





Abstract
BackgroundPulmonary fibrosis is one of the leading indications for lung transplantation. The disease, which is of unknown aetiology, can be progressive, resulting in distortion of the extracellular matrix (ECM), inflammation, fibrosis and eventual death.
Methods13 patients born to consanguineous parents from two unrelated families presenting with interstitial lung disease were clinically investigated. Nine patients developed respiratory failure and subsequently died. Molecular genetic investigations were performed on patients' whole blood or archived tissues, and cell biological investigations were performed on patient-derived fibroblasts.
ResultsThe combination of a unique pattern of early-onset lung fibrosis (at 12–15 years old) with distinctive radiological findings, including 1) traction bronchiectasis, 2) intralobular septal thickening, 3) shrinkage of the secondary pulmonary lobules mainly around the bronchovascular bundles and 4) early type 2 respiratory failure (elevated blood carbon dioxide levels), represents a novel clinical subtype of familial pulmonary fibrosis. Molecular genetic investigation of families revealed a hypomorphic variant inS100A3and a novel truncating mutation inS100A13, both segregating with the disease in an autosomal recessive manner. Family members that were either heterozygous carriers or wild-type normal for both variants were unaffected. Analysis of patient-derived fibroblasts demonstrated significantly reduced S100A3 and S100A13 expression. Further analysis demonstrated aberrant intracellular calcium homeostasis, mitochondrial dysregulation and differential expression of ECM components.
ConclusionOur data demonstrate that digenic inheritance of mutations inS100A3andS100A13underlie the pathophysiology of pulmonary fibrosis associated with a significant reduction of both proteins, which suggests a calcium-dependent therapeutic approach for management of the disease.
Abstract
New evidence links an atypical form of pulmonary fibrosis with digenic mutations in the genes for calcium binding proteins S100A3 and S100A13. This implicates calcium homeostasis in the aetiology and pathogenesis of pulmonary fibrosis.http://bit.ly/2LyUQwb
Introduction
Interstitial lung diseases (ILDs) are a heterogeneous group of disorders of largely unknown aetiology that are characterised by variable types of interstitial and alveolar inflammation, parenchymal remodelling, and fibrosis [1]. The most common ILD is idiopathic pulmonary fibrosis (IPF), a progressive disorder that usually affects individuals >55 years of age. Because of the lack of effective treatments and the rapid progression to respiratory failure and death, pulmonary fibrosis remains one of the leading indications for lung transplantations worldwide [2,3]. Several environmental risk factors have been implicated in the pathogenesis of IPF. A genetic predisposition has been demonstrated [4]; however, the great majority of cases of IPF are sporadic.
Familial pulmonary fibrosis (FPF), defined as idiopathic ILD in two or more first-degree relatives (parent, sibling or offspring), has been attributed to nonsynonymous monogenic mutations in surfactant protein A2 (SFTPA2), surfactant protein C (SFTPC)or ATP binding cassette A3 (ABCA3)[5,6]. In addition, a common variant (rs35705950) in the promoter of the gene encoding mucin 5B (MUC5B)substantially increases its expression and markedly increases the risk of pulmonary fibrosis [7]. These mutations are proposed to converge on activation of the unfolded protein response [8]. Among a plurality of FPF kindred, ∼15% had mutations in telomerase genesTERTandTERC, and exhibited shortened telomeres [9,10]. Telomere shortening was also evident in 25% of patients with sporadic IPF who did not have identifiable mutations inTERTorTERC[11]. Several rare variants of other genes related to telomere maintenance, such asTINF2,DKC1,RTEL1,PARNandNAF1, have also been identified in ∼25% of patients with FPF [12].
Despite these advances, the pathogenesis of sporadic pulmonary fibrosis remains unclear. Here, we have identified two variants in the calcium binding protein genesS100A3(NM_002960) andS100A13(NM_001024210) segregating with the disease in seven siblings screened from two unrelated families with pulmonary fibrosis. The mutations led to lower expression of the proteins, aberrant receptor-mediated intracellular calcium responses, reduced capacity to tolerate external oxidative stress and altered extracellular matrix (ECM) protein expression in cells isolated from patients. These results will improve our understanding of the pathogenesis of fibrosing lung diseases.
Materials and methods
All samples were collected at King Faisal Specialist Hospital and Research Centre (Riyadh, Saudi Arabia) with institutional review board (IRB)-approved, written informed consent. The study was approved by the Research Advisory Council of King Faisal Specialist Hospital and Research Centre (KFSH&RC-RAC 2120 009).
Sequence analysis
Genomic DNA was extracted from whole blood or paraffin-embedded archived tissue of the affected patients, their parents and unaffected relatives (Families 1 and 2) using standard methods. Bidirectional sequencing of the coding regions of known IPF-associated genes (TERT,TERC,ABCA3andSFTPB)was performed in patients and nuclear family members of Family 1A. The full coding regions ofS100A3(NM_002960版本1)和S100A13(NM_001024210) were sequenced for all available members (affected and unaffected of Families 1 and 2) and 28 patients with sporadic pulmonary fibrosis, using standard PCR conditions. Sequence analysis was performed manually using the SeqMan 6.1 module of Lasergene (DNASTAR, Madison, WI, USA).
Linkage analysis and homozygosity mapping
Genotyping of all available family members using the Axiom Genome-Wide CEU 1 Array platform (Affymetrix, Santa Clara, CA, USA) was performed and analysed for homozygosity mapping using AutoSNPa (dna.leeds.ac.uk/autosnpa).Linkage analysis was performed using the Allegro module of easyLINKAGE [13]. Direct sequencing of candidate genes in the linkage interval and exome re-sequencing data of genomic DNA was performed using primer pairs designed to cover the entire coding region of each gene.
Whole exome sequencing
Whole exome sequencing (WES) was first performed on the affected son of Family 1A using the HiSeq2000 platform (Illumina, San Diego, CA, USA) with TruSeq3 chemistry by preparing and enriching the sample according to the manufacturer's standard protocol instructions. The concentration of each library was determined using the QPCR NGS Library Quantification Kit (G4880A; Agilent Technologies, Santa Clara, CA, USA) and the sample was sequenced at a final concentration of 10 nM. Mapping and alignment was performed on read files (FASTQ) generated from the sequencing platformviathe manufacturer's proprietary software and using the human genome (hg19/b37) and the Burrows–Wheeler Aligner (BWA) package version 0.6.1 [14]. Further realignment and variant analysis was performed to eventually determine single nucleotide polymorphism (SNP) novelty against dbSNP (www.ncbi.nlm.nih.gov/snp; Human Build 135) [15–17]. Variants were annotated with gene and gene function from Ensembl (www.ensembl.org/index.html)[18], and further analysis of possible causative variants by filtering the full exome dataset for all deletions, insertions, nonsense and canonical splice site mutations, as well as missense mutations (with a PhyloP score >3.5 of the underlying base change), were performed and reported. Similar WES analysis was performed later on one affected patient from both Families 1B and 2 using the same methodology.
Tissue culture
Skin biopsies were collected from two patients and two controls according to IRB-approved protocols. Fibroblasts were isolated and cultured as previously described [19].
Immunofluorescence and Western blotting
Immunofluorescence staining was performed on lung tissue sections prepared from paraffin blocks collected according to IRB-approved protocols from probands and healthy control lungs (healthy donor lungs before transplantation). Primary antibodies to S100A3 and S100A13 (Abcam; Abdulla Fouad Medical Supplies, Dammam, Saudi Arabia) and fluorescein-conjugated secondary anti-rabbit IgG (ThermoFisher Scientific, Waltham, MA, USA) were used according to the manufacturers' instructions. For fibroblast immunofluorescence, anti-S100A3 was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Images were acquired and analysed using cellSens Dimension version 1.9 (Olympus, Dusseldorf, Germany). Image acquisition was thresholded against controls where the primary antibody was omitted and fluorescence intensity due to the secondary antibody alone was considered nonspecific.
For Western blots, cells were lysed, separated on either 7.5% SDS-PAGE (Bio-Rad, Hercules, CA, USA) or 4–12% gradient SDS-PAGE and transferred onto PVDF membranes (Life Technologies, Carlsbad, CA, USA) or nitrocellulose membranes (Hybond ECL; Amersham, Little Chalfont, UK), and immunoblotted using primary rabbit antibodies against S100A3 (Santa Cruz Biotechnology), S100A13, matrix metalloproteinase (MMP) 2, MMP9, tissue inhibitor of MMP (TIMP)-1, actin (Santa Cruz Biotechnology) or glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Cell Signaling; Danvers, MA, USA), followed by peroxidase-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA, USA). Bands were visualised using chemiluminescence (SuperSignal West Pico; Thermo Scientific/Pierce, Rockford, IL, USA) and exposed to X-ray film or scanned using the ChemiDoc XRS+ Molecular Imager with Image Lab (Bio-Rad, Hercules, CA, USA). Western blot membranes were scanned and bands were processed using the Count and Measure function in cellSens Dimension version 1.9 (www.olympus-lifescience.com), and intensities of appropriate regions of interest were determined and expressed relative to corresponding β-actin band intensities.
Sample preparation for label-free protein in-solution digestion
Cells derived from patient and control samples were lysed using RapiGest MS compatible lysis buffer (Waters, Manchester, UK). A total of 100 µg protein from whole cell lysate of each sample was subjected to proteome analysis by in-solution tryptic digestion as previously described [20,21].
Protein identification by the LC-MSESYNAPT G2 platform
We used label-free quantitative one-dimensional nanoACQUITY liquid chromatography tandem mass spectrometry on SYNAPT G2 (Waters) and generated expression protein profiles between the sample groups. The instrument settings were optimised as previously described [20,22,23].
Expression proteomics data analysis and bioinformatics
All data acquisitions were in triplicate runs with automated data processing and database search using the Uniprot Human specific protein sequence database on the Progenesis QI for Proteomics protein identification platform (Waters/Nonlinear Dynamics, Newcastle, UK). Normalised protein abundance of significantly regulated proteins was considered (ANOVA; p<0.05), and significant expression was defined as a fold change >1.5 and a false discovery rate ∼3%. The significant expression dataset was further evaluated for functional/signalling pathway implications using Ingenuity Pathway Analysis version 8.7 (www.ingenuity.com).
Quantitative reverse transcriptase-PCR
Total cellular RNA was isolated using TRIzol reagent (Ambion, Grand Island, NY, USA) and cDNA was synthesised from 1–5 μg of RNA using the RT2第一个链cDNA工具包(试剂盒,日耳曼敦,医学博士,美国)according to the manufacturer's protocol. Primers for 18S rRNA were purchased from SABiosciences/Qiagen (Valencia, CA, USA). Primers forS100A3(forward: 5′-CCCGAACTGGTCAACTCTCA; reverse: 5′-GCCTGGCAGAGCTTGTATTT), plasmid backbone (forward: 5′-GTGGCGCTTTCTCATAGCTC; forward: 5′-TGTCTTACCGGGTTGGACTC) andS100A13(forward: 5′-CATCTGCTCAAGGATGTGGG; reverse: 5′-TCCTGATCTTCAGGTCTTT) were designed using PRIMER3 (SABiosciences/Qiagen). Quantitative reverse transcriptase (qRT)-PCR was performed on a StepOne Plus PCR system (Applied Biosystems, Carlsbad, CA, USA) using RT2SYBR Green qRT-PCR Mastermix (SABiosciences/Qiagen) according to the manufacturer's directions. For qRT-PCR ofS100A3andS100A13in control and patient samples the primers were: forward: 5′-GGACCCCGACTGAGTTTCG; reverse: 5′-GCTCTGAGGGGCAGTCCTTG and forward: 5′-CATCTGCTCAAGGATGTGGG; reverse: 5′-TCCTGATCTTCAGGTCTTT, respectively. For GAPDH the primers were: forward: 5′-CACCATCTTCCAGGAGTGAG; reverse: 5′-TCACGCCACAGTTTCCCGGA.
Intracellular calcium and mitochondrial integrity measurements
Cytosolic calcium measurements were performed on patient or control fibroblasts (from unaffected donors) as previously described [24]. Mitochondrial calcium measurements were performed using Rhod-2 AM as previously described [19]. Receptor-mediated changes in intracellular fluorescence intensity in response to fibroblast growth factor (FGF)-2 (Sigma, St Louis, MO, USA; 10 ng·mL−1), bradykinin (Sigma; 50 µM) and ionomycin (Sigma; 2 µM) were followed using the LSM 510 META laser scanning confocal system (Carl Zeiss MicroImaging, Jena, Germany). Mitochondrial staining was performed using MitoTracker Red CMXRos (Invitrogen, Carlsbad, CA, USA; 1 µM, 5 min at 37°C) and viewed under the Yokogawa Spinning Disk confocal microscopy system (Carl Zeiss MicroImaging).
Transmission electron microscopy and flow cytometry
For transmission electron microscopy (TEM), cells were fixed with 2.5% glutaraldehyde in cacodylate buffer (0.1 M, pH 7.4) for a minimum of 48 h. Osmication was performed using reduced osmium (1:1 mixture of 2% osmium tetroxide and 3% potassium ferrocyanide). After pre-embedding in 1% agar, samples were dehydrated in ethanol series and embedded in epoxy resin. Thin sections (70–100 nm) were collected on copper grids and contrasted with lead citrate. Imaging was performed using a transmission electron microscope operating at 300 kV (Titan Cryo Twin; FEI Company, Hillsboro, OR, USA). Images were recorded on a 4 k×4 k CCD camera (Gatan, Pleasanton, CA, USA). For flow cytometry, cells (1×106 cells·mL−1)were labelled with MitoTracker Green FM (1 µM) for 45 min on ice, washed (PBS, pH 7.2), fixed in 1% paraformaldehyde and analysed using a FACSCalibur flow cytometer (BD Biosciences).
Statistical analysis
ANOVA with Holm–Sidak's multiple comparison test was used to measure statistical significance using Prism (GraphPad, La Jolla, CA, USA). The unpaired two-tailed t-test was used when appropriate. A p-value ≤0.05 was considered significant.
Results
Brief case description
Family 1A
Three siblings (figure 1a), one boy (F1:IV-1) and two girls (F1:IV-2 and IV-3), from a total of seven children were born healthy to consanguineous parents (F1:III-6 and III-7) after normal pregnancies and deliveries. The parents did not report any developmental delay or medical problems until all three children developed pulmonary fibrosis at a young age. The boy developed pulmonary fibrosis at 12 years of age and both girls at 13 years of age. All three affected siblings had an identical clinical presentation and course (figure 1b).All developed dyspnoea in their early teens and, aside from the lung abnormalities, medical examinations did not reveal any abnormalities in appearance, developmental milestones, laboratory findings or other organ systems. Fibrosis was revealed by chest computed tomography (CT) imaging, and characterised by traction bronchiectasis, intralobular septal thickening and shrinkage of the secondary pulmonary lobules mainly around the bronchovascular bundles. The core of the lungs was extensively involved, suggesting bronchocentric interstitial fibrosis (figure 1band c), and pulmonary function testing indicated severe restriction and impaired oxygen transfer (figure 1f).Extensive medical investigations ruled out autoimmune and occupational causes of pulmonary fibrosis. Respiratory symptoms worsened with age and all three patients developed chronic type 2 respiratory failure (elevated blood carbon dioxide levels [25]), requiring noninvasive ventilation. Two of the siblings underwent lung transplantation at ages 23 years (F1:IV-1) and 22 years (F1:IV-2), but died due to primary graft dysfunction. Both received routine immunosuppression and their cross-match was negative. The third sibling (F1:IV-3) died from respiratory failure at age 23 years without having a lung transplant. Analysis of lung tissue obtained at autopsy from one of the subjects (F1:IV-2) revealed areas of interstitial inflammation, mild fibrosis involving the alveolar walls, mild subpleural fibrosis and advanced fibrosis sparing the subpleural space (figure 1g).
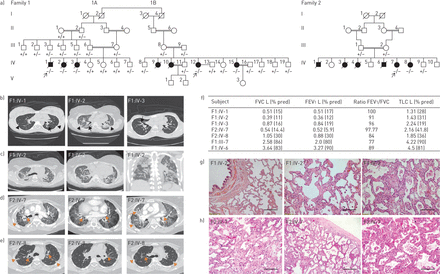
Clinical characteristics of patients affected with pulmonary fibrosis. FVC: forced vital capacity; FEV1: forced expiratory volume in 1 s; TLC: total lung capacity; CT: computed tomography. a) Pedigrees of all families with pulmonary fibrosis with subsequent genotype analyses. Arrows indicate the proband from each family. Circles: females; squares: males; white symbols: not included in the study; white symbols with genotype: unaffected; black symbols: pulmonary fibrosis affected; +: wild-type “C” allele ofS100A3/wild-type sequence ofS100A13; −: mutant “T” allele ofS100A3(c.229C>T)/4 bp deletion ofS100A13(c.238–241delATTG). b) CT scans at initial presentation of the three patients (F1:IV-1, IV-2 and IV-3) showing central traction bronchiectasis (long arrow). The distribution of fibrotic changes was peribronchovascular. The periphery of the lungs was spared. Global volume loss was seen with retracting subpleural fat in the lateral portions of the fissures (short arrows). c) CT scans late in the disease course of patient F1:IV-2: upper chest axial view (left), lower chest axial view (middle) and coronal view (right). There was progression of the peribronchovascular fibrotic changes and volume loss. Patches of ground-glass densities were randomly distributed. d, e) High-resolution CT scans of patients d) F2:IV-7 and e) F2:IV-8 showing central traction bronchiectasis (long arrows). The distribution of fibrotic changes is peribronchovascular and central. The periphery of the lungs was spared. Global volume loss is seen evident by retracting subplural fat in the lateral portions of the fissures (short arrows). f) Pulmonary function tests of five patients and family two members heterozygous for both the p.R77C and p.I80Gfs*13 variants inS100A3andS100A13, respectively. g) Pathology of one affected patient (F1:IV-2): generalised interstitial inflammation with fibrosis. The inflammation mostly consists of lymphocytes in a background of moderate interstitial fibrosis. No advanced lung fibrosis with honeycombing is identified. No granulomas, microgranuloma or vasculitis are noted. Scale bar: left image 100 µm, other images 50 µm. h) Pathology of another affected patient (F2:IV-7): interstitial inflammation with fibrosis in a diffuse pattern with no temporal heterogeneity. Advanced fibrosis seems to be sparing the subpleural space. No granulomas, microgranuloma or vasculitis are noted. Extensive sampling did not reveal a usual interstitial pneumonia-like pattern. Scale bar: left image 100 µm, other images 50 µm.
Family 1B
Four affected girls from a total of nine children born healthy to consanguineous parents (F1:III-8 and III-9) were enrolled. The mother of this family is the second cousin of both of the parents of Family 1A. Two of the girls (figure 1a: F1:IV-9 and IV-10) died of respiratory failure in their late 20s. Detailed clinical information is not available as they died prior to the family presenting to our clinic. Similar to Family 1A, an extensive workup of the living patients currently aged 31 years (F1:IV-12) and 34 years (F1:IV-15), with symptoms beginning at age 12 and 15 years, respectively, revealed no known autoimmune and occupational causes of pulmonary fibrosis. CT imaging revealed identical findings of fibrosis and pulmonary function tests showed a severe restriction pattern (images not shown). No developmental or extrapulmonary findings were documented. Lung transplantation workup has been initiated for both sisters.
Family 2
第二个家庭面对simila引人注目r clinical pattern to Family 1 (A and B). Family 2 was comprised of a total of 10 children with six affected siblings (four girls and two boys) born to healthy consanguineous parents. Extensive pedigree analysis confirmed that they were unrelated to Family 1. Four of the affected children (two girls and two boys) had died previously due to lung disease (between the ages of 25 and 32 years). Detailed clinical information for the deceased is not available. Both of the surviving sisters developed respiratory symptoms, at 13 years of age in one (F2:IV-7) and estimated as late teenage years in the other (F2:IV-8). For both sisters, pulmonary function tests showed a severe restriction pattern. CT imaging was very similar to Family 1 (figure 1d–f and h). Like the affected individuals in Family 1, the affected sisters in Family 2 showed normal appearance, development and laboratory findings, and autoimmune and environmental causes of lung fibrosis were excluded. One of the sisters (F2:IV-7) underwent a lung transplant at age 27 years, and is currently aged 35 years and in good health. The second sister (F2:IV-8) is currently on the waiting list for a lung transplant.
Identification of a novel pulmonary fibrosis region on chromosome 1
Homozygosity mapping using Families 1 and 2 defined a single 34 Mb critical interval (chromosome 1: 120 127 864–154 749 047 bp) shared by all seven affected individuals (F1:IV-1, IV-2 and IV-3, F1:IV-12 and IV-15, and F2:IV-7 and IV-8) (figure 2a), defined proximally by rs10802117 and distally by rs11808053 containing over 363 annotated genes (supplementary figure S1b).A combined parametric multipoint linkage analysis using a total of 17 individuals (seven affected and 10 unaffected) from Families 1 and 2 revealed a single significant peak with a maximum logarithm of the odds (LOD) score of 5.28 corresponding to chromosome 1p12-q21.3 (figure 2b).Initial Sanger sequencing of three candidate genes in this region (based upon function and/or association with ILD) failed to identify a causative mutation.
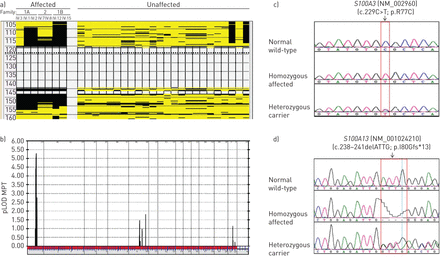
Molecular analyses in pulmonary fibrosis of Families 1(A and B) and 2. a) A single run of homozygosity as a result of homozygosity mapping shared by all seven affected patients between rs10802117 and rs11808053 confirming linkage analysis. In addition, a total of 24 unaffected family members displayed no homozygosity for this region of interest. b) Linkage analysis using a total of 17 individuals (seven affected and 10 unaffected) from the two families resulting in a peak where the maximum multipoint parametric logarithm of the odds score (pLOD MPT) was 5.28, corresponding to chromosome 1p12-q21.3 on the x-axis. c, d) Sequence chromatograms indicating the wild-type, homozygous affected and heterozygous carrier forms of c) the C to T transition at position c.229 changing the arginine residue to cysteine at position 77 of the S100A3 protein (c.229C>T; p.R77C) and d) the c.238–241delATTG (p.I80Gfs*13) inS100A13.Mutation name is based on the full-lengthS100A3(NM_002960) andS100A13(NM_001024210) transcripts.
Exome sequencing
Initially, WES was performed in the proband of Family 1A (F1:IV-1) (figure 1a).After filtering for homozygous nonsynonymous single nucleotide variants (SNVs) within the linkage interval, that were either novel or had either low or unknown minor allele frequency in dbSNP, only three previously described variants, rs3795737 inISG20L2, rs143224912 inSETDB1and rs138355706 inS100A3, and one novel variant inS100A13, were identified. TheISG20L2andSETDB1variants were excluded based on their frequencies in normal population cohorts. Sanger sequencing of Family 1 showed that both rs138355706 inS100A3(c.229C>T, missense causing a p.R77C mutation) and a 4 bp deletion inS100A13(c.238–241delATTG causing a frameshift p.I80Gfs*13) segregated completely with ILD in Family 1 based upon recessive inheritance (figure 2cand d), were in total linkage disequilibrium, and were present in acisconformation. Allele frequency for rs138355706 within the population was calculated from exome sequencing of 2000 individuals and was found to be 0.1% (unpublished data from the Saudi Human Genome Project;www.saudigenomeprogram.org).Furthermore, 500 ethnically matched normal controls were genotyped for this change by re-sequencing ofS100A3.这三个人是杂合的,布鲁里溃疡t none was homozygous for rs138355706. Sequencing of theS100A3intronic and 5′-flanking sequences was performed in the affected patients and no other variants were identified (data not shown). The novel truncated variant inS100A13was not found in our “in-house” Saudi exome data (unpublished data from the Saudi Human Genome Project), 1000 Genome and gnomAD databases. The c.229C>T (p.R77C) variant inS100A3and c.238–241delATTG (p.I80Gfs*13) mutation inS100A13also segregated fully with ILD in Families 1B and 2.
Haplotype analysis
Haplotype analysis carried out using eight markers (four microsatellite markers flankingS100A3,S100A13and three further intragenic markers) (supplementary figure S1a)confirmed that all affected individuals from both families shared a specific disease haplotype on both chromosomes that was not present in the unaffected individuals, suggesting a shared extended haplotype from a common founder.
Consequences of theS100A3c.229C>T andS100A13variants
TheS100A3c.229C>T variant resulted in an arginine to cysteine substitution at residue 77 within the second of the two EF-hand calcium binding motifs in the protein. The predicted impact of this variant on protein structure/function was evaluated using PolyPhen-2 version 2.2.2 (genetics.bwh.harvard.edu/pph2), SIFT prediction (sift.bii.a-star.edu.sg)and Combined Annotation Dependent Depletion (CADD;cadd.gs.washington.edu)software. PolyPhen-2 and SIFT predicted minor effects of the mutation on protein structure/function with scores of 0.004 and 0.21, respectively, and a CADD PHRED score of 17.65. Since microRNAs can modify translation efficiency by binding to coding sequences as well as 3′-untranslated region sequences, we sought to determine whether the c.229C>T mutation altered any known microRNA binding sites using MicroSNiPer release 19 (http://vm24141.virt.gwdg.de/services/microsniper)and a minimum 7 nucleotide seed sequence. We found no effect of the SNV on predicted microRNA binding sites. Moreover, analysis of predicted protease cleavage sites using PeptideCutter (web.expasy.org/peptide_cutter)did not reveal any effect on protease cleavage sites inS100A3.However, Human Splicing Finder version 3.1 (www.umd.be/HSF3)predicted an alteration in the exonic splicing enhancer ofS100A3[26] and creation of a new exonic splicing silencer site, which is known to play a role in constitutive and alternative splicing [27,28]. TheS100A13loss-of-function variant is, as expected, predicted to be “disease causing” by MutationTaster (www.mutationtaster.org).
Effect of mutations onS100A3andS100A13expression
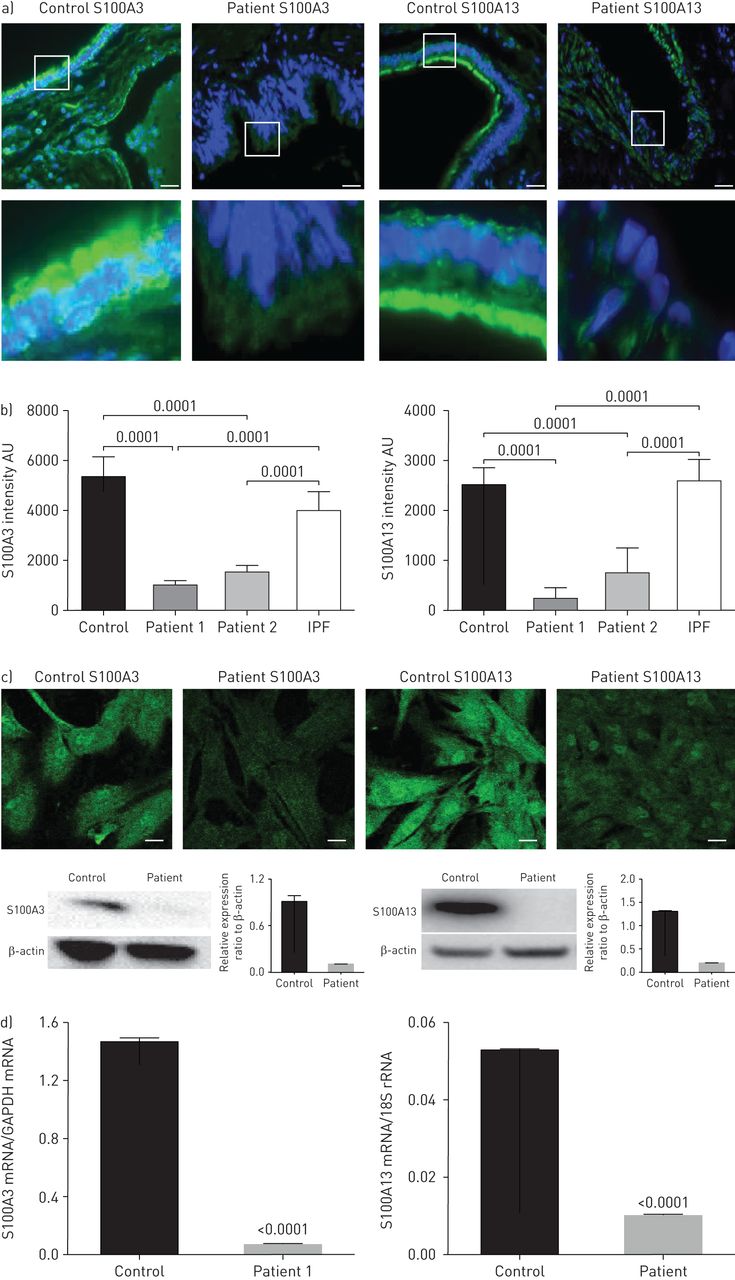
Indirect immunofluorescence staining of thin sections obtained from healthy lung tissues (donor lungs before transplantation) demonstrated the presence of both proteins (figure 3aand b). The staining was consistent with reports in The Human Protein Atlas demonstrating expression of S100A3 and S100A13 in the lungs (www.proteinatlas.org/ENSG00000188015-S100A3/tissueandwww.proteinatlas.org/ENSG00000189171-S100A13/tissue).In normal lung tissue, S100A3 and S100A13 expression was distributed along the ciliary edges and the apical surfaces of the bronchiolar epithelium of the small airways. In contrast to control lung tissue and lung tissue from a patient with sporadic IPF, lung tissue from affected family members demonstrated greatly reduced S100A3 and S100A13 expression (figure 3aand b). Low S100A3 and S100A13 expression levels were also found in patient-derived skin fibroblasts when compared with skin fibroblasts from normal controls (figure 3c, upper). Western blotting confirmed significantly reduced expression of S100A3 and S100A13 in skin fibroblasts isolated from patient cells compared with cells from healthy controls (figure 3c, lower). The reduced expression of the mutant proteins in patients was paralleled by reduced expression of mRNA levels of both variants compared with control (figure 3d).
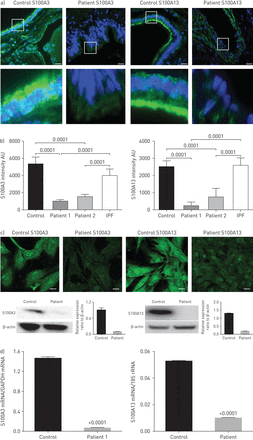
Effect ofS100A3andS100A13突变对蛋白表达。盟:任意单ts; IPF: idiopathic pulmonary fibrosis; GAPDH: glyceraldehyde 3-phosphate dehydrogenase. a) Upper images show immunofluorescence micrographs demonstrating reduced expression of S100A3 and S100A13 proteins in lung tissue from a normal control and an affected family member (F1:IV-2). Scale bar: 10 µm. Magnified areas of the indicated portions are shown in the lower images. b) Relative protein expression of S100A3 and S100A13 in normal control and lung tissues from two independent patients shown together with relative protein expression in an IPF patient. Histograms are mean±sdintensity of multiple fields in the stained samples. c) Confocal fluorescence laser scanning micrographs showing the reduced expression of S100A3 and S100A13 proteins in skin fibroblasts isolated from patients compared with controls and the corresponding Western blots. Data are representative of three independent experiments with cells isolated from two patients and two controls. Scale bar: 20 µm. d) Relative expression of S100A3 and S100A13 mRNA in skin fibroblasts isolated from normal controls and patients. Data are representative of at least three independent experiments. p-values are indicated when appropriate.
Effect ofS100A3andS100A13mutations on intracellular calcium signalling and mitochondrial structure and function
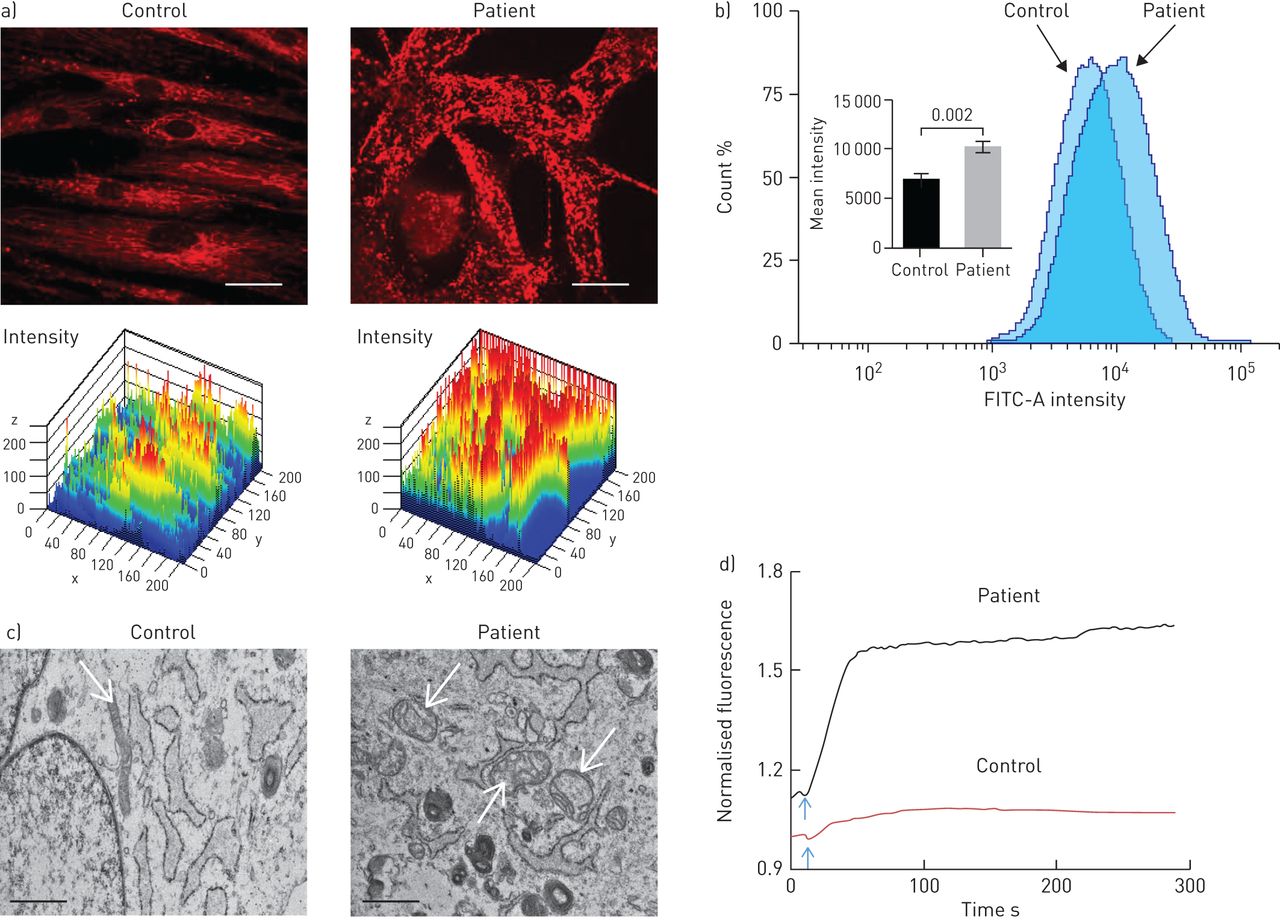
Since theS100A3andS100A13genes encode calcium binding proteins, we explored the possibility that the mutations may affect intracellular calcium homeostasis. We measured intracellular calcium changes in response to bradykinin (50 µM) and FGF-2 (10 ng·mL−1)in skin fibroblasts isolated from healthy controls and patients. Receptor-mediated calcium release was significantly reduced in fibroblasts from patients compared with control fibroblasts (figure 4aand c). The increase in calcium signal following bradykinin stimulation was reduced in patient cells compared with control cells (1.47±0.14-foldversus2.34±0.07-fold; p<0.0001) (figure 4b).FGF-2 induced a 1.19±0.02-fold increase in calcium signal in patient cellsversus1.41±0.06-fold increase in control cells (p=0.017) (figure 4d).Ionomycin-induced calcium release was also significantly reduced in patient fibroblasts compared with control fibroblasts (p<0.02) (figure 4e).Since mitochondria accumulate calcium and shape the temporal and spatial calcium changes in many cell types [29,30], we analysed the bradykinin-induced increase in intramitochondrial calcium levels, which was reduced in patient fibroblasts compared with normal control fibroblasts (p=0.015) (figure 4f).Furthermore, compared with mitochondria in control cells, the mitochondria in patient cells exhibited aberrant morphology and a more punctate MitoTracker Red CMXRos fluorescence pattern (figure 5a).In addition, patient cells appear to have increased mitochondrial staining compared with control cells (figure 5a), which was confirmed by flow cytometry analysis of MitoTracker Green MF-stained cells (p=0.002) (figure 5b).TEM showed evidence of mitochondrial damage with reduced cristae and reduced rough endoplasmic reticulum in patient samples compared with controls (figure 5c).The functional integrity of the mitochondria in both patient and control cells was further investigated by measuring the effect of external oxidative stress induced by treating the cells with hydrogen peroxide (0.03%).Figure 5dillustrates the ability of control cells to resist oxidative stress compared with patient cells.
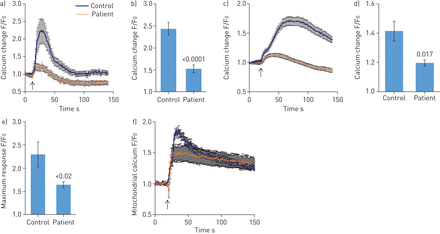
Effect ofS100A3andS100A13mutations on intracellular calcium changes. a) Intracellular calcium changes following stimulation of cultured skin fibroblasts isolated from a healthy control or a patient. Cells were stimulated with bradykinin (50 µM) (arrow). b) The histograms show maximum response to bradykinin. Experiments were performed on live single cells using confocal laser scanning microscopy. Data are expressed as mean±sem(n=23 and 24 for control and patients cells, respectively). Data are expressed as normalised fluorescence intensity ratio (F/F0)relative to the averaged three images obtained prior to the addition of the stimulus and are representative of three independent experiments. c, d) Fibroblast growth factor-2 (10 ng·mL−1)-stimulated cells, with results presented similar to a) and b). Data are expressed as mean±sem(n = 10和7为控制和患者细胞,尊重ively). Data are representative of 116 and 102 cells used in eight and 12 independent experiments from patient and control fibroblasts, respectively. e) Relative maximum calcium response to ionomycin (2 µM) in skin fibroblasts from controls and patients. Data are expressed as mean±sem(n=28 and 17 for control and patient cells, respectively). f) Mitochondrial calcium changes following stimulation of skin fibroblasts isolated from a healthy control or a patient with bradykinin (50 µM). Arrow indicates addition of bradykinin. Experiments were performed in live single cells using confocal laser scanning microscopy. All data are representative of cells isolated from two patients from the two unrelated families and two controls. p-values are indicated.
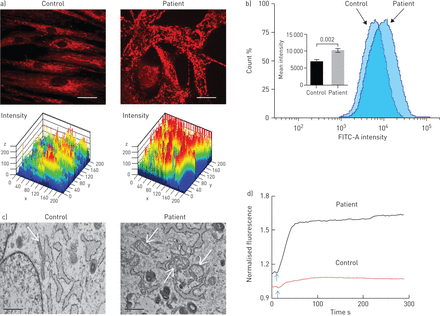
Effect ofS100A3andS100A13mutations on mitochondria. FITC: fluorescein isothiocyanate. a) Confocal fluorescence micrographs of isolated skin fibroblasts labelled with MitoTracker Red CMXRos (1 µM) and the corresponding three-dimensional intensity maps colour coded so that warm colours indicate high intensity and cold colours indicate low intensity. Scale bar: 20 µm. b) Flow cytometry of skin fibroblasts isolated from patient and control cells stained with MitoTracker Green FM. The inset shows mean±sem病人和荧光强度的控制cells. Experiments were performed in triplicate and are representative of at least three independent experiments using 106cells per sample. p-value is indicated. c) Transmission electron micrographs of cells isolated from healthy control and patient cells depicting differences in mitochondrial size (arrows) and loss of cristae. Scale bar: 1 µm. d) Effect of externally added oxidative insult (hydrogen peroxide 0.03%, arrow) on patient and control cells labelled with MitoTracker Red CMXRos. Data are representative of three independent experiments.
Effect ofS100A3andS100A13mutation on ECM remodelling
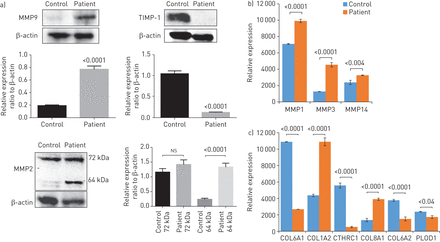
Since pulmonary ECM remodelling is the hallmark of IPF [31,32], we compared the expression of matrix-related proteins in patient and control fibroblasts. Western blot analysis demonstrated increased expression of two MMPs (MMP2 and MMP9) and reduced expression of TIMP-1 in patient fibroblasts compared with control cells (figure 6a).Proteomic analysis of ECM components demonstrated increased expression of MMP1, MMP3 and MMP14 in patient fibroblasts compared with control cells (figure 6b).This was paralleled by differential expression of collagen I (COL1A2), collagen VI (COL6A1 and COL6A2), collagen VIII (COL8A1), collagen triple helix repeat-containing protein 1 (CTHRC1) and procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 (PLOD1) (figure 6c).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect ofS100A3andS100A13mutations on extracellular matrix (ECM) components. MMP: matrix metalloproteinase; TIMP-1: tissue inhibitor of MMP-1; COL6A1: collagen α-1(VI) chain; COL1A2: collagen α-2(I) chain; CTHRC1: collagen triple helix repeat-containing protein 1; COL8A1: collagen α-1(VIII) chain; COL6A2: collagen α-2(VI) chain; PLOD1: procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1. a) Western blots of MMP2, MMP9 and TIMP-1 expression by skin fibroblasts isolated from healthy controls and patients. Relative expression is depicted in the accompanying histograms. b, c) Differential expression of b) matrixins MMP1, MMP3 and MMP14, and c) ECM-associated proteins COL6A1, COL1A2, CTHRC1, COL8A1, COL6A2 and PLOD1. Normalised protein abundance of significantly differentially expressed proteins between patient and control samples is shown (fold change >1.5 and false discovery rate ∼3%). Yeast alcohol dehydrogenase standard (P00330) at a concentration of 200 fmol per injection was used for “Hi3” absolute quantifications of all identified proteins. The histogram bars correspond to the average protein expression between the two sample groups using the label-free liquid chromatography-mass spectrometry expression analysis system on the Progenesis QI for Proteomics platform. Data are expressed as mean±sem(n=3). p-values are indicated.
Discussion
S100A3 and S100A13 are members of a large family of acidic, low-molecular-weight calcium binding proteins. More than 20 different members have been identified with diverse functions [33–36]. The proteins are found exclusively in vertebrates. S100 family members exist as homodimers, heterodimers and multioligomers, displaying cell- and tissue-specific patterns of expression [37] with significant structural similarities to calmodulins [38,39]. Although the proteins function intracellularly, secreted members exhibit cytokine-like effects through binding to various receptors, including RAGE (receptor for advanced glycation end-products) [40]. Of all S100 proteins, S100A3 has the highest content of cysteine and the highest affinity for zinc. It is expressed in human hair cuticles, where it is citrullinated and tetramerised to improve its calcium binding ability, and it contributes to the endocuticle rigidity of aged hair [41]. S100A13 is associated with a nonclassical pathway of interleukin (IL)-1α and FGF-1 secretion [42,43]. It was demonstrated to form a heterotetrameric complex with IL-1α [43].
In this study, we provide evidence implicating S100A3 and S100A13 in pulmonary fibrosis. We describe an atypical form of lung fibrosis radiologically characterised by traction bronchiectasis, intralobular septal thickening and shrinkage of secondary pulmonary lobules mainly around the bronchovascular bundle with a bronchocentric interstitial fibrosis. The disease is early onset leading to type 2 respiratory failure, likely attributed to the bronchocentric nature of the fibrosis around major airways causing airflow limitation. Histological analysis demonstrated advanced fibrosis and microscopic honeycombing sparing the subpleural area. The disease segregates in patients from two unrelated families in which digenic mutations inS100A3andS100A13lead to significant reduction in the expression of both proteins.
The mutation inS100A3introduces an 11th cysteine residue which replaces the arginine residue at position 77. The p.R77C mutation is classified as a SNP having an allele frequency of 0.0009635 (267 out of 277116) and has been found in the homozygous state (four homozygotes in the South Asian population) in the gnomAD database (http://gnomad-old.broadinstitute.org/variant/1-153520235-G-A).有趣的是repla S100A3精氨酸的残渣ced in the patients is completely conserved among orthologues in five mammalian species and it is located within one of the two EF-hand calcium binding motifs of the protein, suggesting more severe consequences for the protein function than was predicted by PolyPhen-2 and SIFT. The frameshift mutation ofS100A13is expected to result in a truncated form of the protein. Sequencing of the full intronic regions and 3′-untranslated region ofS100A3andS100A13excluded any other disease-associated variations ofS100A3orS100A13in affected members of both families. Interestingly, sequencing of the full coding region of both genes in 28 patients with sporadic IPF did not reveal any mutations in either of these genes. However, no other cases of FPF were genetically analysed. The mutations in families from this study did not alter any predicted microRNA binding sites that could affect protein expression.
Whilein silicoand functional evidence clearly support pathogenicity associated with co-inheritance of both variants, it remains unclear if either variant alone is sufficient to cause disease. Population data would suggest that theS100A3variant, although rare, may be too frequent in normal individuals to cause disease independently. Conversely, no population data exist for theS100A13variant and there are no instances to date of symptomatic or asymptomatic individuals with null mutations ofS100A13.As this variant has currently only been observed in strong linkage disequilibrium with an essentially nullS100A3variant, it is not possible to know if it independently causes disease. However, the question arises as to a possible modulating role of theS100A3variant on the expression of S100A13. Given theS100A13variant is a frameshift mutation resulting in premature truncation and loss of function, any modulating role of S100A3 if present is unlikely to contribute to disease predisposition in this instance. Indeed, the population frequency of theS100A3variant argues against it being pathogenic. Accordingly, it is unlikely that it impactsS100A13expression in a deleterious manner. It is possible, however, that the activity of S100A3 is complemented by S100A13 andvice versa(this is particularly pertinent since both proteins are involved in the calcium signalling pathway), which might explain the digenic inheritance of the disease in the families studied.
Both mutations result in a significant reduction in the levels of corresponding proteins, which is paralleled by reduced mRNA levels. The underlying mechanism(s) behind the reduced mRNA levels is yet to be determined. Whereas the reducedS100A13mRNA levels due to frameshift mutation might be explained by post-transcriptional nonsense mediated mRNA decay, the reducedS100A3mRNA levels are difficult to reconcile especially sincein silicoinvestigations predicted no effect of the mutation on predicted microRNA binding sites. However, a possible potential alteration of splicing mechanism(s) which was predicted by Human Splicing Finder may account for the low mRNA levels seen in our patients' samples. This is yet to be confirmed.
适当的细胞内钙稳态是paramount to certain stimulus response coupling, in which engagement of cognate receptors evokes the correct cellular response. Inappropriate signalling can lead to aberrant cellular behaviour underlying many diseases. Here, we demonstrate that the digenic mutation is associated with aberrant calcium changes in response to two independent agonists,i.e.bradykinin and FGF-2, suggesting a pivotal role for S100A3/S100A13 in receptor-induced calcium transients. A direct effect of S100 proteins on intracellular calcium is not uncommon. It is noteworthy that S100A1 regulates voltage sensing and calcium release necessary for excitation contraction coupling in mouse skeletal muscle [44]. Together with calmodulin, S100A1 has been shown to bind and modulate ryanodine receptor-dependent calcium release [45]. In addition, action potential-induced calcium transients are suppressed in skeletal muscle cells from S100A1 knockout mice [46]. Whether S100A3/S100A13 proteins behave in a similar way to S100A1 is yet to be determined.
Central to receptor-mediated calcium transients is mitochondrial calcium uptake, which plays a crucial role in intracellular calcium signalling by shaping and buffering calcium transients [30]. Here, we demonstrate that cells isolated from patients carrying theS100A3/S100A13mutations display increased numbers of mitochondria and an aberrant response to oxidative challenge with signs of autophagy, which might underpin the aberrant calcium response seen in patient fibroblasts. Moreover, direct measurement of mitochondrial calcium showed a significantly different response to bradykinin stimulation between patient and control fibroblasts.
IPF has been shown to be associated with increased levels of type I collagen and elevated levels of MMPs in blood and lung samples [31]. Since MMP expression, structure and activity are modulated by calcium, we measured MMP levels in patient-derived cells. Our study demonstrates elevated levels of MMP1, MMP2, MMP3, MMP9 and MMP14, and reduced levels of TIMP-1, which were paralleled by differential expression of collagens and other ECM-containing proteins in cultured cells isolated from our patients.
Finally, in this study we demonstrate for the first time a co-inheritance of two mutations leading to diminished expression of two calcium binding proteins in patients with an atypical form of lung fibrosis. We provide evidence that altered calcium homeostasis is associated with differential expression of ECM components in cells isolated from patients with pulmonary fibrosis. A direct link between calcium homeostasis and lung function was also reported in bleomycin models of lung fibrosis [47]. Furthermore, agonist-induced calcium oscillations have been demonstrated in pulmonary fibroblasts where they are hypothesised to control fibrotic gene expression [48]. Our data lend support to this hypothesis and suggest a “calcium-based therapy” be considered for the management of this as yet incurable disease.
Supplementary material
Supplementary Material
Please note:supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary figure S1. Haplotype analysis. a) The pedigree of families 1 and 2 with haplotype and genotype analyses; disease haplotype is highlighted in yellow. Genotype of variants in genesS100A3: c.229 C>T transition (rs138355706) causing p.R77C andS100A13: c.238-241delATTG causing p.I80Gfs*13 is denoted in red text. b) Circular ideogram of the genome (Agile MultiIdeogram; http://dna.leeds.ac.uk/) showing the exclusive region of homozygosity (ROH) between the affected individuals in all families (dark blue) on chromosome 1 (chr1:120,127,864-154,749,047 bp; UCSC genome browser, build hg19), that is not shared with any of the unaffected individuals (pink). Light blue and pink blocks denote ROHs present in affected and unaffected members, respectively.ERJ-02041-2018.Figure_S1
Acknowledgements
We are thankful to Rachid Sougrat (Abdullah University of Science and Technology, Jeddah, Saudi Arabia) for performing TEM, Mohammed Akhtar (Dept of Laboratory Medicine and Pathology, Hamad Medical Corporation, Qatar) for help with histology images, Amer Almzroua (Flow Cytometry Core Facility, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia) for flow cytometry experiments, and Turfah AlSheddi (Dept of Genetics, King Faisal Specialist Hospital and Research Centre) and John Schneider (Cyclotron and Radiopharmaceutical Dept, King Faisal Specialist Hospital and Research Centre) for technical assistance. The authors are indebted to the Saudi Human Genome Project (King AbdulAziz City for Science and Technology, Riyadh, Saudi Arabia), Research Centre Administration, Training and Education Office and Office of Research Affairs, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia.
Footnotes
This article has supplementary material available fromwww.qdcxjkg.com
Author contributions: E.A. Al-Mutairy, S.P. Atamas, J.D. Hasday, F.A. Imtiaz, M. Khalid and F. Al-Mohanna: conceptualisation, investigation, resources, supervision and writing; S. Al Qattan, S. Saleh, L.M. Mahmoud, M. Al-Saif, L. Al-Haj, A. Al-Enazi, A.M. Abdelsayed, S.F. Mohammed, A.F. Mobeireek, M.A. Chisti, I.G. Luzina, M. Al-Owain, K. Ramzan, I. Weheba, F.A. Imtiaz, E.A. Al-Mutairy, K.S. Khabar and F. Al-Mohanna: investigation, data curation, formal analysis and methodology; L.J. Janssen, B.F. Meyer, W. Conca, A. Alaiya, K.S. Collison, S.P. Atamas, K.S. Khabar, J.D. Hasday, K. Ramzan, F.A. Imtiaz, E.A. Al-Mutairy and F. Al-Mohanna: writing, review and editing.
Conflict of interest: E.A. Al-Mutairy has a patent “Method for treating pulmonary fibrosis using S100A3 protein” pending and a patent “The use of S100A13 in the diagnosis and treatment of pulmonary fibrosis” pending.
Conflict of interest: F.A. Imtiaz has nothing to disclose.
Conflict of interest: M. Khalid has a patent “Method for treating pulmonary fibrosis using S100A3 protein” pending and a patent “The use of S100A13 in the diagnosis and treatment of pulmonary fibrosis” pending.
Conflict of interest: S. Al Qattan has nothing to disclose.
Conflict of interest: S. Saleh has nothing to disclose.
利益冲突:L.M.艾哈迈迪没有thing to disclose.
Conflict of interest: M.M. Al-Saif has nothing to disclose.
Conflict of interest: L. Al-Haj has nothing to disclose.
Conflict of interest: A. Al-Enazi has nothing to disclose.
Conflict of interest: A.M. AlJebreen has nothing to disclose.
Conflict of interest: S.F. Mohammed has nothing to disclose.
Conflict of interest: A.F. Mobeireek has nothing to disclose.
Conflict of interest: K. Alkattan has nothing to disclose.
Conflict of interest: M.A. Chisti has nothing to disclose.
Conflict of interest: I.G. Luzina has nothing to disclose.
Conflict of interest: M. Al-Owain has nothing to disclose.
Conflict of interest: I. Weheba has nothing to disclose.
Conflict of interest: A.M. Abdelsayed has nothing to disclose.
Conflict of interest: K. Ramzan has nothing to disclose.
Conflict of interest: L.J. Janssen has nothing to disclose.
Conflict of interest: W. Conca has nothing to disclose.
Conflict of interest: A. Alaiya has nothing to disclose.
Conflict of interest: K.S. Collison has nothing to disclose.
Conflict of interest: B.F. Meyer has nothing to disclose.
Conflict of interest: S.P. Atamas has nothing to disclose.
Conflict of interest: K.S. Khabar has nothing to disclose.
Conflict of interest: J.D. Hasday has nothing to disclose.
Conflict of interest: F. Al-Mohanna has a patent “Method for treating pulmonary fibrosis using S100A3 protein” pending and a patent “The use of S100A13 in the diagnosis and treatment of pulmonary fibrosis” pending.
- ReceivedOctober 25, 2018.
- AcceptedApril 14, 2019.
- Copyright ©ERS 2019
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










