





文摘
肺动脉高血压流行病学证据链接(多环芳烃)和甲状腺疾病,但缺乏机械的解释这种关联。
因为血管重塑的核心标志在肺动脉高压腔内皮细胞生长和消亡,因为甲状腺激素被认为是血管生成,我们提出,甲状腺激素在控制中发挥作用的内皮细胞增殖实验PAH的老鼠。
甲状腺功能减退引起全甲状腺切除术,治疗丙基硫氧嘧啶(PTU)在大鼠实验后多环芳烃暴露于血管内皮生长因子受体抑制和缺氧(Sugen-chronic缺氧(SuHx)模型)。小计甲状腺切除术预防和PTU治疗逆转的发展严重的实验多环芳烃。甲状腺素恢复饱满的PAH表型thyroidectomised SuHx老鼠。多环芳烃的预防甲状腺切除术与减少的细胞营业额,减少细胞外signal-regulated蛋白质磷酸化激酶1和2,并降低α的表达vβ3整合蛋白,纤维母细胞生长因子(FGF) 2和FGF受体。甲状腺切除术减轻低氧诱导肺动脉高压,但是这种影响并不是与肺血管阻力下降有关。
这些数据表明,甲状腺激素允许在多环芳烃的内皮细胞增殖。甲状腺疾病和之间的因果关系的发作或恶化血管重塑PAH患者仍有待确定。
慢性肺动脉高压(PAH)的特点是肺血管重塑涉及媒体和增长的平滑肌细胞表型的改变内皮细胞(1,2]。多种形式的严重慢性PAH耐火血管舒张药治疗药物(3)和总体死亡率仍然很高。在许多PAH患者看来,pre-capillary小动脉由apoptosis-resistant内皮细胞消失通过一种类似血管生成的过程(4]。严重形式的多环芳烃被分为特发性、遗传、药物和toxin-induced和相关(5,6]。相关的形式的例子有多环芳烃在结缔组织疾病,艾滋病毒感染、结节病和镰状细胞病(7- - - - - -9]。大量的PAH患者也有甲状腺疾病(10),但目前还不清楚这是否多环芳烃和甲状腺疾病之间的联系是巧合或者相关病原学的甲状腺激素的作用在多环芳烃的发展。甲状腺激素刺激内皮细胞生长(11- - - - - -13和增强缺氧性肺血管收缩14),因此我们提出甲状腺疾病可以影响肺血管重塑的慢性多环芳烃的设置。我们使用了Sugen-chronic缺氧严重angioproliferative PAH (SuHx)大鼠模型(15- - - - - -17),这取决于两个冲击:初始肺内皮细胞凋亡的诱导血管内皮生长因子(VEGF)受体酪氨酸激酶抑制剂SU5416,和促进细胞生长慢性缺氧。这里我们首次展示,甲状腺激素是高度宽容对肺动脉高压(PH),形成腔造成肺血管病变。
方法
小计thyroidectomised Sprague-Dawley老鼠用于本研究从哈伦购买实验室(Frederic,医学博士,美国)。5毫克的甲状腺素在60天准备释放颗粒(美国佛罗里达州萨拉索塔,美国创新研究)。严重angioproliferative PAH和右心衰在男性Sprague-Dawley诱导大鼠(体重200克,年龄6周)通过他们接触VEGF受体拮抗剂SU5416 SuHx,像前面描述的那样16,17]。thyroidectomised老鼠的一个子集是鉴于SuHx组合开始2周后甲状腺切除术手术,另一个是由于甲状腺素丸皮下注射2天前从SuHx曝光。在另一个的子集SuHx动物、丙基硫氧嘧啶(PTU);10 mg·公斤−1每天;σ,圣路易斯,密苏里州,美国)皮下注射(南卡罗来纳州。)5天·周−14周2周后开始建立PAH模型。最后,甲状腺切除术的效果进行了研究肺血管重塑和慢性缺氧暴露4周后血液动力学(吸入氧气分数10%)。心脏超声检查,血液动力学的测量,组织收获和加工进行了如前所述。心输出量和中风被索引的身体表面积体积(从体重计算18]),心脏指数(CI)和中风指数,分别。肺血管阻力指数(PVRI)计算:

其中mP巴勒斯坦权力机构平均肺动脉压力,LVEDP左心室舒张末期压力。在这些情况下,进入肺动脉导管进步是不可能的(大约每8个老鼠),的意思P巴勒斯坦权力机构估计从右心室收缩压(RVSP),如前所述[19]。动物模型,进一步的细节评估angioproliferative血管损伤,免疫组织化学、免疫印迹分析和统计分析,可在网上补充材料。
结果
PH值和肺血管重塑SuHx鼠模型中严重的PH值由甲状腺切除术预防
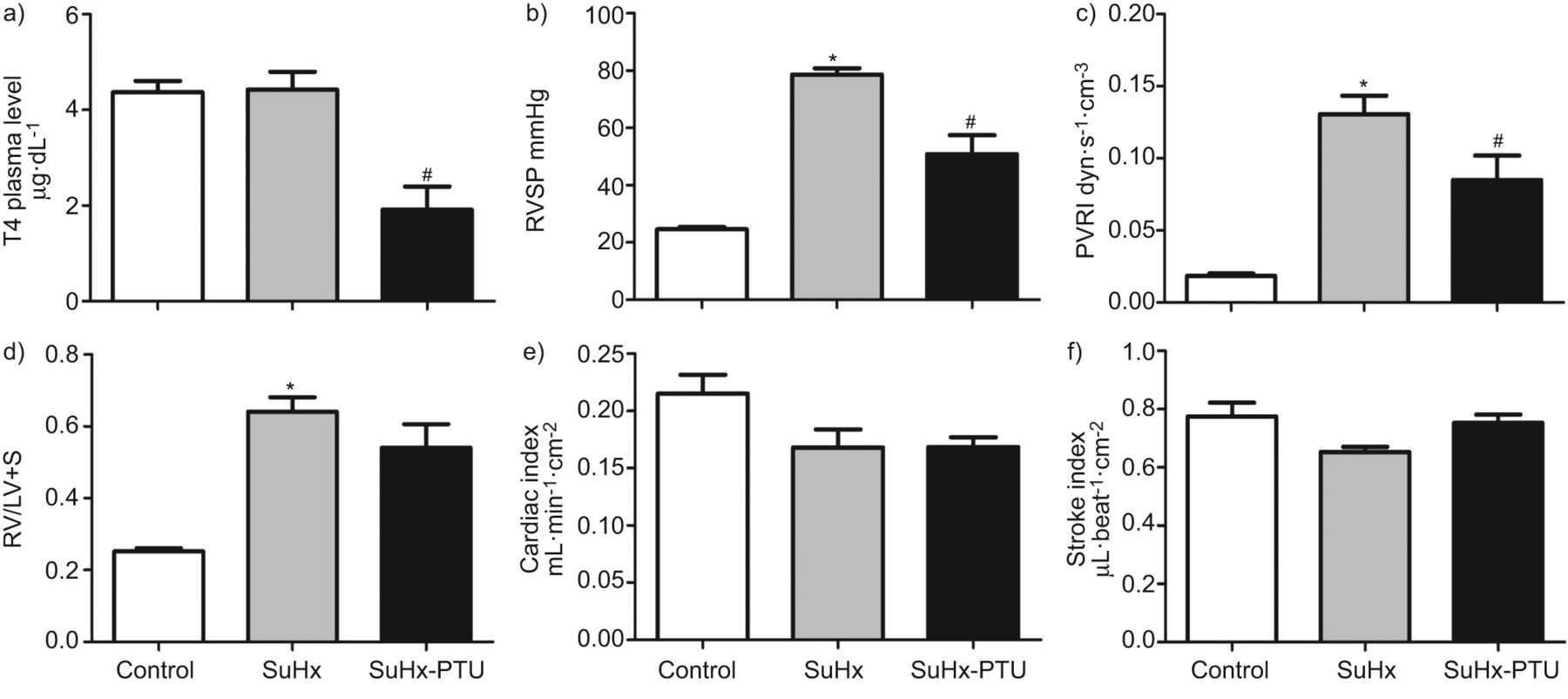
解决的问题是否甲状腺激素有助于发展的多环芳烃和肺血管重塑在SuHx模型中,我们评估是否全甲状腺切除术预防的发展这个模型的血流动力学和肺组织学变化特征。Euthyroid和thyroidectomised老鼠暴露在单一剂量的VEGF受体酪氨酸激酶抑制剂SU5416·公斤(25毫克−1南卡罗来纳州。4周)和缺氧,足够的曝光时间来产生严重vaso-obliterative PAH (15。我们可以看到图1,甲状腺切除术预防RVSP回应SuHx的增加。甲状腺切除术后减少RVSP SuHx老鼠不仅与CI下降,预期发现甲状腺动物,但也在很大程度上,归因于PVRI下降。甲状腺素水平下降,正如预期的那样,全甲状腺切除术组和甲状腺素水平与RVSP值密切相关。发展的PH值与异常多普勒信号在肺动脉和肺动脉的进步缩短加速时间(图S1)。甲状腺切除术预防这些多普勒信号的变化。预防的PH值thyroidectomised SuHx老鼠不是预防右心室(RV)扩张和肥大。补充甲状腺素恢复生理的甲状腺素水平thyroidectomised SuHx老鼠,导致恢复RVSP和PVRI的水平euthyroid SuHx老鼠。而大量的肺小动脉要么是完全或部分阻挡从SuHx老鼠SuHx处理(图2一个和b),甲状腺切除术导致肺组织学特点是专利但肌小动脉(图2 c和d)。补充甲状腺素的thyroidectomised SuHx动物允许PAH-characteristic血管闭塞(图2 e和f)。部分专利和完全腔闭塞血管的数量(每分)所示图2 g和专利的比例船只与RVSP (图2 h)。因为缺氧会影响甲状腺激素的分泌,也会导致血浆甲状腺素水平的变化(20.- - - - - -22),我们测试是否SU5416 +管理的结合甲状腺素(然而,没有缺氧)引起的肺血管重塑和博士我们的数据表明,慢性甲状腺素单独管理只引起轻微的PH值在SU5416-treated老鼠(图S2)。因此,慢性甲状腺素补充没有缺氧不复制肺血管疾病SuHx模型中观察到。
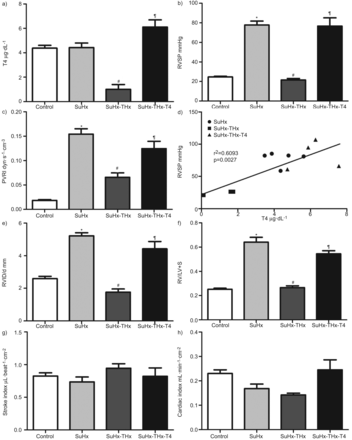
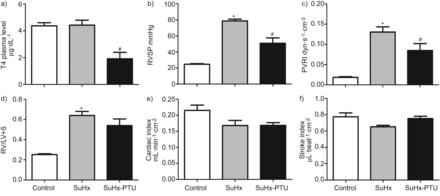
小计的影响甲状腺切除术(谢谢)和小计甲状腺切除术与甲状腺素替代(THx + T4)在Sugen-chronic缺氧(SuHx)大鼠血浆T4水平,b)右心室收缩压(RVSP), c)肺血管阻力指数(PVRI), e)右心室直径,以舒张(RVID / d), f)右心室肥大(右心室重量/左心室+室间隔重量,或RV / LV + S), g)中风指数和h)心脏指数。d)与RVSP血浆T4水平之间的关系。数据意味着±se(n = 5 - 8)。*:p < 0.05与控制;#:p < 0.05与SuHx;¶:p < 0.05与SuHx-THx。
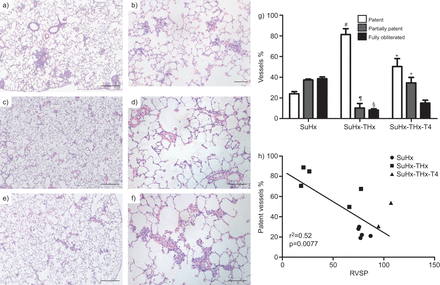
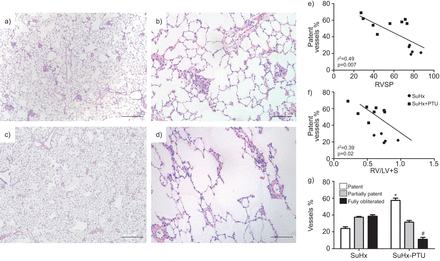
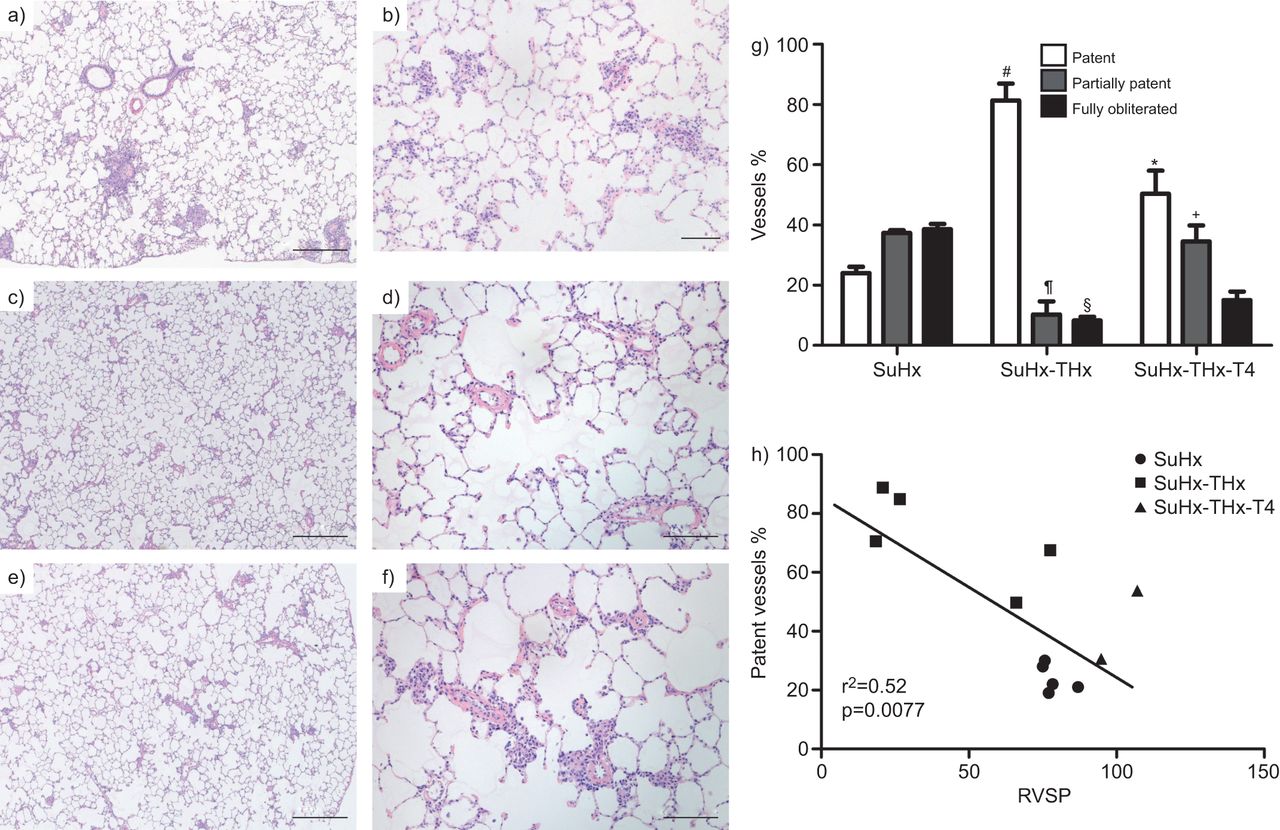
f)代表的苏木精和伊红染色肺部分显微照片,b) Sugen-chronic缺氧(SuHx)老鼠,c和d) SuHx鼠甲状腺切除术(谢谢)和e, f) SuHx大鼠甲状腺切除术+甲状腺素(T4)替代。a、c、e)比例尺= 500μm;和b, d, f) = 100μm规模酒吧。g)的比例小肺动脉(< 80直径μm)列为专利,部分专利和完全闭塞。h)的百分比之间的关系专利船只和右心室收缩压(RVSP)。*:p < 0.05与比例的专利船只与甲状腺切除术SuHx老鼠的肺;#:p < 0.05与比例的专利船只SuHx老鼠的肺;¶:p < 0.05与比例的部分闭塞血管SuHx老鼠的肺;+:p < 0.05与与甲状腺切除术的比例部分闭塞血管SuHx老鼠的肺;§:p < 0.05与的比例完全闭塞血管SuHx老鼠的肺。数据意味着±se(n = 3 - 5)。
甲状腺切除术在缺氧性肺血管重塑的影响
存在的一定程度的内侧甲状腺切除术后壁增厚(图2)认为缺乏甲状腺激素主要影响肺血管的闭塞thyroidectomised SuHx老鼠。也确定甲状腺激素影响的程度muscularisation,我们随后暴露正常和thyroidectomised老鼠独自慢性缺氧(内侧壁增厚的典范,但不是血管遮挡),没有伴随SU5416管理局(图3)。甲状腺切除术减少媒体壁厚和降低RVSP缺氧暴露后,但是PVRI在统计学上没有显著的减少,降低房车肥大。慢性低氧暴露导致预期增加血球容积计20 - 30%的增加并没有受到甲状腺切除术的影响。
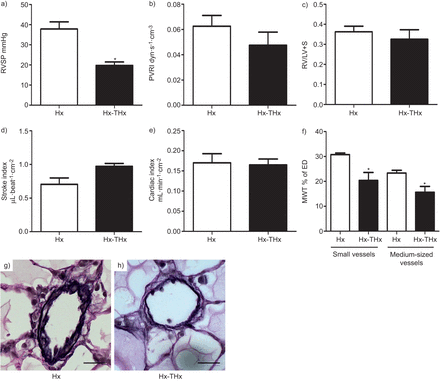
小结甲状腺切除术(谢谢)在大鼠暴露于慢性缺氧(Hx)导致)降低右心室收缩压(RVSP)和b)肺血管阻力指数较低的趋势(PVRI),而c)右心室重量索引为左心室重量加上隔(RV / (LV + S)), d)中风指数和e)心脏指数保持不变。媒体壁厚(MWT)比例的外部直径(ED)中小肺血管甲状腺切除术后减少。数据意味着±se(n = 5)。g)和h)代表肺小血管的图像。酒吧= 20μm规模。*:p < 0.05与Hx。p-Erk:磷酸化细胞外signal-regulated激酶。
甲状腺切除术减少或抑制细胞增殖和细胞死亡标记的表达在肺部SuHx动物
评估是否甲状腺切除术预防病变细胞生长我们用免疫组织化学(包含IHC)本土化增殖细胞核抗原(PCNA)的表达(图S3)和肺组织蛋白溶菌产物进行分析通过免疫印迹(图4)。因为SuHx angio-obliterative病变的发展模式还取决于肺细胞死亡(16),我们评估了裂解半胱天冬酶3表达在肺组织包含IHC S3(图)和免疫印迹(图4)。PCNA-positive细胞被发现在SuHx肺部的检查组织部分包含IHC和免疫印迹显示PCNA的表达减少肺从thyroidectomised SuHx动物相比,肺从euthyroid SuHx老鼠。此外,甲状腺切除术的表达降低裂解半胱天冬酶3 SuHx肺、甲状腺素补充thyroidectomised SuHx老鼠与PCNA的表达增加裂解半胱天冬酶3 (无花果4和S3)。
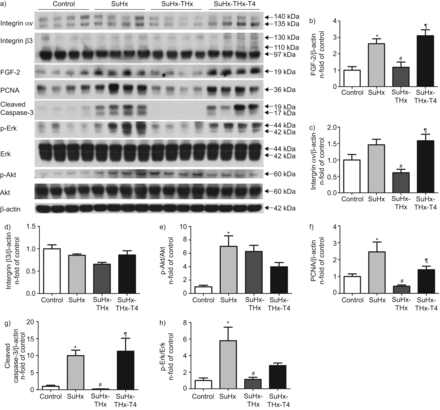
免疫印迹分析整合素)代表αv,整合素β3,纤维母细胞生长因子(FGF) 2、增殖细胞核抗原(PCNA)、裂解caspase-3, phosphorylated-extracellular signal-regulated激酶(p-Erk),兵,phospo-Akt (p-Akt),一种蛋白激酶和β-actin肺蛋白提取物控制,Sugen-chronic缺氧(SuHx) SuHx与甲状腺切除术(谢谢)和SuHx甲状腺切除术,取而代之的是甲状腺素(T4)。酒吧图表显示b) FGF-2的比率,c)整合素αv, d)整合素β3,e) p-Akt / Akt比率,f) PCNA, g)裂解caspase-3和h) p-Erk / Erk蛋白表达相对比例控制。数据意味着±se(n = 4)。*:p < 0.05与控制;#:p < 0.05与SuHx;¶:p < 0.05与SuHx-THx。
甲状腺激素的宽松的效果在SuHx angio-obliteration老鼠与整合素改变αvβ3和纤维母细胞生长因子2信号
因为一个远端信号通路的细胞膜甲状腺激素受体涉及血管生成纤维母细胞生长因子(FGF) 2和整合素αvβ3 [11,12),这是在激活内皮细胞中表达的高度23,24),因为FGF-2与严重的多环芳烃(有关25- - - - - -27),我们评估了这种生长因子和整合素的表达在肺部αvβ3从动物发达国家严重的多环芳烃。甲状腺切除术的蛋白表达降低FGF-2,αvβ3整合蛋白(图4模拟)和phosphorylated-extracellular signal-regulated激酶(Erk)在肺SuHx动物(图4 a股和h)。甲状腺素补充在thyroidectomised SuHx动物恢复FGF-2的表达和αvβ3整合素蛋白质SuHx动物中观察到的水平(图4)。图5和数字S4-S7证明FGF-2和αvβ3蛋白过表达在肺SuHx老鼠和超表达不发生在thyroidectomised动物。肺组织FGF-2蛋白表达和αv整合素,和PCNAαv整合素相关(图S3i和j),表明严重的PH值设置的细胞增殖,FGF-2表达和整合素表达可能是同步的。
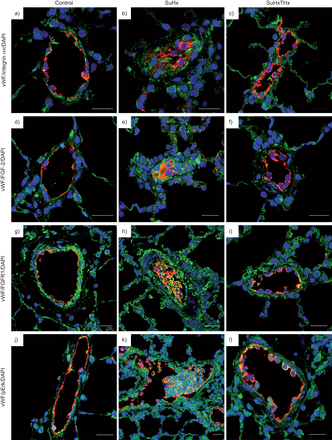
代表获得了共焦显微镜的光学部分血管性血友病因子双重免疫荧光染色(vWF;红色),整合素αν(绿色),纤维母细胞生长因子(FGF) 2(绿色),FGF受体(R) 1(绿色)和phosphorylated-extracellular signal-regulated激酶(p-Erk) 1/2(绿色)。核与DAPI对比染色(蓝色)。酒吧= 20μm规模。a - c) vWF /整合素αν双重免疫荧光染色法显示增加αν整合素在腔阻塞vWF染色+)细胞,媒体层细胞和周围细胞渗透和肺泡细胞b) Sugen-chronic缺氧(SuHx)动物,而少αν整合素染色中发现和周围血管的)控制和c) SuHx / thyroidectomised(谢谢)动物。d-f) vWF / FGF-2双重免疫荧光染色主要演示了高架FGF-2染色在腔阻塞vWF+细胞,而且在媒体层细胞和周围细胞的渗透和肺泡细胞e) SuHx动物,而FGF-2是发现在和周围血管的d)控制和f) SuHxTHx动物。胃肠道)vWF / FGFR1双重免疫荧光染色法显示广泛的腔阻塞vWF FGFR1的免疫反应性+媒体层细胞和周围细胞浸润,肺泡细胞h) SuHx动物,而少FGFR1染色中发现和周围血管的g)控制我)SuHxThx动物。j-l) vWF / p-Erk1/2双重免疫荧光染色清楚地显示多个腔阻塞vWF+细胞表现出强大的核和cytoplasmatic p-Erk1/2染色k) SuHx angioproliferative病变的动物,而更少的vWF+/ pErk1/2+细胞在血管j)控制和l) SuHxThx动物。注意媒体的高度增加p-Erk1/2染色层细胞浸润,肺泡细胞SuHx动物比控制和SuHxThx动物。还要注意在内皮(vWF的区别+,红色)之间的形态控制动物(核)和SuHxThx动物(圆形,激活核)图像一个l)。
PTU治疗大鼠建立了PH值
建立了甲状腺切除术防止angioproliferative PAH SuHx模型的发展,我们下一个检查是否PTU会影响SuHx模型中建立了多环芳烃。4周的每日剂量PTU治疗减少了RVSP, PVRI和房车肥大(图6)。这些结果表明,甲状腺素水平降低也不仅能够防止逆转,至少在某种程度上,PH值及其对RV的后果。图7 gydF4y2Ba表明PTU治疗减少的数量完全或部分阻挡在肺小动脉。实际上,专利船只的数量与RVSP,阻塞血管的数量也与房车肥大的程度有关。图7 g显示了专利和闭塞的小动脉的百分比。类似于thyroidectomised SuHx动物,PTU治疗导致增殖细胞核抗原阳性细胞的数量减少(图S8)。的表达FGF-2αvβ3蛋白减少的但不是PTU治疗动物的肺组织(图S9)。因此,PTU治疗SuHx老鼠与现有的多环芳烃和肺小动脉的闭塞增长和apoptosis-related因素的组织蛋白表达改变的方向类似于甲状腺切除术。
的影响丙基硫氧嘧啶(PTU)治疗Sugen-chronic缺氧(SuHx)大鼠在)血浆甲状腺素(T4)水平,b)右心室收缩压(RVSP), c)肺血管阻力指数(PVRI), d)右心室重量(左心室重量加上隔索引,或RV / (LV + S)),心脏指数和f)中风指数e)。数据意味着±se(n = 5 - 8)。*:p < 0.05与控制;#:p < 0.05与SuHx。
代表显微照片的苏木精和eosing-stained肺部分、b) Sugen-chronic缺氧(SuHx)老鼠和c, d) SuHx老鼠接受丙基硫氧嘧啶(PTU)。a、c)酒吧= 500μm规模;b, d) = 100μm规模酒吧。专利的比例关系船舶和e)右心室收缩压(RVSP)和f)右心室(RV)肥大(RV重量/左心室(LV) +隔(S)重量,或RV / (LV + S))。g)的比例小肺动脉(< 80直径μm)列为专利,部分专利和完全闭塞。数据意味着±se(n = 5 - 8)。*:p < 0.05与比例的专利船只SuHx老鼠的肺;#:p < 0.05与完全闭塞血管SuHx老鼠的肺。
讨论
基于知识的甲状腺激素刺激内皮细胞生长(11- - - - - -13和增强缺氧性肺血管收缩14),我们假设,肺血管重塑的开发过程中严重的多环芳烃是甲状腺激素依赖。在这里,我们表明,诱导甲状腺功能减退可防止和逆转的肺血管闭塞SuHx老鼠和降低缺氧性肺血管重塑。其他重要的发现是,甲状腺切除术和PTU治疗减少了高的过度生长因子FGF-2和整合素αvβ3蛋白质,描述SuHx肺,多环芳烃的预防或治疗通过调节甲状腺素导致大大减少房车肥大和扩张。
而甲状腺疾病协会与特发性和对于形式的PH值已经被认可,目前没有说假设试图解释甲状腺激素如何影响肺血管疾病的发病机理。一些出版物文档∼30%的甲状腺功能减退的发生率PAH患者(28- - - - - -30.]。由于明显的重叠特发性肺动脉高压(IPAH),自身免疫性疾病,一个假设是,IPAH患者的甲状腺功能减退的原因是自身免疫性甲状腺炎(31日]。也有一个强大的协会甲状腺机能亢进和PH值之间(表S1)。甲状腺亢进患者目前尚不清楚PH值与左心脏病、高动力性的循环或宽松的甲状腺激素对肺血管重塑的影响(11,12]。我们的发现可能的临床翻译如下:1)我们发现没有证据表明甲状腺功能减退之间的因果关系和多环芳烃;2)在甲状腺甲状腺激素替代PAH患者应该被谨慎地执行,以避免增强血管重塑;和3)甲状腺机能亢进可能加速pre-capillary形式的PH值。
机制肺动脉muscularisation和管腔闭塞SuHx老鼠无疑是复杂和多细胞的相互作用。这里我们集中我们的注意力在细胞凋亡和细胞生长信号表达的内腔填充细胞和血管周的细胞群。在thyroidectomised SuHx老鼠,只有少数细胞的支架表面涂层的细动脉包含IHC PCNA表达裂解半胱天冬酶3(图S2)相比,肺部分肺高血压SuHx动物。这些结果支持分析整个肺组织PCNA的裂解半胱天冬酶3蛋白表达(图4)。结合数据来源于thyroidectomised SuHx老鼠收到补充甲状腺素(图2),我们的数据确实表明,甲状腺素驱动腔消灭设置的细胞生长严重的多环芳烃。缺氧性肺血管重塑的缓解甲状腺切除术后表明,甲状腺素也宽容肺动脉平滑肌细胞肥大。我们的研究没有设计确定的确切性质平滑肌和内皮细胞的相互作用实验PAH SuHx模型。减少的趋势在thyroidectomised PVRI缺氧大鼠没有追踪的房车肥大的减少,这是意想不到的但可能与甲状腺功能减退对心肌细胞肥大的影响是不可预测的。
我们假定FGF-2血管生成因子和信号通过Erk可能参与甲状腺与荷尔蒙相关的血管重塑的机制在动物模型(11]。随机检查肺组织样本,FGF-2的表达和p-Erk显然是甲状腺激素依赖。通过包含IHC分析我们发现FGF-2, FGF受体1 (R)和p-Erk是肺血管病变细胞中表达,这些蛋白的表达在病变影响甲状腺切除术和甲状腺素替代治疗。以类似的方式,肺组织整合素的表达蛋白αv减少thyroidectomised SuHx鼠肺和表达恢复后甲状腺素替代(图4摄氏度),而β3链的表达变化不明显(图4 d)。在观察甲状腺激素依赖性肺血管重塑变化thyroidectomised老鼠,我们评估了PTU治疗效果建立了多环芳烃和肺血管重塑。我们已经表明,PTU治疗减少了RVSP PVR,从而减少房车肥大,尽管PTU的影响大大小于预防甲状腺切除术的影响(图5)。PTU治疗重新开放了小动脉,专利器皿在肺部从PTU-treated SuHx肺了,这更大程度的船与RVSP明显相关(图6)。而PTU治疗明显减少血管生成因子的表达FGF-2, SuHx肺PTU治疗的效果在αvβ3表达式是微不足道的(图S9),这表明也许在SuHx肺FGF-2表达取决于甲状腺激素的作用。潜在的致病的作用FGF-2我提出的在多环芳烃zikki等。(26)和FGF-2表达在人类肺血管病变一直在前面描述的(27]。在这里,我们表明,VEGF受体封锁的设置和慢性缺氧,FGF-2肺组织样本中高度表达,建议FGF-2 SuHx模型中的血管生成多环芳烃。测量血浆甲状腺素浓度显示甲状腺切除术和PTU治疗甲状腺素水平降低,SU5416结合慢性缺氧不增加血浆甲状腺素水平和甲状腺素替代thyroidectomised SuHx大鼠甲状腺素水平恢复正常或略高于正常。因此SuHx动物,血浆甲状腺素水平与RVSP (图1 d)。
甲状腺激素在我们的血管生成行为SuHx鼠模型angio-obliterative严重的多环芳烃可能解释道通过甲状腺激素细胞膜受体信号(32]。慢性条件下VEGF受体封锁(SU5416所致),FGF-2变得过度和整合素αvβ3可能作为信号调制器对FGF-2 [33)和甲状腺激素(11]。在细胞实验中,FGF-2的增长促进行动和甲状腺素的抑制增殖作用蛋白激酶抑制剂(pd - 98059) [11]。特别是,B纯砂沙漠et al。(12]表明,整合素αvβ3包含甲状腺素的细胞表面受体结合位点与map激酶激活,而另一个绑定域名的αvβ3整合素与磷酸肌醇3-kinase和可能导致细胞生长的信号通过激活核低氧factor-1α[32]。这些细胞是否信号事件的甲状腺素在培养内皮细胞(11,12,32)解释甲状腺素的血管生成的行为确实是适用于甲状腺激素依赖性angioproliferation SuHx模型是目前未知的和将会在未来的实验研究。
研究的局限性
描述性的本质我们的研究排除了明确的结论关于甲状腺与荷尔蒙相关的确切机制angioproliferative改造。因为小鼠模型PH值缺乏一致的angioproliferative组件(34),我们的研究必要性,使专用的鼠模型,从而限制使用转基因的干预措施的可能性。与我们的研究提供的强有力的证据对甲状腺激素的作用在最初发展angioproliferative改造,甲状腺激素的作用在疾病维护尚不明朗。PTU部分逆转肺动脉压力的增加,但影响相对较小的相比,预防甲状腺切除术的效果。这项研究没有提供的解释经常报道甲状腺功能减退和博士之间的联系,结果表明这种关联的结果更可能是这两种情况之间的联系和自身免疫31日]。
总之,在这个报告中我们首次展示,在实验PAH,甲状腺激素是高度宽容的发展严重angio-obliterative PAH和导致肺血管的损伤反应时肺血管内皮细胞凋亡(16]。甲状腺激素可归属angioproliferative组件可能不需要血浆激素水平升高,而是可能取决于促进甲状腺激素细胞膜受体(αvβ3整合素)信号(32]。
确认
作者希望感谢诉Kraskauskasiene帮助甲状腺激素测量和d·加德纳(无论是在医学部门,弗吉尼亚联邦大学,里士满,弗吉尼亚州,美国)对批判性阅读手稿。
脚注
可以从本文的补充材料www.www.qdcxjkg.com
支持声明
支持本研究从维多利亚获得约翰逊肺阻塞性研究中心在弗吉尼亚联邦大学,里士满,弗吉尼亚州,美国。
感兴趣的语句
一份声明中对N。F Voelkel可以找到www.www.qdcxjkg.com/site/misc/statements.xhtml
- 收到了2011年11月11日。
- 接受2012年4月20日。
- ©2013人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}