





摘要
新型ICS / FAST-ONSET Laba Relifive治疗方案在轻度哮喘中的实际研究描述http://ow.ly/WE3Yp
给编辑:
本文描述了吸入皮质类固醇(ICS)/快速起效长效β受体激动剂(LABA)缓解轻度哮喘的Novel START(信比可妥buhaler Asthma relief Therapy, Symbicort Turbuhaler)随机对照试验的原理和设计。
哮喘临床研究和管理的主要焦点是中度或重度哮喘患者。很少有人注意到“沉默的大多数”哮喘谁经历所谓的间歇性和轻度持续哮喘。这些患者通常单独使用吸入短效β激动剂(SABA)治疗,以缓解症状。然而,尽管单独使用SABA疗法确实能缓解症状,但几乎没有强有力的证据支持单独使用SABA治疗轻度哮喘的长期有效性和安全性,或指导临床医生和患者何时开始常规低剂量ICS疗法。此外,当症状不常见时,医生和患者很难认识到对ICS治疗的需要,在这种情况下,对ICS的依从性较差是常见的。这导致了对间歇性或轻度哮喘的常规ICS处方的替代方案的考虑。
包含ICS和快速缺口Laba的组合吸入器,其单独使用,其仅作为释放治疗,单独优于SABA释放治疗,并且可能是常规IC的替代性,具有间歇性或轻度哮喘患者的SABA缓解疗法[1- - - - - -4].联合ICS/快速起病的LABA缓解疗法允许根据症状滴定ICS治疗。这可能会增加过度依赖SABA和维持型ICS治疗依从性差的患者对ICS的使用,并且可能实际上代表了常规联合ICS/快速起病LABA治疗的患者在“现实世界”中所接受的治疗[5,6].
这项新的START试验将调查ICS/快发型LABA作为间歇性或轻度持续性哮喘患者的唯一缓解疗法的有效性和安全性。本研究假设,在间歇性或轻度哮喘的成人患者中,ICS/快速起病的LABA缓解疗法比SABA缓解疗法和维持性ICS和SABA缓解疗法更有效。主要目的是比较ICS/快发LABA缓解疗法与其他两种治疗策略的疗效:仅采用SABA缓解疗法和维持ICS联合SABA缓解疗法。参与者将是成年患者,仅使用SABA单药治疗,不使用其他哮喘药物。次要目标是比较每个方案的安全性,检查吸入器使用模式与随机方案,并探讨基线临床特征(如t辅助细胞(Th) 2谱)是否预测随机治疗的优先反应。
该研究设计是52周,开放式标签,并行组,多中心,第III期,随机对照试验(RCT),以便在新西兰,英国,意大利和澳大利亚的网站中进行。实验治疗将是Budesonide,Formoterol富马酸二水合物(Symbicort Turbuhaler; Astazeneca,Sodertalje,Sweden)200μg/6μg,一种吸入,用于根据需要缓解症状。代表当前临床实践的两种比较剂治疗将是首先是Salbutamol加压计量剂量吸入器(ventolin; ventolin; glaxosmithkline,伦敦,英国)100μg,两个吸入,以便根据需要缓解症状;其次是Budesonide(Pulmicort Turbuhaler; Astrazeneca)200μg,每日一次吸入,Salbutamol PMDI(ventolin)100μg,两个吸入,用于根据需要缓解症状。参与者将提供哮喘行动计划,其中包括何时寻求恶化哮喘的医学审查。所有研究吸入器都将具有记录使用的电子监控设备,使得识别药物的使用模式,如前所述[5].
该研究旨在招募675名接受SABA缓解治疗的医生诊断的哮喘患者。招募潜在参与者的条件如下:1)报告在过去12个月内发生了严重的病情恶化,并自我报告在过去4周内平均每天≤2次使用SABA;或2)在过去12个月内没有出现严重恶化,在过去4周内平均使用SABA≥2次自我报告[7],但在前4周内平均每天不超过2次。病人在每次情况下通常采取的刺激次数将被记录下来。有关纳入和排除标准、研究程序和方案的详细信息,包括统计分析计划,可在澳大利亚新西兰临床试验注册中心(https://www.anzctr.org.au/trier/registration/trialreview.aspx?id=369311&isreview=true.);试验设计总结为图1.
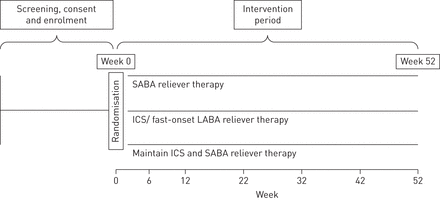
研究设计。萨巴:短效β受体激动剂;ICS:吸入激素;腊八:长效β受体激动剂。
主要结果将是哮喘加重率,表示为每位患者每年的加重次数。
研究对哮喘恶化的定义是:1)哮喘恶化导致紧急医疗复查(初级保健就诊、急诊(ED)就诊或住院);和/或2)哮喘恶化,导致持续使用全身皮质类固醇,如强的松;和/或3)哮喘恶化导致高β激动剂使用发作,定义为24小时内>16激活沙丁胺醇或>8激活布地奈德/福莫特罗。
主要次要成果包括:
1)临床结果:
a)三个标准中的每一个定义的加剧的比例,至少一个加剧的患者的比例以及第一次加剧的时间。
b)根据美国胸科学会/欧洲呼吸学会标准定义的严重加重率和首次严重加重时间[188bet官网地址8].
全身使用皮质类固醇至少3天。
因哮喘住院或急诊,需要全身皮质类固醇。
c)因治疗失败而退出的患者比例,定义为:
一个严重的恶化。
三个加剧,分开至少7天。
不稳定的哮喘导致随机治疗的改变。
d)哮喘控制问卷(ACQ-5评分)。
e) 1秒用力呼气量(FEV)1) % 预料到的。
f)呼出的一氧化氮的分数。
2)研究用药:
a)平均每天ICS剂量(布地奈德μ g·d)-1);无ICS使用的天数,最长的无ICS使用时间。
b)口服皮质类固醇总剂量。每年口服皮质类固醇的疗程数;每年综合全身皮质类固醇暴露,其中每年总ICS剂量转换为口服强的松-肾上腺功能全身影响的等效剂量[9如前所述,添加到每年口服泼尼松剂量中[5].
c) β-激动剂高使用发作包括至少一次高使用发作的参与者比例;48小时、7天和14天内无医疗检查的高使用率发作比例。
d) 24小时内β-激动剂的最大激活数。
e)如前所定义,在严重病情加重前14天内使用研究药物[10.].
3)不良事件:
不良事件。
b)严重不良事件。
4)成本效益。医疗费用(药物、急诊和急诊部就诊、住院)和非医疗费用(休假)。收集到的成本效益数据可以用来推断未来的定价模型。
5)病人的态度:
a) ASK-12问卷[11.].
b)在120名参与者的子集中进行定性访谈,以评估药物的可接受性和效用以及药物对患者信仰的影响。这将以10名参与者组成,由随机待遇和国家选择。
统计分析将是“意向治疗”。初级分析是每年通过泊松回归对每年的恶化率的比较,以抵消观察日的偏移,以及招聘前的SABA使用和先前严重恶化的数量的固定效果。在分析之前将评估过分分散,并且在必要时施加校正的分析。将进行两个敏感性分析,以考虑潜在重要预测因子的不同分布。随着Kaplan-Meier Plots和Cox比例危害的生存分析将用于计算危险比首次加剧的时间。
对于基于药物使用的次要结果变量,通过泊松回归分析事件的计数,随着时间的观察时间和校正,如有必要,对超分散的校正。将通过计算相对风险或差距和适当的置信区间来分析事件的比例。连续变量,如ACQ-5分数和FEV1将通过t检验和混合线性模型进行比较,以解释重复测量并检查随时间变化的模式。将进行预先指定的亚组分析,以评估特定特征是否影响治疗反应。
每个治疗组的样本量为225个,占20%的退出,有80%的效力,α 5%,相对加重率为0.75,从每个患者每年1.2到0.9。
数据安全监测委员会将在400名患者随机化后审查所有严重的临时安全统计分析。
本研究是第一个独立的调查员启动和管理的RCT进入ICS / FAST-ONSEL LABA吸入器,作为哮喘患者哮喘患者的唯一释放治疗,并将提供关于所有三种治疗的安全性和疗效的强大数据战略。该研究具有务实的开放式标签设计,使得两个潜在的“现实世界”优势ICS / FAST-ONSEL LABA REVIFEVER治疗方案,即。在临床实践中,使用单个吸入器并不要求正常吸入器使用。避免双伪,双盲设计和广泛的纳入标准,将确保调查结果可以推广到临床实践。亚组分析以确定是否具体特征,例如对随机处理的Th2轮廓影响响应,将提供个性化治疗可能是基于的数据。
我们将解决临床问题:“在单独用SABA缓解疗法治疗的间歇性或轻度哮喘的患者中,SABA救济疗法的比较疗效是什么(继续他们目前的处理)与维护IC和SABA Reliever疗法(按照指南推荐)[7]与ICS / FAST-ONSET Laba Reliever治疗(新型治疗方案)?“虽然该研究尚未旨在验证指导方针,但它将评估在前4周内至少有两次使用SABA的建议应接受常规ICS治疗,除了具有严重恶化的人过去12个月,无论缺少SABA使用吗?7].
主要结果,哮喘加剧率,是一种复合措施,包括如前所述的高β-激动剂使用发作[5],基于β-激动剂使用水平的阈值,要求在标准动作计划中进行医学审查,并由6μg蛋白质醇的短期支气管胆管等当量,以急性哮喘中重复给药6μg甲酚至200μgsalbutamol [12.,13.].经过验证的电子监控设备,可记录吸入器用于识别其他未记录的恶化,以及使用行动计划,这些计划提供有关如何识别恶化和采取行动的书面指示,允许稳健识别恶化。主要结果措施对应于“中度至严重的加剧”;还将分析严重的恶化作为重要的二次结果。
根据需要的SABA组每年每年每年1.2患者的假定加剧率是保守的,源自两个安慰剂控制的RCT,其每年举报安慰剂维持和SABA救济治疗的加剧率为每年0.77-1.63的速度为0.77-1.63 [1,14.].不同研究对加重的定义不同,但最重要的区别是,我们纳入了高β-激动剂使用时期,通过研究用药电子监测确定,应该改善对加重的识别[5].提名的临床有意义的治疗效果为0.75的相对加油率也是保守的。
总之,这项新的START研究将提供ICS/快发LABA缓解方案与SABA缓解方案以及维持ICS和SABA缓解方案在成人轻度哮喘患者中的疗效和安全性的数据。
脚注
临床试验:本研究注册于www.anzctr.org.au使用标识符ACTRN12615000999538.universal试用号:U1111-1170-2118。
支持声明:本研究由AstraZeneca的授权资助(参考号ESR 14-10452)。在与Astrazeneca咨询之前,调查人员启动和设计了该研究。该研究的全球赞助商是新西兰的医学研究所(惠灵顿,新西兰)(参考号:MRINZ / 15 / A1),这将对研究行为,监测和数据管理进行全面责任。本文的资助信息已存入FundRef.
利益冲突:可以在本文的在线版本旁找到披露www.qdcxjkg.com
- 已收到2015年10月12日。
- 接受2015年12月12日。
- 版权所有©2016











{kind=link}
{kind=link}