





摘要
慢性阻塞性肺疾病(COPD)是一种由多种潜在机制引起的综合诊断,需要分子亚表型来开发分子诊断/预后工具和有效的治疗。
这些研究的目的是研究多组学整合是否能提高小群体COPD分子分类的准确性。
采用相似性网络融合(SNF)方法,对52名女性的9个组学数据块(包括mRNA、微RNA、蛋白质组和代谢组)进行整合。多组学整合显著提高了从健康的不吸烟者和肺功能正常的吸烟者分类COPD患者的准确性,在95%的功率下,将所需的组大小从n=30减少到n=6。四到七个组学平台的七种不同组合达到了>95%的准确率。
首次在九个组学数据块上证明了多组学数据整合与数据驱动分类能力准确性之间的定量关系。整合五到七个组学数据块,使COPD诊断的正确分类率达到100%,分组人数少至n=6人,尽管当前吸烟会产生强烈的混杂效应。这些结果可以作为未来基于系统的多组学研究设计的指南,表明整合来自多个分子水平和解剖位置的五到六个数据块足以促进小队列中的无监督分子分类。
摘要
多组学整合显著改善COPD的无监督分子预测;100%准确率,亚组n=6http://ow.ly/EeiY30iWB2f
介绍
慢性阻塞性肺疾病(COPD)是一种综合性诊断,目前仅通过肺活量测定和症状来定义。然而,慢性阻塞性肺病有多种病因,包括环境暴露、遗传易感性和发育因素。全基因组关联研究表明,多基因变异可以部分解释两组1秒用力呼气量(FEV)变异1.)和钒铁1./强制肺活量[1.],丰富了参与肺功能调节的发育和炎症途径[2.]然而,大量的生物途径似乎控制着肺功能的发展和下降,遗传变异对表型的影响有限[3.].很明显,下游分子级联的多个调节介质,源自几个不同的解剖室,参与了慢性炎症和慢性阻塞性肺病的特征结构变化。
由于大量COPD亚表型导致相似的临床特征,COPD的分子亚表型是识别和分类这些亚群的重要第一步,然后才能为相应的患者亚群建立诊断/预后工具和治疗选择ale分析,即基因组学、蛋白质组学、脂质组学、代谢组学和呼吸组学为阐明复杂炎症疾病(如COPD)的整体变化提供了手段。然而,由于不同分子水平和解剖位置的失调可能在不同的疾病亚群中占主导地位,多组学整合可能有必要促进诊断和理解潜在COPD疾病亚群中涉及的疾病机制[4.–8.].最近的一些整合两三个组学数据块的研究表明,整合来自多个分子水平的数据可以提高生物标志物的识别[9,10,亚表现型预测[11,12和机械理解[13慢性阻塞性肺病。虽然这些具体的例子为组学整合的优势提供了令人信服的证据,但除了双组学和三重组学整合之外,还没有对统计能力增益的定量评估发表。
在这里,我们提出了一个定量评估的9个多分子水平组学数据块(基因分型、mRNA、microRNA (miRNA)、蛋白质组和代谢组)的整合,这些数据块来自Karolinska COSMIC队列的多个解剖位置(气道上皮细胞、肺部免疫细胞、气道渗出液、外泌体和血清)。主要目的是评估与单组或双组学调查相比,基于网络的三到七个组学数据块的整合是否能提高小群体群体分类的统计能力和准确性。
作为组学整合的框架,我们使用了相似网络融合(SNF)[14],它代表了一类新的集成方法[15].大多数网络集成方法都是为了聚类变量(例如基因,信使rna,蛋白质)来确定已知组之间的生物相互作用或机制。相比之下,SNF是一种基于研究对象的方法,利用研究对象之间整体分子谱的相似性,将他们聚为以前不知道的组。SNF代表了一种独特的网络方法,首先为每一个单组数据块创建一个相似网络,然后为每一对组数据块创建一个相似网络,等,以迭代的方式,直到所有概要文件都被表示出来。最重要的是,这个过程是在无监督的情况下进行的,这意味着没有关于疾病状态或其他群体分类的信息被包括在内。这是慢性阻塞性肺病分类的一个重要方面,因为潜在的假设是,目前的临床特征并没有提供足够的分辨率来促进慢性阻塞性肺病亚表型[16].
目前的研究提出了一个协调一致的工作流程,旨在最大限度地利用获得的分子信息,同时最大限度地减少获得强大统计能力所需的个体数量。该方法为在小群体中进行基于系统医学的研究提供了可行的策略,代表了一个重要的进步这将有助于设计和开展呼吸系统疾病分子亚表型研究。
材料和方法
方法的详细描述请参见补充材料.
临床队列
本研究使用卡罗琳斯卡宇宙队列(ClinicalTrials.gov ID:NCT02627872),一项三组横断面研究[17–25]与年龄匹配(45-65岁)和性别匹配的健康不吸烟者(“健康”)、肺功能正常的吸烟者(“吸烟者”)和COPD患者(“COPD”;全球慢性阻塞性肺病倡议(GOLD) I-II / A-B阶段;FEV1.51 - 97%;FEV1./ FVC < 70) (表E1).如前所述,收集外周血、支气管肺泡灌洗液(BAL)和支气管上皮细胞(BEC)[17,19].受试者在纳入研究前3个月无过敏或哮喘史,未使用吸入或口服糖皮质激素,>无加重。根据吸烟史(>10包年)和吸烟习惯(>过去6个月每天10支)匹配当前吸烟者。目前的吸烟状况和BAL前8小时>的戒断是通过呼出一氧化碳来验证的[26].该研究得到了斯德哥尔摩地区伦理委员会(案例编号2006/959-31/1)的批准,参与者提供了知情的书面同意。
组学数据块
九个组学数据块(图1)从52名女性受试者(20名健康,20名吸烟者,12名慢性阻塞性肺病患者)中提取:通过微阵列收集BAL细胞的mRNA [22];通过微阵列收集BAL细胞和BAL液(BALF)外体的miRNA[22,27]BAL细胞的差异凝胶电泳(DIGE)蛋白质组学[17]; 通过相对和绝对定量(iTRAQ)质谱(MS)等压标签收集BAL细胞的鸟枪蛋白质组学数据[25,28];通过串联质量标签(TMT)-MS收集BEC的散弹蛋白质组学数据[29];来自血清和BALF的二十烷类分析数据[21];以及血清代谢组学数据[30.]。有关数据收集平台和数据预处理的详细信息,请参阅以前的出版物和补充材料.中提供了缺失的数据矩阵图E1仅包括女性受试者(n=52)的动机是基于每个受试者在组学平台上的最大覆盖率。
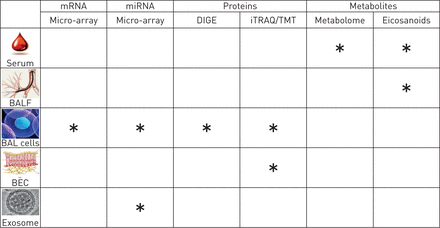
相似网络融合
使用R-package SNFtool(cran.R-project.org/web/packages/SNFtool)进行基于网络的多组数据融合分析和基于主题的聚类[14].简单地说,使用每个单组数据集计算每个受试者的距离矩阵,然后构建每个单组数据集的相似图。本质上,每个组学数据块因此被简化为一个亲和矩阵,其中预测因子的数量取决于分析中的研究对象的数量,而不是变量的数量(即信使rna、蛋白质等)。因此,组学平台之间的变量数量差异很大(例如mRNA数据包含40000个变量与具有100个变量的二十烷类数据)不会以它在其他类型的分析工作流中的相同方式影响SNF分析。来自不同组学平台的亲和矩阵被融合成一个单一的“融合相似性矩阵”,代表每个研究对象与其他研究对象的相似度。因此,输入预测器是来自每个组学数据块的相似性矩阵,而不是原始变量的完整矩阵,因此,可以将不同类型的数据与这些不同数量的变量整合在一起。基于九个组学数据集的所有组合,从双网络到七位网络,构建融合主题相似图。然后使用遗漏一交叉验证(LOOCV)预测群体归属[31]采用随机抽样,使用标签传播(图E2),或使用谱聚类[14].
将SNF参数超参数(alpha)、迭代次数(t)和邻居数量(K)设置为alpha=0.5, t=30, K=5,基于鲁棒性优化(E3−E4的数据).此外,还评估了亚组样本量对多组融合的影响。受试者网络被可视化为一个固定位置网络,根据临床参数(健康、吸烟者、慢性阻塞性肺病)定义的分组进行聚类,以及根据网络相似性对受试者进行聚类[32].所有网络均由Cytoscape 3.1.1生成[33].
网络集成中缺失数据的处理策略
对SNF预测中处理缺失数据块的三种策略进行了评估:1)保守策略包括所有受试者中最全面覆盖组学数据块的24名受试者(图E1);2)等样本量策略包括所有52名受试者(图E1),但在LOOCV中每次迭代训练集的子群大小相等(n=4);3)样本大小不等的策略包括所有52名受试者,允许训练集中有不同的组规模(n= 5-12),从而利用每个组学整合的最大信息(图E1)处理缺失组学数据块的三种评估方法显示出相似的平均性能,对于不等样本量策略,平均性能略高(图E5).考虑到该策略在允许纳入缺少组学数据块的受试者方面也是最自由的,不平等样本量策略的结果如下所示。从其他两种策略的结果在补充材料.
精度和功率计算
根据上述SNF多组学工作流程将受试者正确划分为三个研究组(健康、吸烟者、慢性阻塞性肺病)的比例来计算分组预测/分类的准确性。根据COPD诊断确定正确的研究组(根据GOLD计划;FEV1./FVC<0.70),以及吸烟史和当前吸烟状况(由所有四次临床就诊时测量的呼出一氧化碳来证实[26])得出的归一化互信息(NMI)表示正确分类的受试者的比率,其中0表示所有被错误分类的受试者,1表示所有被正确分类的受试者。评估预测因子数量增加(此处,组学数据块数量增加)导致的准确性提高,在对受试者标签进行排列后重复分析。根据相同的分配样本量(代表卡罗林斯卡宇宙队列使用的研究设计),计算每个组学n元组(整合组学数据集的数量)平均准确度的幂曲线。所需组数(n)在80%和95%的统计功率水平下计算。为了调查COPD患者亚分类的准确性,慢性支气管炎诊断被用作基础事实,用于计算临床诊断和基于SNF的预测之间的NMI,使用谱聚类。慢性支气管炎诊断被确定为自回归咳嗽和咳痰≥3. 至少连续两年中的每个月[34].
结果
多组学整合提高群体预测的准确性
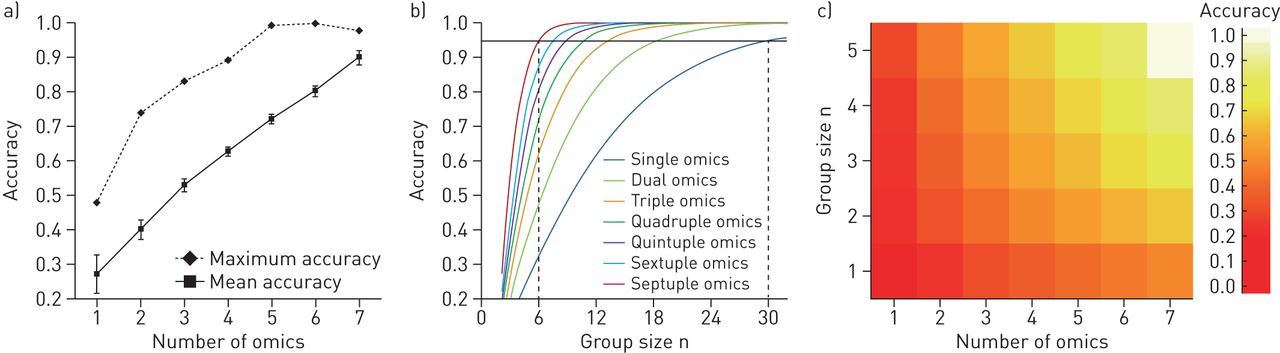
我们研究了多组数据融合是否能在存在诸如吸烟等强混杂因素的情况下提高COPD无监督分子分类的统计能力和准确性。当使用标签传播方法时,组预测(健康、吸烟者、慢性阻塞性肺病)的平均准确性与组学n元组呈线性方式增加,从9个单组学平台的平均准确性0.28增加到7个组学网络的0.90 (图2a,实线)。对于此处使用的小队列,使用LOOCV的组预测似乎比使用光谱聚类的组预测更稳健(图E6A).一项排列测试显示,由于预测因子数目的增加,准确率的提高是偶然发生的(即组学数据块的数量)可以忽略不计,从单个组学增加到七个组学,从0.09增加到0.13 (图E6B).
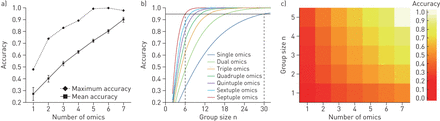
a)使用来自Karolinska COSMIC队列的9组数据集,作为snf介导的组学整合中所包含组学数据集数量函数的组学预测的准确性(见图1).数值显示为平均精度±se(实线)以及每个不同数量的组学平台的所有可能组学组合的最大精度(虚线),范围从单个组学整合到七个组学整合。提供的数据基于不等样本量策略(对于其他抽样策略,请参阅图E5).b)对应于每个组学n元组平均准确度水平的单独功率曲线。图中显示了组大小(n)与每个组元组的组预测精度,范围从单个组到七个组元组的整合。实黑色水平线表示95%的精度水平,虚线表示n在95%的精度水平需要单与七倍的组学整合。c)热图显示每个n元组组学集成的子组规模为n= 1-5所达到的平均精度水平。精度计算为基于相似网络融合的精度,与慢性阻塞性肺疾病诊断分类(根据慢性阻塞性肺疾病全球倡议标准)和当前吸烟状态(由呼出一氧化碳监测定义)相比。
与每个组学n元组的平均准确度相对应的功率曲线表明,在95%的准确度水平下,七组学整合将所需的子组大小从单个组学的n=30减少到七组学的n=6(图2b,表1).在0.80精度水平下,所需的亚组大小从n=18减少到n=4。使用更小的亚组(n= 1-5),与个性化药物或非常罕见的患者亚组相关的每个n元组组学整合所达到的平均准确性显示在图2c.
高性能的网络
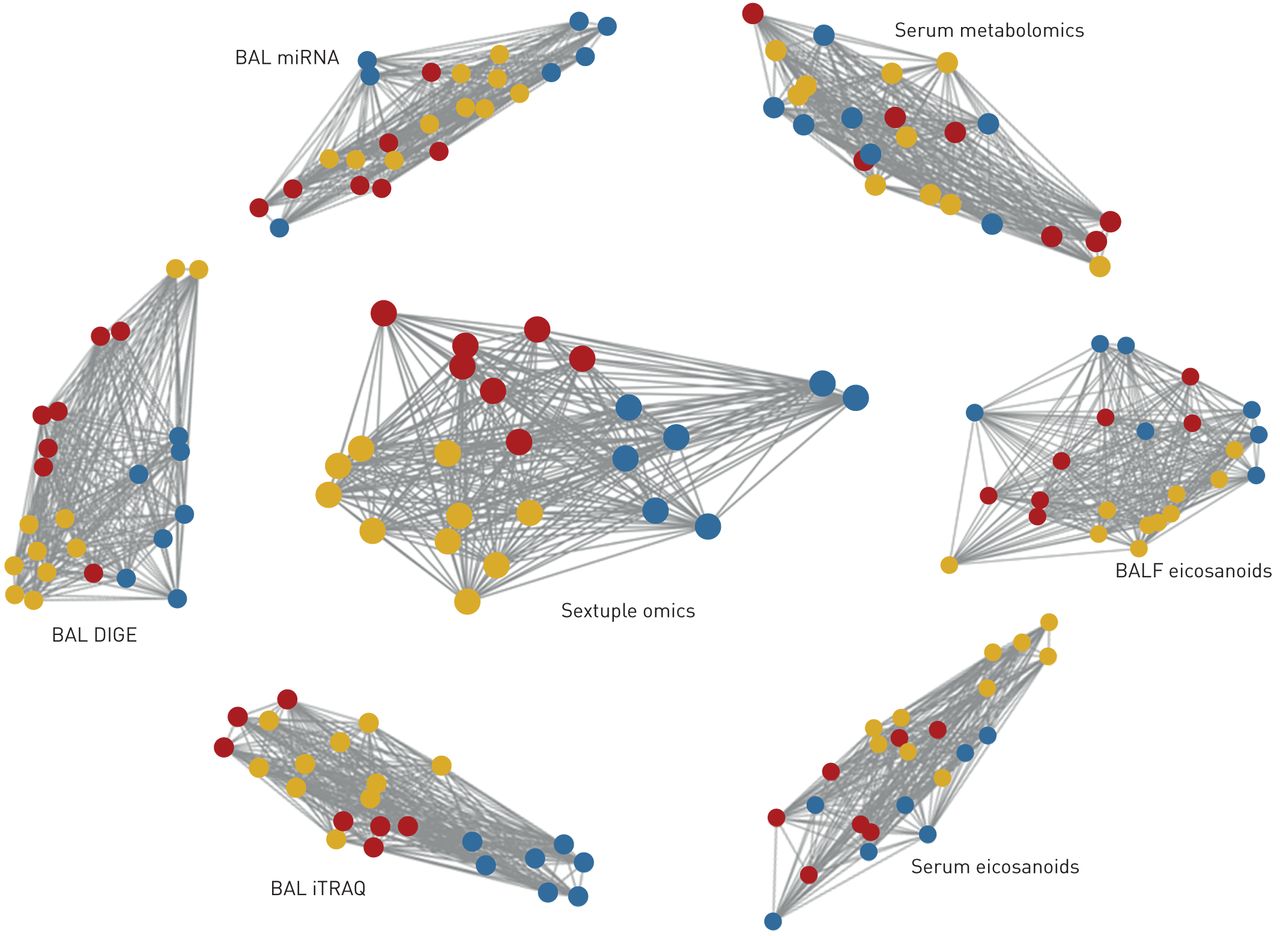
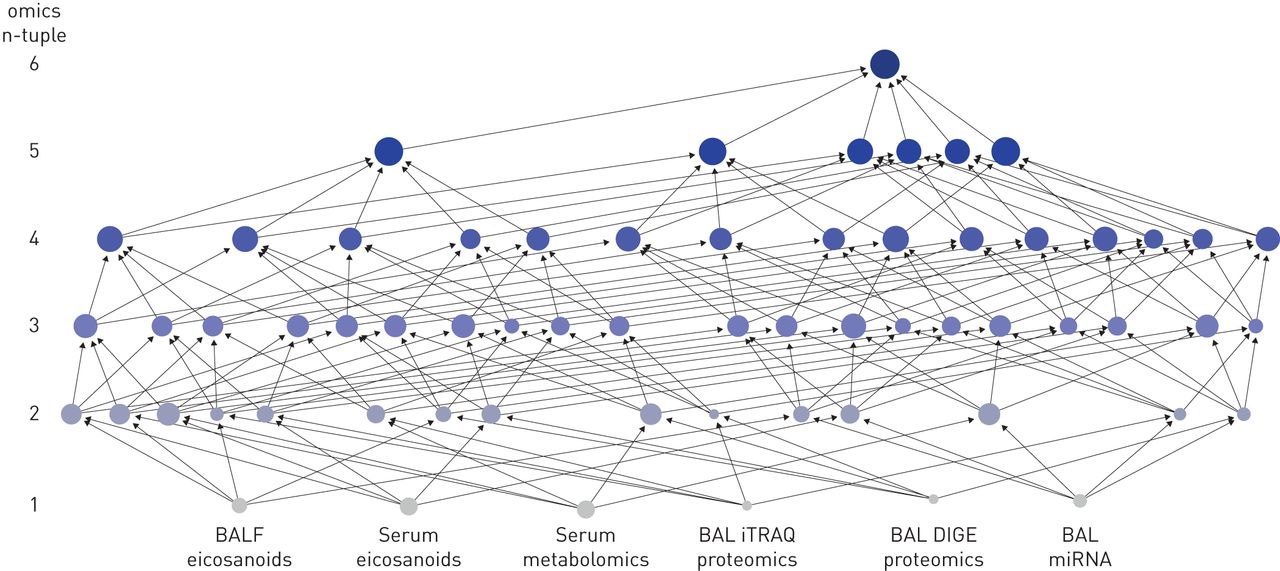
尽管以上描述的平均性能与所包含的组学平台数量高度相关,最多达到90%的准确率,但一些特定的网络组合达到了更好的准确率。最值得注意的是,由BAL细胞microRNA、BAL细胞DIGE蛋白质组学、BAL细胞iTRAQ蛋白质组学、血清代谢组学、BALF二十烷类化合物和血清二十烷类化合物组成的六组组学组合,在COPD诊断和吸烟状态方面对所有受试者进行了100%的正确预测(图3,图E7).作为对比,BEC TMT蛋白质组数据在无监督下获得了最佳的单组预测,结果的NMI=0.46。这些结果是在最小样本量n=6的情况下获得的。将6个单独的单组相似性网络与融合的六组相似性网络进行比较,结果表明,通过聚合多种类型的分子数据,提高了性能,降低了噪声;在当前吸烟状态的混杂影响下,六组整合区分了吸烟相关COPD的分子改变。没有一个单一组学数据块有能力分离当前吸烟状态和COPD诊断。增加的预测信息流轨迹与所有可能的n元组组融合为六组组网络提供100%的准确性显示图3所示图4.
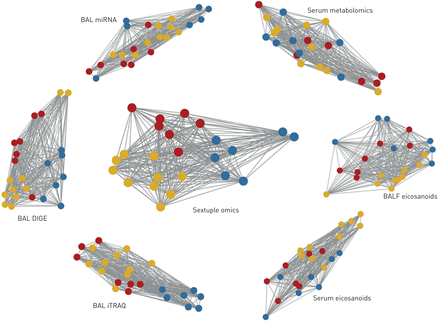
表现最好的受试者相似网络,由六组学整合相似网络融合相似网络(中心)组成,提供了100%正确的健康、吸烟者和慢性阻塞性肺病三个主题组的分类。为每个纳入的单组数据(外围)的相似网络显示供参考。节点代表对象。网络显示时,根据网络相似性对主题进行聚类。100%的准确率是基于10000次漏一交叉验证排列检验,使用训练数据,从每组中迭代选择6个样本。同一网络显示为固定位置网络,并根据六组融合网络保留所有七个网络的聚类,以方便视觉比较图E7.蓝色:健康never-smoker;黄色:吸烟者肺功能正常;红色:患有慢性阻塞性肺病的吸烟者。拜尔港:支气管肺泡灌洗;DIGE:二维差异凝胶电泳蛋白质组学;iTRAQ:相对定量和绝对定量蛋白质组学等压标签;BALF:支气管肺泡灌洗液。
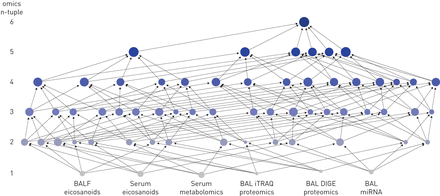
相似性网络融合中组学n元组提高精度的实例。预测的准确性由淋巴结大小表示,范围从最小的(5%的准确性;支气管肺泡灌洗液(BAL)等压标签用于相对和绝对定量蛋白质组学(iTRAQ)单个组学)到代表100%准确性的最大值(六元组学)。融合后的组学数据集的n元组从单组(底部)到六组组(顶部)显示。n元组也由灰色到蓝色的颜色编码表示。支气管肺泡灌洗液;DIGE:2-D差异凝胶电泳蛋白质组学。
使用保守抽样策略的最佳预测网络是通过一个七元网络实现的,群体预测的准确率为91% (图E8).
在303种可能的单到七组组组合中,25种不同的四倍到七组组组合的预测精度达到了>85%,其中7种网络组合的预测精度达到了>95% (图5),表明在选择组学平台进行最佳分类时具有一定的可塑性。

30种不同的相似网络融合多组学网络组合达到了组预测的准确率>85%,计算为与慢性阻塞性肺疾病(COPD)诊断和吸烟状态(三组:健康、吸烟者、COPD)相比的归一化互信息。七种网络组合达到了>95%的准确率(灰线)。a)各自组合的预测精度;B)组学数据集包含在特定的融合网络中,对应于上面所示的柱状图。请注意,每条线代表一个单一的,特定的网络的准确性,因此没有误差条。BAL:支气管肺泡灌洗细胞;DIGE:二维差异凝胶电泳蛋白质组学;BALF:支气管肺泡灌洗液;iTRAQ:相对定量和绝对定量蛋白质组学等压标签;外泌体:BALF分离的外泌体; BEC: bronchial epithelial cells; TMT: tandem mass tag proteomics.
讨论
当前研究的主要目标是研究从多个分子水平和解剖位置整合大规模组学数据是否会增加复杂疾病的分子分类的力量和准确性,这里以COPD为例。我们的概念验证调查应用SNF网络集成[14从卡罗林斯卡COSMIC队列的52名女性受试者的9个组学数据块清楚地表明,整合多组学数据极大地提高了无监督分类的准确性。在303个评估网络组合中,COPD、肺功能正常的吸烟者和不吸烟者的三组预测的平均准确性以一种近线性的方式增加,从单组学平台的平均准确性28%增加到七组学整合的90%。然而,根据所包含的特定组学平台,观察到很大程度的差异。更大的n元组组学并不等同于对特定组学组合的更好预测,通过六组组学整合,可以实现100%的准确分类(图3,图E7).25种不同的四倍到七倍组学组合的准确率达到了>85%,这表明在优化预测准确性所需的组学数据组合中有很大程度的可塑性(图5).值得注意的是,>95%的预测准确率的7个网络都包含了来自不同分子水平(miRNA、蛋白质组、代谢组和类生物)和解剖位置(BAL细胞、BAL液和血清)的组学数据集,这意味着结合来自不同解剖部位和分子水平的数据是有优势的。
仅吸烟就会导致肺中多达50%的生物分子发生显著变化,这在同一队列的BAL细胞和BEC蛋白质组中得到了证实[28,29].因此,在这个队列中真正的挑战是区分与慢性阻塞性肺病病理相关的微妙的分子效应和当前吸烟慢性阻塞性肺病患者中急性吸烟的混杂效应。没有一个单一组学数据块能够以无监督的方式从当前吸烟的混杂效应中分离出轻到中度COPD造成的分子改变(图3,图E7).在我们之前对单组数据块的研究中[17,21,30.]与大多数慢性阻塞性肺病调查一样,根据当前吸烟状况进行分层并结合监督分析是识别慢性阻塞性肺病相关改变的强制性方法。相比之下,五倍到七倍组学整合提供了从从不吸烟者和当前吸烟控制中对慢性阻塞性肺病诊断进行100%准确分类的能力y(图2a).应该强调的是,这是在无监督、数据驱动的方式下实现的,子组小到n=6。在整合两个或更多组学平台后,基于慢性支气管炎诊断的COPD组具有100%准确率的亚表型能力,使用小至n=4名受试者(图E9)进一步证明了这些方法的临床实用性。因此,此处采用的整合方法可能使基于系统医学的方法能够在小型、重点人群中进行,鉴于在大型人群中进行非耦合组学分析的高昂成本,这是可取的来自多个隔间的离子可以提供从统计上相对较小的队列中检测罕见的疾病分子亚表型的能力,这是翻译性多组学研究的一般情况。
为这些原则性证据调查选择的同质COPD人群,包括没有合并症或治疗的轻度至中度COPD女性患者,反映了某种人为的情景,这限制了将结果推断到更一般的疾病人群的能力。COPD是一种hete由10-15种不同的分子亚表型组成的遗传性疾病[35–37],其中很少有定义至今[16].很明显,目前的临床特征方案并不能提供必要的解决方案来分类甚至识别这些疾病亚群[16]。虽然本文介绍的无监督工作流程的最终目标是对所有或至少几个现有COPD亚型进行分子亚分类,但任何评估该方法性能的研究都必须绝对关注与我们的COPD亚型分子相似的COPD亚型。fema的存在在对单个组学数据块的监督分析中,我们的发现已经很好地确定了卡罗林斯卡宇宙队列中以le为主的COPD分子表型[17,21,23,30.]因此,卡罗琳斯卡宇宙队列的女性部分提供了一组分子上不同的基础真相研究小组,以评估无监督分类。
在人类受试者研究中,数据缺失是一个常见问题,特别是在多组学、多室研究中,如卡罗林斯卡COSMIC研究。由于各种原因,组学数据块在给定的主题中丢失是很常见的,例如。出于患者的安全考虑,或出于质量控制标准,将单个组学实验排除在外,因此省略了对选定生物样本的取样。结果是一个有间隙的数据矩阵,其中每个受试者都有一些缺失的组学数据块(图E1).原始SNF数据集成方法不适应缺失的数据块[14].因此,开发处理网络建设中缺失数据的方法是本研究的第二个目的。在三种评估方法中,在允许纳入缺失数据的受试者方面,三种方法中最自由的方法(不平等样本量策略;图E5)表现最好。因此,尽管存在数据块缺失的问题,但这种策略似乎提供了一种稳健的方法,允许纳入所有受试者和收集的数据,这在这种在炎症部位进行侵入性取样的转译研究中是不可避免的。
尽管所使用的交叉验证设计确保准确度独立于训练集进行估计,但该队列的相对同质性和有限样本量带来了一些限制。更大的队列可能有助于实现多组学数据整合的真正目标:识别先前未知的、分子上不同的亚表型这些原则性证据调查的结果可能为未来系统医学研究的设计提供一些指导,其中从互补的分子水平和解剖位置收集五到七个组学数据块,队列规模为六到十名受试者的预期数量的六到十倍分子亚群可能代表复杂疾病分子亚表型的良好起点。例如,目前COPD的诊断标准代表一系列亚表型,受性别、吸烟史、早产、环境暴露等因素的影响等.如果我们假设这些病因导致15个分子上不同的表型,每个表型由不同的生物标志物和药物靶点子集表示,那么基于该队列显示的亚群大小(表1),使用一个组学数据集进行的调查需要每个COPD亚组30名研究对象,即450名患者加上相关的对照组。七组组学整合所提供的更高的分子分辨率将需要识别的研究对象数量减少到每个亚组n=6个。因此,假设的旨在识别15个分子上不同的COPD亚组的研究设计,将足以包括90名COPD患者。实现研究目标所需的患者数量的大幅减少将大大增加分子表型研究的临床可行性。临床研究的一个重要瓶颈往往是及时招募足够的患者群体的能力。使用七组组学整合可以大大减少队列收集所需的时间,促进未知分子亚表型的鉴定。
总之,通过整合来自卡罗林斯卡COSMIC队列的9个组学数据块,这些例子证明了通过整合来自多个分子水平和解剖位置的数据,在统计能力和群体分类的准确性方面有了非凡的提高。我们首次量化了多组学集成所带来的统计能力的提高,七组学集成所带来的分类能力平均从28%提高到90%。从计算系统医学的角度来看,疾病的机制不是由传统单变量统计所识别的基因/蛋白质/代谢物的独立子集引起的,而是由它们的相互作用引起的,这使得发展对疾病的系统级理解至关重要。正如这里所展示的,桥接和整合来自多个分子水平和与同一个体的疾病病理相关的解剖区域的数据,最终可以提供无监督分类的统计力量。无监督方面是必须的,以促进识别复杂疾病的未知分子亚表型,如COPD和哮喘。在无监督的情况下,对分子上不同的疾病亚群的识别是阐明可治疗特征和生物标志物亚群的第一步,也是至关重要的一步。SNF识别的分子不同亚群可以通过监督的多变量建模方法来询问诊断或预后生物标志物的手印,即对潜在结构的正交投影,提供感兴趣变量的过滤器。此外,我们目前正在开发路径层面的多组学整合方法。通过在识别与已识别的患者亚组相关的机制特征和可治疗特征的下游步骤中整合多个分子水平,这些利用增加的力量。结合已确定的生物标志物子集或“疾病手印”[38]从多个分子水平来看,可能会带来人们期待已久的精确和个性化医学范式转变。
补充材料
脚注
这篇文章的补充材料可从www.qdcxjkg.com
Karolinska COSMIC队列注册于ClinicalTrials.gov与标识符NCT02627872.
作者贡献:概念和设计:C-X。李,点惠洛克;分析和解释:C-X。李,点惠洛克;草稿稿:C-X。李,点惠洛克;系统医学、蛋白质组学和转录组学负责人:Å.M。 Wheelock; principal investigator of clinical segment, including clinical characterisation, bronchoscopies and sample collection: C.M. Sköld; principal investigator of metabolomics and eicosanoid segments: C.E. Wheelock.
支持声明:卡洛林斯卡宇宙研究由瑞典心脏肺基金会资助,瑞典战略研究基金会(SSF),Vimina(VNMER),欧盟FP6玛丽居里,卡罗林斯卡学院,AFA保险,国王奥斯卡II禧年基金会,国王古斯塔夫V和维多利亚女王的共济会基金会,瑞典研究理事会,区域医学培训和临床研究协议(阿尔夫)在斯德哥尔摩县议会和卡罗林斯卡学院、过敏研究中心、卡洛林斯卡研究所和阿斯利康翻译科学联合研究项目。C·X·L.由EER/EU玛丽居里ReistiR2博士后奖学金资助。C.E. Wheelock由瑞典心肺基金会资助。由瑞典心肺基金会高级研究员职位资助。这篇文章的资助信息已经存入CouthReF基金会注册处。
利益冲突:无声明。
- 收到2017年9月22日。
- 认可的2018年3月8日。
- 版权所有©ERS 2018











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}