严重恶化和第一集的哮喘恶化进行了分析使用Cox比例风险回归模型与“治疗”作为影响安装。安全分析是描述性的。
样本大小是决定使用一个保守的两群t以及电力的80%和2.5%的错误概率(单边)。假设420毫升的标准差,是确定每个治疗组125例患者被要求检测的差异在峰值FEV 150毫升1 (0-3h)响应。
结果
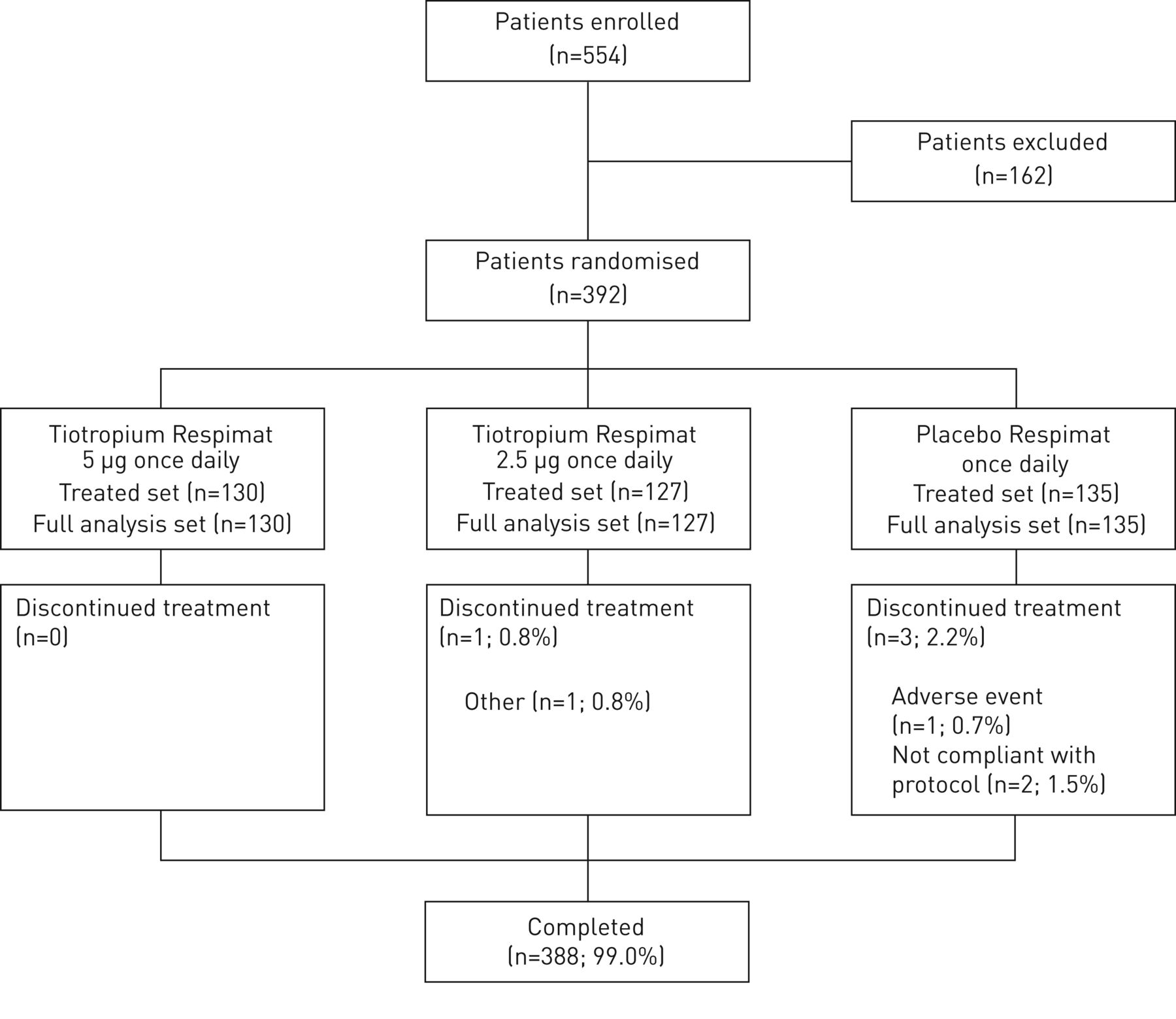
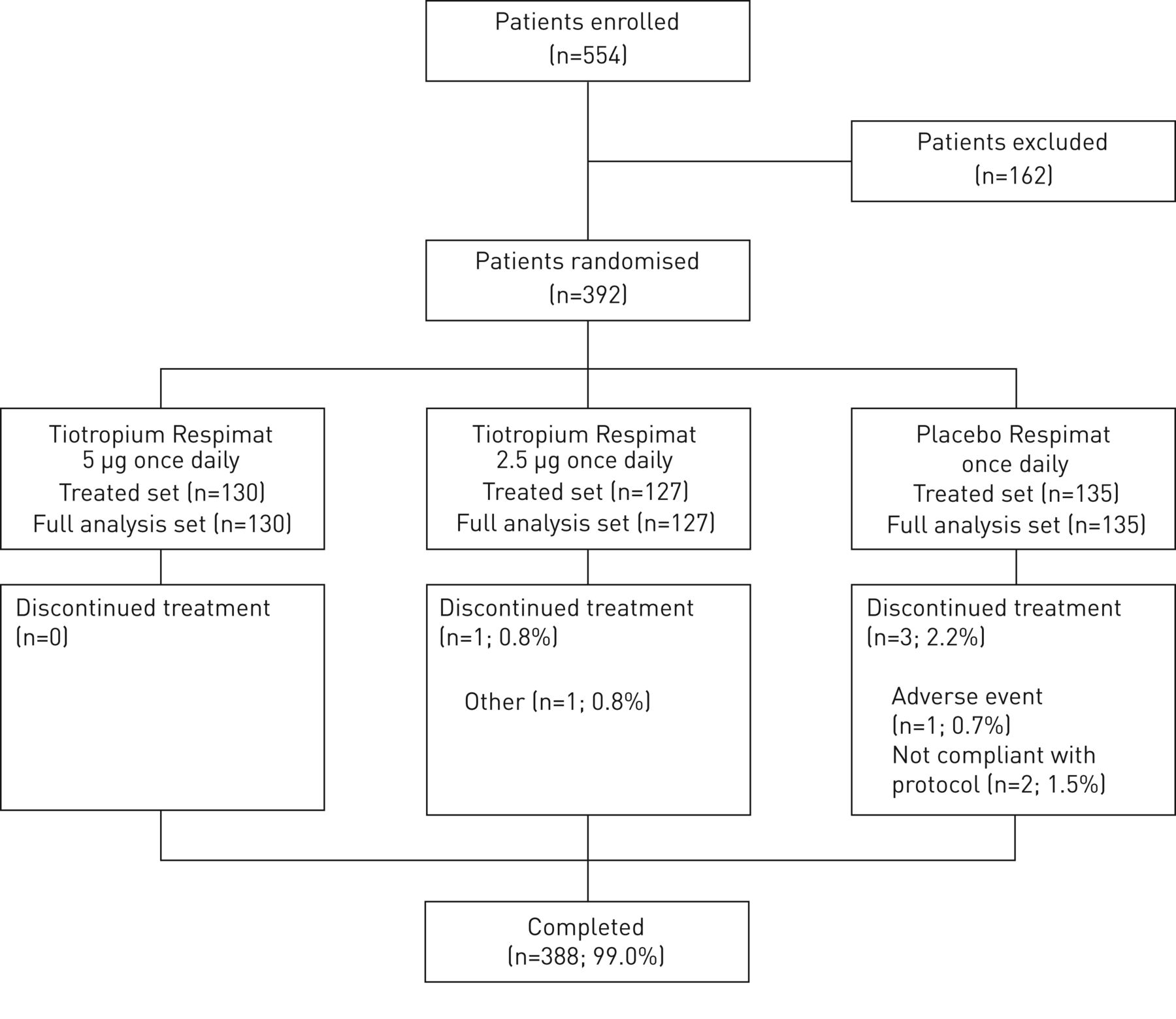
392名患者被随机。其中,388名患者(99.0%)完成了12周的治疗期和4名患者(1.0%)过早停用研究药物(图2)。
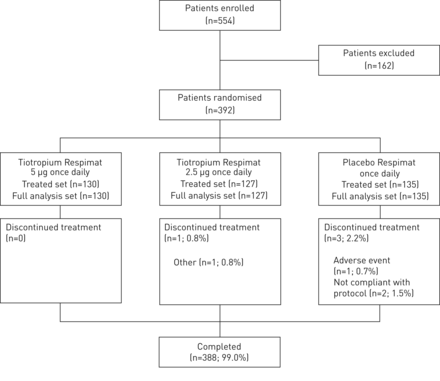
配偶关系图。
基线患者人口统计学和疾病特征
总的来说,基线患者人口平衡的整个治疗组(表1)。有更多的男性病人(61.7%);53.8%的患者12 - 14岁和46.2%是15 - 17岁;哮喘的平均持续时间为7.8年;和6.1%的患者已经暴露于二手烟。在基线,124名患者和268名患者使用两个和三个控制器疗法,分别,除了他们的ICS治疗。在筛选前的3个月,所有患者与ICS已经接受治疗,83.2%的患者服用长效β2受体激动剂,80.4%已经采取茶碱白三烯受体拮抗剂和6.1%。伴随药物治疗期间发表在在线补充表S1。
功效
主要和关键二级终端
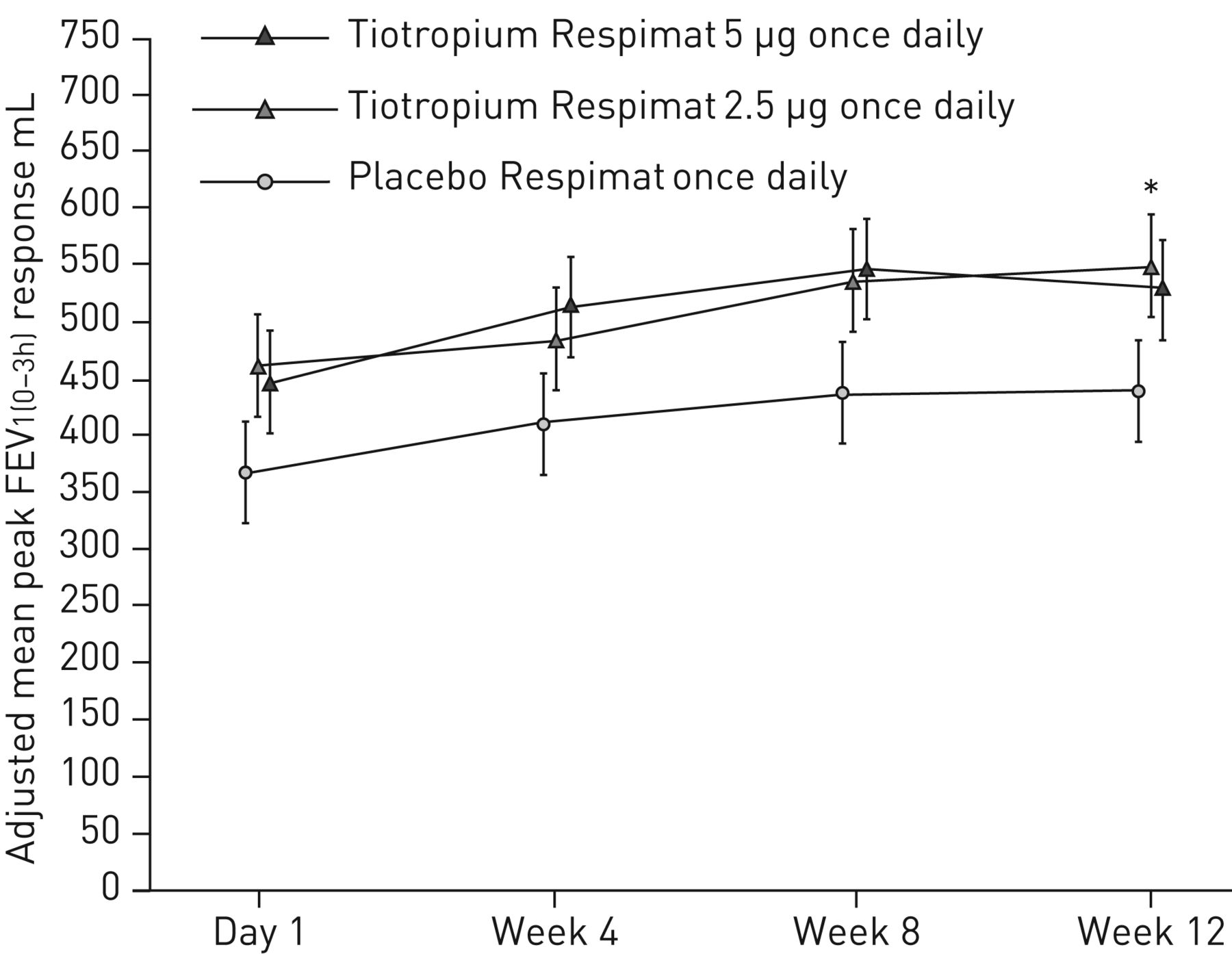
调整后的平均差峰值FEV的安慰剂1 (0-3h)响应与tiotropium 5µg没有统计学意义,虽然数值改进观察(90毫升,95% CI−19 - 198;p = 0.104) (图3)。有一个峰值FEV显著改善1 (0-3h)与2.5µg剂量响应(111毫升;95%可信区间2 - 220;p = 0.046),但由于tiotropium 5µg /安慰剂的疗效无法证明,因此审判的主要终点是不满足,所有进一步治疗比较被认为是描述性只控制错误。
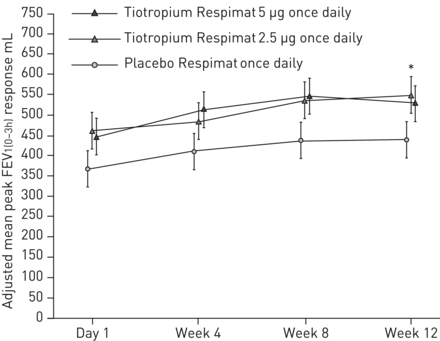
用力呼气量峰值在1 s 3 h post-dosing (FEV1 (0-3h)在12周内)响应。全分析集。常见的基线意味着±sdFEV12525±618毫升。数据意味着±se。*:p < 0.05。
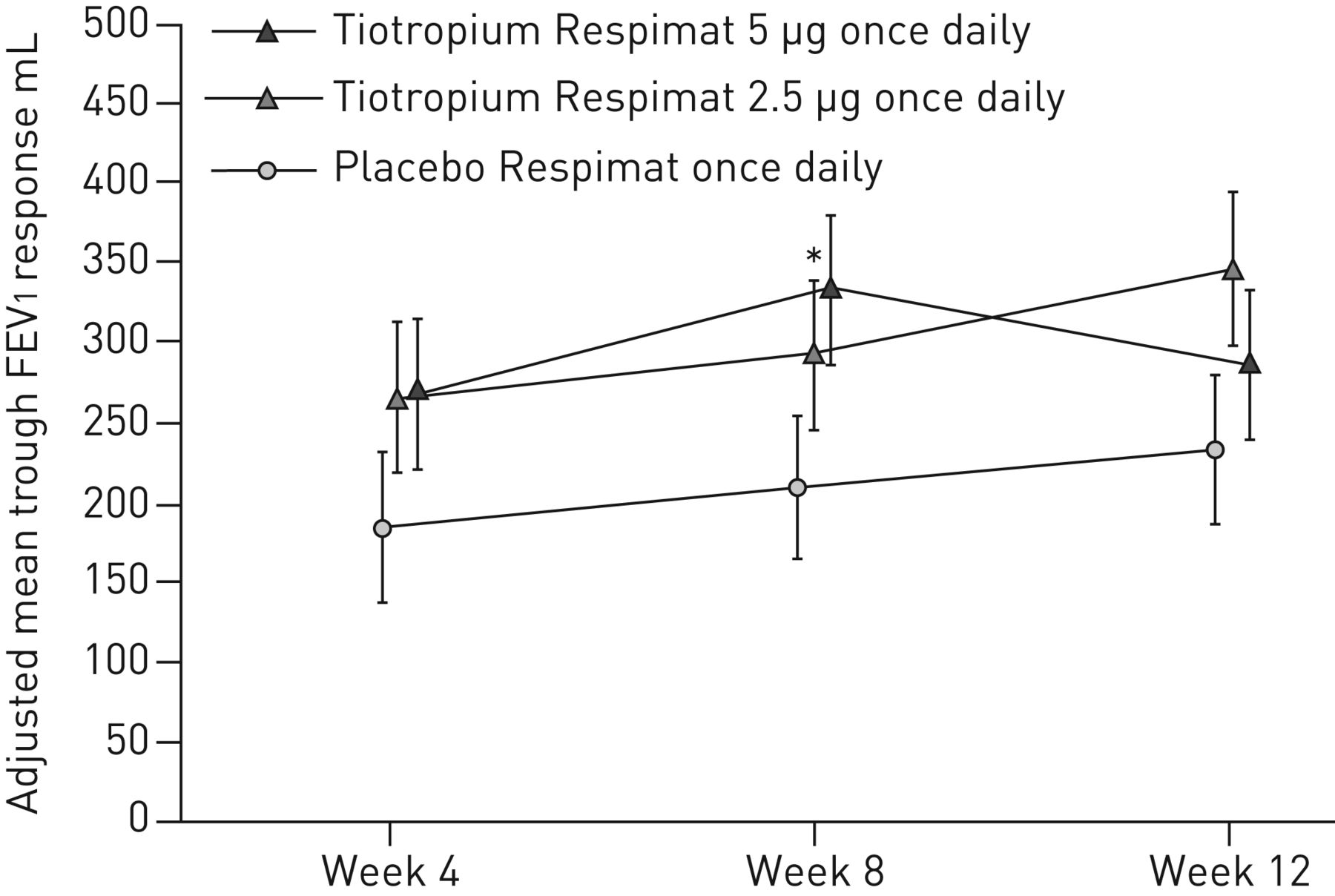
数值的改进关键二级终点,槽FEV1反应在12周,观察与tiotropium 5µg(54毫升;95%可信区间−61 - 168;p = 0.361)和2.5 tiotropiumµg(115毫升;95%可信区间0 - 231;与安慰剂相比,p = 0.051) (图4);未达到统计上的显著水平,改善剂量。
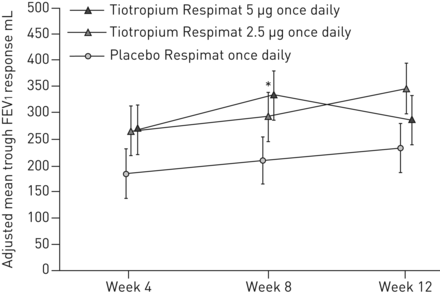
槽在1 s (FEV用力呼气量1在12周内)响应。全分析集。常见的基线意味着±sdFEV12525±618毫升。数据意味着±se。*:p < 0.05。
事后不同年龄组的亚组分析显示数值在峰值FEV的改进1 (0-3h)(在线补充表S2)和槽FEV1(在线补充表S3) 12 - 14岁的病人和15 - 17年,是统计学意义与tiotropium 2.5µg 12 - 14岁的患者只对FEV高峰(p = 0.0071 (0-3h)并为槽FEV p = 0.0181)。改进被证明是独立的年龄组(p = 0.656 treatment-by-subgroup交互FEV峰值1 (0-3h)并为槽FEV treatment-by-subgroup交互p = 0.4841)。
额外的辅助和进一步的端点
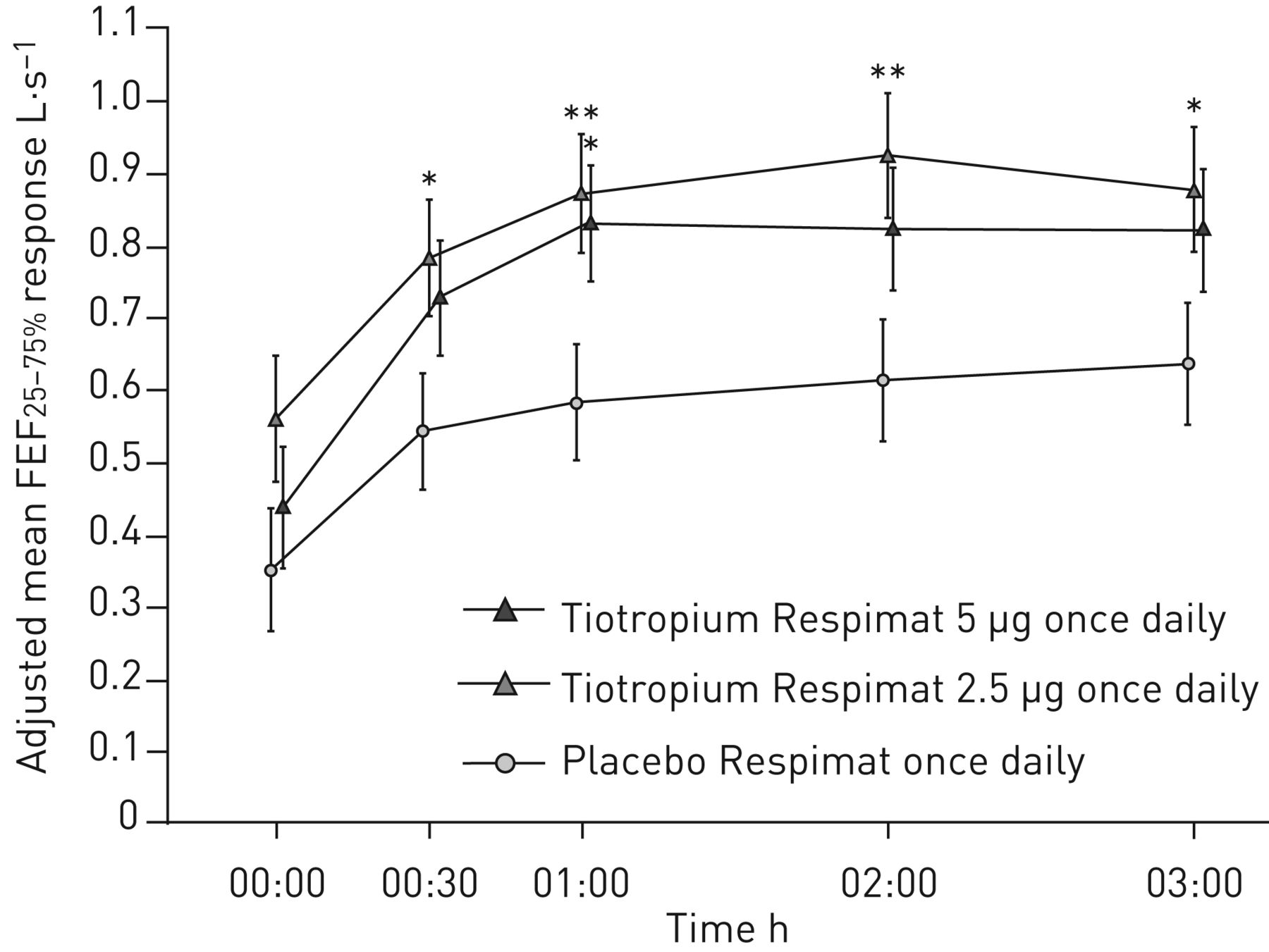
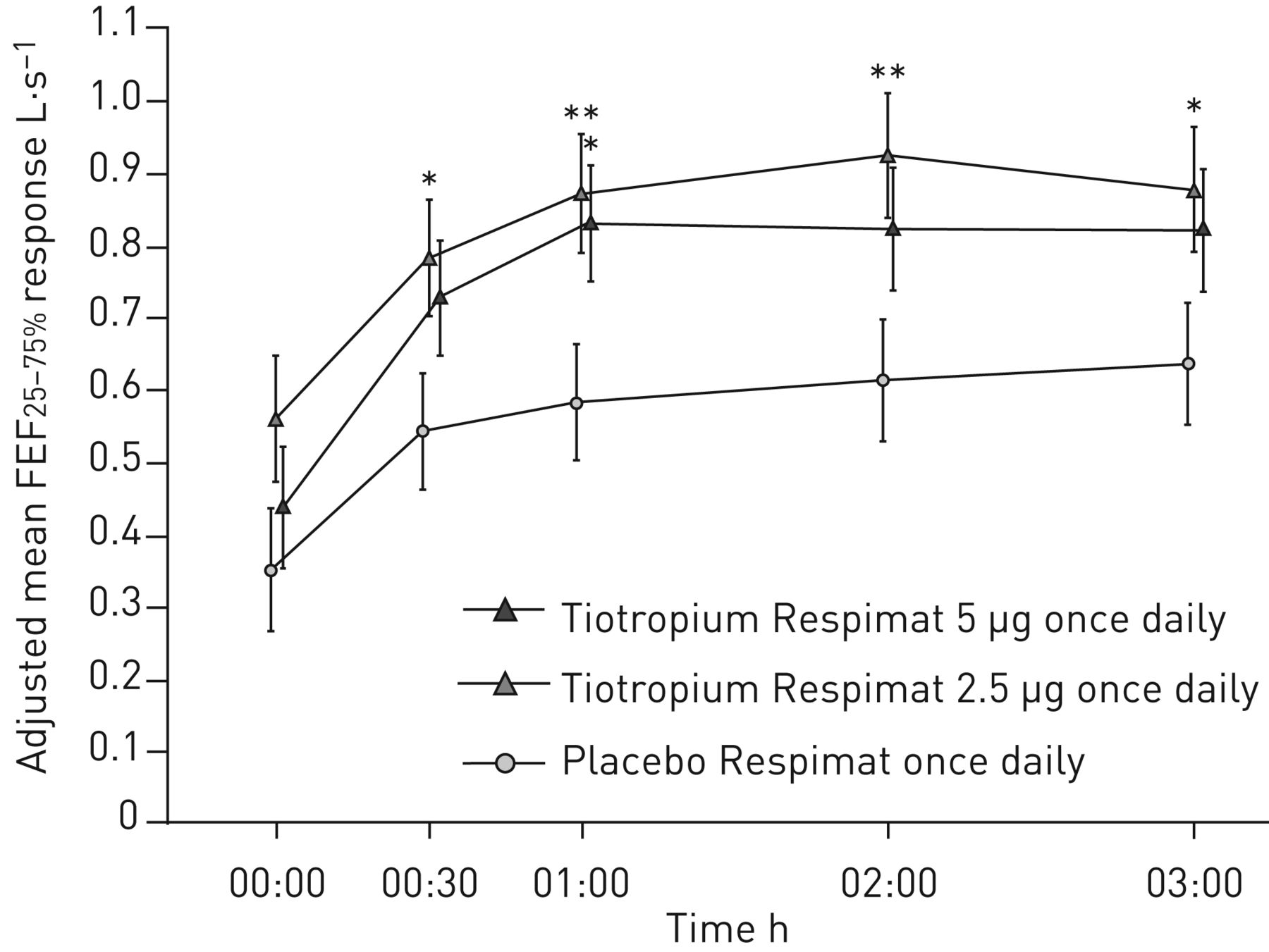
额外的辅助,进一步在第12周的疗效端点响应提出了表2。治疗tiotropium导致数值FEV的改善1AUC(0-3h)tiotropium 5µg和所有措施FVC(高峰、低谷和AUC(0-3h))与tiotropium剂量;FEV的改善1AUC(0-3h)有统计学意义,与安慰剂相比,2.5只µg剂量(p = 0.034)。Pre-dose早晚PEF反应tiotropium 5µg组比tiotropium 2.5µg组和安慰剂组最小;调整的差异意味着pre-dose早晚PEF反应tiotropium和安慰剂之间只和5µg剂量显著(p = 0.005 pre-dose清晨PEF和p = 0.004 pre-dose晚上PEF)。对于FEF25 - 75%、治疗差异显著tiotropium 2.5µg最多时间点,与安慰剂相比,和tiotropium 5µg 1 h post-dosing,与安慰剂比较(图5)。
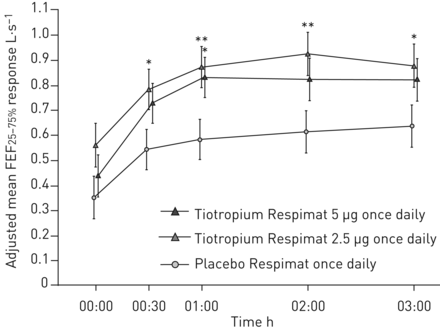
用力呼气流量在用力肺活量(FEF的25 - 75%25 - 75%在12周内)响应。全分析集。常见的基线意味着±sd2.23±0.96 L·s−1。*:p < 0.05;* *:p < 0.01。
每周的平均数量的泡芙救援药物使用在白天,夜晚,24小时内降低在所有治疗组在12周的治疗期,但之间的差异在统计学上没有显著意义tiotropium剂量和安慰剂。白天,整个完整的24小时内,救援药物的使用最低tiotropium 5µg治疗组(在线补充表S4)。
调整意味着ACQ-6和ACQ-7分数提高(降低),tiotropium患者比较,差异(95% CI)为0.053(−0.119 - -0.226)和0.036(0.123−-0.196)点,分别5µg和安慰剂治疗组之间和0.118(−0.055 - -0.292)和0.058(0.102−-0.219)点,分别在2.5µg和安慰剂治疗组在12周。响应中没有统计上的显著差异(列为响应者,没有变化或恶化)的ACQ-6 (Wilcoxon等级和测试:5µg p = 0.926;2.5µg p = 0.839)和ACQ-7 (Wilcoxon等级和测试:5µg p = 0.952;2.5µg p = 0.802)之间的分数tiotropium剂量和安慰剂在第12周(在线补充图S1)。
一个病人在每个tiotropium 2.5µg(0.79%)和安慰剂治疗组(0.74%)和两个病人(1.54%)tiotropium 5µg治疗组在12周的治疗期间经历了严重的哮喘恶化。至少一个集哮喘恶化的报告15例(11.5%)患者tiotropium 5µg组,18例(14.2%)患者在tiotropium 2.5µg组和安慰剂组25例(18.5%)患者。中位数时间第一次严重哮喘恶化和时间第一集哮喘恶化不能计算,作为<事件被报道在每个治疗组50%的患者。
讨论
在这三期临床试验,我们调查了每日一次tiotropium附加ICS +至少一个控制器治疗12 - 17岁的青少年患者患有严重哮喘症状。的主要和关键二级终端并不满足,FEV峰值与改善1 (0-3h)和槽FEV1在第12周的反应与tiotropium 5µg没有达到统计学意义,与安慰剂相比。因此,所有其他分析被认为是描述性的,按照逐步分层测试方法。这些数据有些意想不到的多个其他调查的tiotropium附加至少ICS治疗青少年(16)和成人(9,13与哮喘),这表明5µg剂量提供了显著的改进FEV波峰和波谷1。有趣的是,在目前的研究FEV1改进,与安慰剂,5µg剂量甚至高于观察2.5µg剂量在周4和8,所以对患者肺功能的下降在第12周收到5µg剂量不符合结果在之前的时间点。我们相信这一观点可能与治疗剂量无关,认为这是一个机会发现。
我们观察到显著改善在pre-dose早晚PEF tiotropium 5µg。脉动电场计算的每周平均每日价值和可能因此被认为比FEV提供更可靠的数据1测量,它代表的是单一的值了一天在诊所外的病人的真实环境。改善FEF25 - 75%是统计学意义与tiotropium 2.5µg最多时间点,但在统计上显著的1 h post-dosing只有5µg剂量。FEF25 - 75%测量可能反映了更大的影响tiotropium小型航空公司在青少年患者中,尽管药物高水平的控制器。哮喘控制的积极趋势评估观察治疗手臂,减少救援药物的使用和改善ACQ得分观察;然而,与tiotropium差异,与安慰剂相比,未达到统计上的显著水平。严重的急性加重和哮喘发作的发病率低,恶化的安全性和耐受性tiotropium被发现与安慰剂相当。
我们研究的结果可能已经受到限制病人的人口和试验设计。首先,观察明显的安慰剂反应的发生,造成小房间之间的分化治疗组和可能的改进初始坚持背景ICS治疗临床试验的设置。坚持哮喘治疗在临床试验环境在儿科人口,特别是贫穷的病人和他们的父母或照顾者影响依从性(19- - - - - -21]。数据从我们的事后不同年龄组的分析证明了在高峰和低谷FEV显著改善1tiotropium 2.5µg后12 - 14岁的患者,这可能反映了一个更积极的角色的父母和照顾者照顾年轻的青少年患者,可能导致增加的治疗依从性和影响肺功能结果。其次,相对较短的试验持续时间不允许一个结论性的分析的影响tiotropium附加治疗严重恶化,哮喘恶化或哮喘控制。因此,长期在青少年患者的进一步临床试验证实了严重的哮喘症状会是有益的确认和扩大这些试验的初步结果。此外,这次审判是150毫升的动力检测不同峰值FEV的主要终点1 (0-3h)响应,基于420毫升的标准差;然而,观察到的标准差是∼510毫升,略高于预期。因此,这项研究可能是动力不足检测统计上的显著差异,可能是因为青少年的肺容积更跨年龄和性别变量。
虽然目前的试验不符合其主要终点,tiotropium已被证明是有益的在成人和青少年哮喘症状当添加到至少ICS (8- - - - - -16]。此外,tiotropium的比较研究与氟替卡松加沙美特罗作为附加ICS在成人患者中发现急性反应舒喘灵和气道阻塞因素可以帮助预测积极的临床反应tiotropium [22]。处方医生应该考虑trial-level反应未必反映个体层面的反应,必须优化哮喘治疗患者个体的基础上。
总之,显著改善主要终点,FEV峰值1 (0-3h)响应tiotropium 5µg与安慰剂相比,并没有显示在我们的试验中,数值在肺功能指标的改善和哮喘控制观察。从我们的试验结果添加到当前的证据从先前发表的研究tiotropium Respimat附加治疗有症状的哮喘患者,因此,每天换一次tiotropium Respimat附加ICS +一个或多个控制器疗法可能被视为另一种治疗选择青少年患者严重的哮喘症状,安全性和耐受性与安慰剂相比。
补充材料
补充材料
请注意:补充材料并不是由编辑部,编辑和上传已由作者提供。
补充图S1。在第12周的应答率分析ACQ-6 (A)和ACQ-7 (B)。全分析集。常见的基线平均±标准差ACQ-6 = 2.075±0.455。常见的基线平均±SD ACQ-7 = 2.132±0.428。ACQ-6: 6-question哮喘控制调查问卷;ACQ-7: 7-question哮喘控制问卷。erj - 01100 - 2016 - _figure_s1
披露的信息
补充材料
确认
作者要感谢斯坦利·j·Szefler(科罗拉多州儿童医院呼吸研究所的极光,有限公司,美国)的输入和建议在开发这个手稿。医疗援助,写作形式的准备和修改的手稿,是由丽芬妮年轻(完成HealthVizion,马格斯菲特,英国)作者的概念方向,基于作者的反馈。作者感谢所有调查者支持审判(在线补充材料的完整列表)。
脚注
可以从本文的补充材料www.qdcxjkg.com
这项研究是在注册clinicaltrials.gov标识号NCT01277523。
支持声明:本研究支持勃林格殷格翰集团制药GmbH & Co .融资信息本文已沉积的资助者打开注册表。
利益冲突:披露可以找到与这篇文章www.qdcxjkg.com
- 收到了2016年6月1日。
- 接受2016年9月4日。
- 版权©2017人队。
这个版本分布在创作共用署名非商业性许可证的条款4.0。











![Study design. Usual background therapy was defined as high-dose inhaled corticosteroids (ICS) plus one or more controller therapies (e.g. a long-acting β2-agonist or leukotriene receptor antagonist) or medium-dose ICS plus two or more controller therapies (e.g. a long-acting β2-agonist and/or leukotriene receptor antagonist and/or sustained-release theophylline). High-dose ICS was defined as >400 µg budesonide or equivalent in patients aged 12–14 years and 800–1600 µg budesonide or equivalent in patients aged 15–17 years. Medium-dose ICS was defined as 200–400 µg budesonide or equivalent in patients aged 12–14 years and 400–800 µg budesonide or equivalent in patients aged 15–17 years [6]. #: two puffs of 2.5 μg; ¶: two puffs of 1.25 μg; +: two puffs.](http://www.qdcxjkg.com/content/erj/49/1/1601100/F1.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}