





文摘
遗传素质对肺纤维化的发现证实了几个基因突变导致肺纤维化。尽管基因测序的家族性肺纤维化(基维辛迪例是嵌入到常规临床实践在几个国家,许多中心还将基因测序在间质性肺病(ILD)服务和适当的国际共识尚未建立。国际和多学科专家工作组(位肺脏,遗传学家,儿科医生,病理学家,遗传顾问,病人代表和图书管理员)回顾了文献在1945年至2022年之间,并达成共识的下列问题:1)哪些患者可能受益于基因测序和临床咨询吗?2)所谓的自然历史基维辛迪?3)通常是哪些基因测试?4)端粒长度测量的证据是什么?5)的角色是什么常见的遗传变异(多态性)诊断?6)基维辛迪的最佳治疗方案是什么?7)家庭成员有资格获得基因测序?8)临床筛查和随访参数可能被视为家庭成员? Through a robust review of the literature, the Task Force offers a statement on genetic sequencing, clinical management and screening of patients with FPF and their relatives. This proposal may serve as a basis for a prospective evaluation and future international recommendations.
文摘
间质性肺疾病的遗传倾向(ILDs)是众所周知的。声明旨在协助卫生专业人员的单基因肺纤维化和家庭ILD病人和他们的亲属。https://bit.ly/3h9hvQ6
介绍
肺纤维化遗传素质是由疾病患病率增加10倍的家庭患者诊断为特发性肺纤维化(IPF) [1- - - - - -3]。初步研究家族性聚集的间质性肺病(ILD)导致基因突变的发现与端粒内稳态(telomere-related基因(采用))和表面活性剂体内平衡(surfactant-related基因(SRGs)) (表1和补充表S1)[3,4]。这些研究表明,家庭的疾病表型不限于IPF,但包括频谱潜在的进步肺纤维化,包括非特异性间质性肺炎(NSIP)、过敏性肺炎、风湿性arthritis-associated ILD [5- - - - - -10]。此外,ILD形态模式往往是不确定的或包含重叠的特性和不一定适合建立诊断ILD类别(11]。另一方面,在采用突变的存在或SRGs与临床特点有关,如年龄、性别、肺表型,肺外疾病和生存。明显的家族遗传的存在,与单个基因变异有关,如采用或SRGs,隔离ILD的存在,强烈支持这样的结论:ILD在这种情况下是一种单基因(基因)障碍。
由于财富的科学报告和公众意识的增强遗传疾病,基因测序肺纤维化患者在临床常规,找个地方可能对病人的治疗产生深远影响。然而,2018年和2022年国际指南IPF不解决这个问题(12,13]。2017年法国国家实用指南IPF首先建议家族性肺纤维化患者的基因测序(基维辛迪)14]。在其他国家,个别医生和ILD诊所在日常实践中实现基因测序或提供基因测序的研究背景(15]。当前缺乏共识声明或建议导致了大量的意见和方法,没有适当的国际共识在基维辛迪基因测序。
众多的单基因疾病可以与ILD有关。然而,目前的声明是有限的两组基因,采用和SRGs基维辛迪的最常见原因。囊性肺疾病患者,Hermansky-Pudlak综合征、肺肺泡蛋白质沉积症,lysinuric蛋白不耐受,溶酶体储存障碍,interferon-related基因突变和肺肺泡microlithiasis出现罕见但具体症状,需要专门的独立的语句(补充表S1)。
这个声明的目标受众包括成人和儿科位肺脏,遗传咨询师和临床遗传学家照顾ILD患者。通过回顾现有文献在ILD基因的发现,我们的目标是大纲的诊断,预后和治疗基因测序的价值,并提供一个声明基维辛迪的管理。
方法
这个工作组是一个国际和多学科的努力支持的欧洲呼吸学会(人)。188bet官网地址9名成员是位肺脏,两个是遗传学家,一个儿科医生,一个病理学家,一个遗传顾问,一个病人代表和一个图书馆员(Nienke van der Werf)。椅子(r . Borie和c . van Moorsel)选择其他工作组成员根据他们的专长在ILD和遗传学。利益冲突的声明的所有工作组成员和管理根据人政策(完全披露,请参阅补充材料)。早期的会议椅发生在2020年1月。在这次会议上,建议在创建三个工作组在工作组和特定的叙事(或non-PICO(人口、干预、比较器、结果)16组(内)需要解决的问题表2)。随后,相关研究的文献检索和回顾在1945年和2020年之间在执行工作小组利用MEDLINE和Embase数据库(补充图)。搜索限于文章中可用英语,对人类的研究报告。二次搜索了相关文章的引用列表。从2020年开始,工作组成员被要求提供额外的关键文献他们意识到。在第二次工作组会议(2020年10月),个人评价的结果被整个工作组提出和讨论。基于这些讨论,每组组装最重要的语句(“索赔”)解决一个特定的区域,和语句专责小组所有成员进行评估的正确性和重要性。没有明显的共识,两个网络致力于特定问题调查,包括1)定义基维辛迪和2)筛查无症状的亲属。绝大多数的选票的定义是保留(补充表S2和S3)。尽管工作组成员最初支持“家族ILD”这个词,因为并不是所有的家族ILD纤维化在演讲,随后投票显示偏好“家族性肺纤维化(基维辛迪)”一词,作为最常用的文献中,避免混淆。
在此基础上选择,手稿写的初稿。这个草案是在第三和第四小组讨论会议(2022年1月到5月);声明一起敲定的要点和研究包括第一次工作组会议后发表的。最终的文档结合一个基于证据的方法,依靠审查出版物,工作组成员的临床专业知识,可以与最近美国肺纤维化基金会基因测试工作组的建议(17]。
结果
叙述问题1:哪些患者可能受益于基因测序和临床咨询吗?
声明
在接下来的临床情况下,项目组成员通常提供基因测序:
总结的证据
虽然没有一致的定义,研究制定基维辛迪通常被定义为一个纤维ILD的情况下与一个或多个亲戚ILD的一种纤维(19- - - - - -22]。在此声明我们基维辛迪定义为:任何纤维ILD至少在两个血相对第一——或者二级家庭成员。工作组也被认为是“基因肺纤维化”尤其是对病人携带致病变种导致ILD没有任何家庭相对与肺纤维化。尽管这个词是对未来的承诺,目前不经常使用,没有明确定义。目前,专责小组选择“基维辛迪”这个词,也帮助位肺脏承认的优势表型和理解它对家庭成员的影响。我们定义它包括ILD的散发病例发现变异或涉嫌的遗传变异。家族病的存在进步的可能性增加肺纤维化(尽管发生管理适合单独的实体)。对疾病的影响进化的亲戚non-fibrotic ILD的形式,如结节病或non-fibrotic过敏性肺炎,还没有被研究过,但观测情况下一系列家族性结节病和non-fibrotic过敏性肺炎不显示偏好对进行性纤维化ILD [23- - - - - -25]。工作组成员通常限制对纤维化ILD的基因测序。
成人基维辛迪本质上是没有区别的从零星的患者的临床表现、影像学和组织病理学2,26- - - - - -28]。然而,他们通常是年轻和疾病的发展朝着进步fibrosing ILD有限生存(2,26- - - - - -28]。特发性常见的遗传背景,对于非ILD报道(5- - - - - -7,29日]。事实上所有形式的纤维ILD在年轻的成年人,具有象征意义的特发性或对于可能的丹或分析突变(6,8,10,11,30.]。
在丹突变患者和肺纤维化,贫血出现在17 - 27%,大红细胞症在8 - 54%[24 - 41%,血小板减少症8,10,31日]。DKC1中,TINF2和,TERC突变携带者似乎更容易开发血液学的参与叔,PARN狂欢dF4y2BaRTEL1突变携带者(10),但还需要进一步的研究来评估是否可能与一个军团之间的年龄差距。建立脊髓发育不良和急性白血病可能更暗示丹在年轻患者突变载体地位,尽管少(8,32,33]。的确,在ILD患者,原因不明的大红细胞症、血小板减少症、脊髓发育不良和急性白血病具有象征意义的迹象可能丹突变(表3)。
肝与丹相关表型变异是异构的,从无症状的肝酶报告在5 - 27%增加到不明原因引起的肝硬化或二级(8,10,34]。G狂欢等。(35)强调了高频hepato-pulmonary综合征与丹突变有关。确实存在不明原因引起的肝酶升高,hepato-pulmonary综合症或肝硬化(特发性或不)代表一个可能的突变。
丹在角化病突变最初报告congenita,罕见的多系统疾病,包括特定的皮肤的三合会(表1),通常骨髓衰竭。大部分的丹载体ILD患者不存在皮肤的三合会,但15 - 40%的突变携带者早期头发老龄化(30岁前)8,36]。虽然没有共识的定义的年龄和早期的头发老龄化比例考虑丹突变,重要的岁头发头发花白的30年是指示性的突变。
短端粒综合症可能还包括原发性免疫缺陷等表现,视网膜或神经紊乱37]。基因测序的必要性应该在多学科团队会议讨论相关罕见表现怀疑丹突变(18,38]。鉴于相对年轻的年龄大多数丹突变患者被诊断为ILD(平均年龄58岁),肺移植往往是讨论和提供给这些病人8,10,30.,39,40]正如报道患病率11.8%和24.2%的罕见变异丹在262年和149年两个群IPF病人肺移植(表4)[41,42]。还需要更多的研究来确定是否所有纤维ILD病人应该进行端粒长度测量和肺移植前基因测序。基维辛迪肺移植患者潜在的候选人,但可能需要一个定制的免疫抑制方案的一个潜在的短端粒综合症(参见叙事问题6)。
告知患者家族性疾病和基因测序的影响已成为护理在家族性疾病的一个重要组成部分,并使共享和明智的决策。项目组成员在基因测序之前通常提供遗传咨询。遗传咨询是一个过程,帮助人们理解和适应医疗、心理和家族基因对疾病的影响。有广泛的国际法律的变化,卫生系统和文化有关遗传咨询(43]。在一些国家培训顾问被分配到的任务,而在其他国家,它的任务是治疗医生。这句话之前,项目组组织进行了调查的患者,亲戚和位肺脏的经验和需要在基因测序ILD评估当前形势并确定未来的策略。在大多数情况下,位肺脏照顾病人的基因测序和相对的信息和沟通的结果。病人、家属和位肺脏同意需要更多信息和意识44]。
叙事问题2:所谓的自然历史基维辛迪?
声明
家族性或单基因纤维化ILD成年人通常是进步的和经常与不良预后相关,独立于疾病实体。
在儿童,ILD通常是单基因和疾病过程依赖于基因和突变。
总结的证据
的主要表型基维辛迪是一种慢性进行性纤维化ILD [2,26- - - - - -28,45]。病人的临床表现进行因果变异对于成人和儿科ILD类似于零星ILD,呼吸困难,咳嗽,吸气发出爆裂声,在少数,夜总会。
肺功能下降基维辛迪一直与零星的IPF相比,虽然不可避免的少量基维辛迪病人在每个研究估计不精确的。在46基维辛迪病人的一项研究中,54.3%有明确的通常的间质性肺炎(摘要),21.8%可能的摘要和23.9%与摘要不一致,不同利率的用力肺活量(FVC)和传播肺活量的肺一氧化碳(DLCO)下降被认为根据不同的高分辨率计算机断层扫描(HRCT)模式26]。一项研究表明FVC的快速下降的趋势(每年9.9%)21例家族性IPF指数相比,54零星的IPF患者每年4.9%,尽管这种差异没有达到统计学意义(p = 0.12)28]。115年ILD病人带着丹突变,其中46%有IPF,每年下降300毫升FVC被认为无论基因及ILD的实体(10]。
基维辛迪的生存已经在一些研究调查,与相似性成人纤维化基维辛迪,不管底层实体,和零星的IPF (46]。总体存活率很低在家族和gene-specific军团,虽然使用了不同的方法,从而阻碍直接比较。生存中值诊断基维辛迪[2.4 - -7.3年9,26,27,45]。
最近的一项研究包括1262名ILD患者发现家族IPF的明显恶化的生存和non-IPF基维辛迪与零星的同行相比(IPF患者的死亡风险比或移植1.8 (95% CI 1.37 - -2.37;p < 0.001)和2.08 (95% CI 1.46 - -2.96;p < 0.001)对non-IPF病人),而没有区别non-IPF基维辛迪和零星的IPF被发现46]。其他几个零星的回顾性群组没有确定区别ILD和基维辛迪4,27,45]。
类似低生存在gene-specific ILD军团,包括transplant-free生存中值的2.9 - -4.2年丹突变患者确诊后(8,10,30.,36]。
只有少数研究比较不同临床实体之间生存的家族性患者。N尤顿et al。(10]表明,组内telomere-related疾病诊断的患者的平均生存IPF(2.8年)没有什么区别的non-IPF病人组(3.1年)。虽然这些发现表明,在基维辛迪,生存是独立于ILD的实体,需要更多的验证数据。
ILD的童年有几个年长的小案例系列篮子军团与肺纤维化(47- - - - - -50]。10 - 30%的儿童ILDs有纤维化的证据(51]。群潜在的纤维化和分子定义ILDs从稳定进步的致命的疾病(52- - - - - -56在成年早期和晚期][57,58]。为ABCA3,一个强大的存在genotype-phenotype关联(59,60]。Biallelic零突变导致早期死亡在婴儿期,而突变导致慢性ILD剩余函数,与广泛的临床表现包括演示在成年晚期(59- - - - - -62年]。
叙述问题3:哪些基因通常是测试?
声明
端粒,surfactant-related基因通常是分析。
其他基因的靶向基因测序可以讨论在个案基础上,根据表现型,超出了本文的范围。
总结的证据
诱发突变基维辛迪包括采用突变和SRGs (表1)。
Telomere-related基因
端粒是非编码DNA序列(TTAGGG)位于染色体的结束。他们在染色体完整性维护中发挥作用。端粒缩短每次细胞分裂,最终导致细胞衰老。端粒缩短与老化有关。抵消端粒缩短,增加了端粒酶核糖核蛋白端粒酶复杂重复。端粒酶是指集团复杂的蛋白质和RNA催化作用的核苷酸重复染色体末端。有很多组件端粒复杂,包括端粒酶逆转录酶(叔)和端粒酶RNA组件(TERC),对于正常端粒功能和完整性。丹突变往往与较短的端粒(63年]。丹突变导致短端粒综合症已经与许多疾病有关表现,包括进行性纤维化ILD [63年]。
叔和,TERC突变是第一个采用在基维辛迪[64年]。基维辛迪病例的大量研究和他们的家族已经确定了各采用突变,和超过12采用目前基维辛迪(表1)[65年]。的确,个人或家族的存在肺外的短端粒综合症的迹象已被证明与丹的速度突变零星或家族ILD (表4)。
胸部CT最初是一个典型的摘要模式在多达74%的ILD例丹突变,但后来发现只有46 - 55%的情况下(8,10,31日]。不寻常的特性中发现13 - 20%的病例包括upper-lung优势的纤维化,血管优势或pleuro-parenchymal fibroelastosis (PPFE)模式(8,10,31日,66年]。总的来说,14 - 40%的病例显示合并肺纤维化和肺气肿(CPFE)模式(67年]。CT扫描和组织学模式可能会有所不同和重叠,甚至在同一个家庭,摘要之间NSIP、过敏性肺炎、PPFE和不可归类的纤维化。事实上所有形式的纤维ILD可以关联到一个丹突变(6,8,10,30.]。
与丹有关的肺外表型变异是异构(表3)。
Surfactant-related基因
表面活性剂是由II型肺泡上皮细胞合成和分泌。表面活性剂是由90%的油脂和10%的蛋白质(68年]。Surfactant-associated蛋白(SP)——对肺和SP-D重要防御,而疏水蛋白质SP-B和SP-C对表面活性剂的功能很重要。相应的基因SFTPA,SFTPB,SFTPC和SFTPD(68年]。层状体内SP-B SP-C和表面活性剂脂质。后者被运送到层状的身体通过ATP结合盒家族,成员3 (ABCA3)转运蛋白编码ABCA3(69年]。甲状腺转录因子1 (TTF1)编码NKX2.1,是一个转录因子表达的肺、甲状腺和中枢神经系统调节蛋白质和表面活性剂ABCA3基因转录(70年,71年]。
纯合子或复合杂合的SFTPB突变已被确认为新生儿呼吸窘迫综合征的原因,但没有被确认在成人ILD [68年]。SFTPD突变尚未与肺部疾病有关。
儿科和成人ILD与SFTPC突变,但他们的频率通常是在基维辛迪(< 5%表5)[3,58,72年,73年]。传播是常染色体显性遗传。新创突变是频繁的,也许可以解释50%的情况下56]。虽然纯合子和复合杂合的ABCA3突变通常是与新生儿呼吸衰竭(59],一些成人ILD病人运营商的纯合子/复合杂合的ABCA3突变已报告(61年,62年,74年,75年]。
杂合的NKX2.1突变是经典与三合会的相关肺部疾病(新生儿呼吸窘迫综合征、复发性支气管炎或肺炎和ILD),甲状腺功能减退和神经系统异常(肌张力减退、发育延迟和舞蹈病)[76年,77年]。这些突变可能与ILD没有甲状腺功能减退或神经异常在多达三分之一的情况下,包括成年人案件中,最常见的HRCT模式是典型的摘要(76年]。
BiallelicABCA3突变和杂合的NKX2.1,SFTPA1,SFTPA2和SFTPC突变成人可以分享相似的临床和放射学表现。最常见的辐射模式通常被描述为不可归类的肺纤维化的特点是主要分散毛玻璃的透明和肺纤维化(11,58]。小叶间隔增厚和双边变量大小的囊肿,优惠分布在上部叶和胸膜下区,可以观察到(11,58]。区分肺气肿从囊肿有时是困难的78年]。组织学检查在成人最常见的相关模式是摘要,但NSIP混合的特点,组织肺炎、肺泡蛋白质沉积症或脱皮的间质性肺炎也被报道。可以观察到中度炎症和小叶中心的纤维化(4]。在年轻患者中,组织学显示NSIP、肺泡蛋白质沉积症或脱皮的间质性肺炎单独或结合在同一标本(56,60]。
杂合的SFTPA1和SFTPA2是唯一的种系突变,这些突变与肺癌的患病率高,虽然机制知之甚少(11,58,79年- - - - - -81年]。3例肺癌患者与ILD相关NKX2.1突变已报告(82年]。然而,数据缺乏提供分析测序零星ILD和肺癌患者。SRGs通常基维辛迪患者进行,或与特发性纤维化ILD在50岁之前,作为叙事问题1中讨论。
方法论的注意事项
基维辛迪基因的基因测序面板(采用和SRGs)寻找新的和先前描述件变体编码区域可能是由不同的技术:whole-exome测序(韦斯),全基因组测序(WGS)或下一代测序面板,每个都有各自的优点和局限性:成本、可用性和覆盖率。完整的面板可以针对一次,致病性测定应遵循国际标准(83年]。韦斯或WGS,不仅丹和分析,而且变异出现在其他症状(补充表S1),可能表现为ILD可以确定在一个分析。然而,可用性的技术,国家卫生或保险政策和许多不肯舍弃的一代意义不明的变异可能需要更有针对性的方法。在童年ILD,分析突变是最常见;在成人中,致病性变异丹比SRGs更常见。表1并公布产量最高的基因突变谱显示基维辛迪[41,58]。
叙述问题4:端粒长度测量的证据是什么?
声明
端粒长度有时基因测序提供了额外的信息,有时是在疑似telomere-related疾病时执行。
总结的证据
端粒长度测量
在外周血染色体端粒的长度测量,可以表示为一个绝对的数字(kb)或作为一个相对长度与年龄的人口控制和纠正。后者是首选,如上所述在以下段落。
端粒长度测量存在的几种方法(补充材料和表6)。定量PCR和Flow-FISH目前两个广泛使用的技术,以Flow-FISH报道为更多可再生的。虽然较短的端粒丹致病性变异的逻辑结果,只有少数研究发现端粒长度和丹变体之间的直接相关性,相互矛盾的结果。因为随着年龄的增长,端粒长度减少健康人群,端粒长度应该年龄调整(84年]。年轻患者端粒较短的症状通常出现更严重的表型,如角化病congenita和很短的年龄调整有关端粒长度(不到一百分位)(85年]。然而,成人丹突变患者只有肝或肺部疾病不一定存在明显短端粒长度(85年]。ILD病人携带致病丹变异显示比年龄组平均端粒较短,通常低于25百分位(86年]。然而15%的叔在一个队列(突变携带者呈现正常的端粒长度36]。在另一个群,几乎所有ILD病人,年龄超过60年,叔,,TERC狂欢dF4y2BaRTEL1突变,染色体端粒的长度小于1百分位,50%有端粒长度大于第十百分位(86年]。
短的端粒长度在外周血单核细胞通常是与致病性变异丹(87年,88年]。然而,端粒长度在正常价值经常被观察到在家庭与肺纤维化和端粒长度的收益率信息排除短端粒的存在不足综合征或肺部疾病的风险,尤其是在受试者年龄> 60岁(10,85年,86年,89年- - - - - -91年]。值得注意的是,一些丹变异可能不会影响端粒长度,而是端粒功能,具有相似的临床后果(92年]。端粒长度的测量可以用来筛选突变。在基维辛迪患者没有发现突变,短端粒长度可能识别未知的突变患者丹或拟表型,病人从他们的父母那里继承了很短的端粒(93年]。
较短的端粒长度与一个贫穷的结果在IPF和其他纤维ILDs [7,34,94年]。较短的端粒长度也会影响肺移植术后预后[95年,96年]。
总之,端粒长度可以确定当可用,但正常或不可用端粒长度通常不会阻碍基因测序。
叙述的问题5:什么是常见的基因变异的作用(多态性)诊断?
声明
等待新的研究,大多数工作小组成员不包括与IPF相关的常见基因变异或基维辛迪基因诊断。
总结的证据
常见的基因变异(也称为多态性)存在于基因组的任何地区,有一个等位基因频率> 1%。有些常见变异强劲与特定疾病相关,尽管它们的功能并不总是知道结果。
多个共同多态性引起纤维化ILD已确定,虽然他们的效果通常是低(97年]。的rs35705950MUC5B启动子基因多态性是最广泛的研究。粘蛋白5 b是一种蛋白质组成的呼吸道粘液。小等位基因的启动子变体rs35705950 (11 - 1241221 g - t (GRCh37))中发现约有10%的欧洲人,相当于大约20%的运营商(杂合子)小该等位基因和1%的次要的等位基因。这种微小的等位基因变异明显更频繁的在欧洲零星的IPF患者和基维辛迪(34 - 38%)(22,98年]。的存在MUC5B启动子多态性也与其他纤维ILDs有关,包括风湿性arthritis-associated ILD [5],零星的纤维化过敏性肺炎[7),石棉沉滞症(29日)和进行性纤维化肺间质异常(99年),但不是系统性sclerosis-associated ILD,肺结节病或自身免疫性myositis-associated ILD [One hundred.- - - - - -102年]。
的rs35705950 T-allele MUC5B表达增加有关终端细支气管(103年,104年),一项研究表明这种关联是摘要/ IPF但不是NSIP [101年]。值得注意的是,在IPF患者中,航空公司的MUC5B子变异可能与更好的生存和发展放缓有关(105年- - - - - -108年]。
的MUC5B子变体不是特别相关的家族或早发性肺纤维化的发生。然而,一些团体建议筛查MUC5B启动子变体在基维辛迪变体经典与基维辛迪作为一个单基因疾病(端粒酶/ surfactant-related)不发现3]。
上下文中的筛选IPF患者的亲属的存在MUC5B启动子多态性与间质异常可以预测随后的纤维化和进展(9,109年,110年]。然而,考虑到高的患病率MUC5Brs35705950微小等位基因及其相关的低外显率的pcr变体目前不是基维辛迪基因调查的一部分。还需要进一步的研究来评估是否该启动子多态性有助于变量表现性基维辛迪的单基因疾病。
MUC5B基因分型结果有用分层患者如间质肺异常或类风湿性关节炎的危险发展肺纤维化,有时在临床常规使用。
叙述问题6:基维辛迪的最佳治疗方案是什么?
声明
IPF患者或进行性肺纤维化(尽管标准管理),包括那些基维辛迪或已知的单基因疾病,工作组同意当前国际指南(13)推荐antifibrotic药物。
治疗儿童ILD本质上是基于专家意见。童年ILD应该被包括在治疗试验时可用。
工作小组成员将考虑肺移植患者的单基因疾病或基维辛迪,在适当的时候。在短端粒综合症,移植后免疫抑制方案时应仔细监控和调整。
总结的证据
药物治疗成人
临床试验测试包括在IPF和进步antifibrotic治疗肺纤维化患者独立于家庭或遗传关联。Antifibrotic治疗进行性肺纤维化已获批准,并回顾性分析患者的丹变体处理antifibrotics显示减少FVC治疗后开始下降,其安全性与先前的IPF试验观察(111年]。事后试验数据分析表明pirfenidone治疗是有益的,不管端粒长度(107年]。
antifibrotics与这些令人鼓舞的结果是消极的结果与免疫抑制药物治疗肺患者短端粒或携带突变。在一个事后IPF试验数据的分析,不良结果(死亡、肺移植或FVC下降)与免疫抑制治疗观察优先患者白细胞端粒长度小于第十百分位(112年]。此外,在纤维过敏性肺炎患者的回顾性研究,患者端粒长度的最低四分位数没有受益于与霉酚酸酯治疗,患者相比更长的端粒长度(113年]。成人患者分析突变,没有特定的药物试验。
在这个小数量的研究中,患者携带丹突变或短的端粒长度似乎有不同的疾病进化和免疫抑制治疗的反应不同。然而,它是未知的,如果基维辛迪特定患者炎症特征可能受益于免疫抑制治疗,免疫抑制之间的直接比较,目前缺乏antifibrotic治疗。
儿童药物治疗
在儿童,只有经验数据从案例报告或小系列出版ILD治疗临床上明显。第二阶段的随机对照试验的结果羟氯喹在童年ILD安慰剂和羟氯喹组没有区别在对治疗的反应,评估氧化或其他终端包括与健康有关的生活质量和肺功能(114年]。审判的nintedanib fibrosing童年ILD评估安全性和药物动力学(115年]。至少有一项研究正在致力于与达那唑治疗患者> 15岁肺纤维化与丹突变,结果预计在2023年(ClinicalTrials.gov:NCT03710356)。目前数据不合理处方的达那唑以外的临床试验。ILD的儿童纳入临床试验应该鼓励和所有成年试验应该有一个儿科临床实验的计划。
分析突变患者通常被称为参考中心和专业多学科会议中讨论。基于羟氯喹的随机试验114年),项目组成员逐个进行仔细评估正在进行的药方。在儿童中,欧洲和美国提出的语句,分析突变造成的ILD的治疗可能包括免疫抑制(强的松脉冲或连续治疗,steroid-sparing药物)在一个纯粹的经验基础上,因为没有随机试验在分析类固醇,或大环内酯物长时间使用(38,116年,117年]。
肺移植
在先进的疾病,证据表明,单基因肺纤维化患者可能受益于肺移植,在适当的时候。肺移植后5年生存率分析突变患儿(55%118年),最近的一项研究表明类似的存活率在年轻和年长的孩子119年]。在成人中,肺移植结果基维辛迪患者与零星ILD患者(120年]。因为肺癌发展一直在多达37%的记录SFTPA1/SFTPA2突变携带者(58),工作小组成员将考虑尽早安排和双边移植。
多项研究表明,移植肺纤维化患者丹突变也有类似的结果是没有这种变异的病人(39,41,96年,121年,122年]。血小板减少症和血小板输血的需要频繁,和骨髓增生异常综合征和/或骨髓衰竭报告在一些病人37,96年,121年- - - - - -123年]。最近的一项回顾性研究没有报告血液学的并发症的风险较高叔,RTEL1狂欢dF4y2BaPARN罕见的变异携带者(96年),但还需要确认观测。免疫抑制移植后治疗的标准治疗方案可以不容忍和增加发展中血液学的并发症或肾脏疾病的风险(121年- - - - - -123年]。因此,调整免疫抑制方案建议,以避免细胞毒性药物,如硫唑嘌呤(37,39]。总之,基维辛迪患者被认为是由专责小组成员作为肺移植的潜在候选人,但可能需要一个定制的免疫抑制方案的一个潜在的短端粒综合症(37,39]。
叙述问题7:家庭成员有资格获得基因测序?
声明
为家庭证明单基因疾病,工作组支持根据国家指令或立法提供基因测序。
总结的证据
在遗传性疾病,家庭成员患这种疾病的风险显著增加。基维辛迪通常占主导地位的产业,导致了50%的机会继承有害的等位基因在一级家庭成员(父母、兄弟姐妹和孩子)。基因测序在无症状的家庭成员可以执行的证明单基因疾病(识别(可能)致病突变的影响相对)这可能是紧随其后的是级联基因测序在随后的一级亲属。然而,当地的卫生和保险政策可能确定无症状的基因测序亲戚的可能性。此外,由于基因测序的结果有一个心理,社会和金融影响建议无症状的亲戚之前没有测试他们可以使自己的明智的决定,通常> 18岁(124年]。由于高外显率,疾病发展的基因测序分析预测能力是相对较高的58]。丹突变传输更多的在一个常染色体显性遗传方式不完全外显率和变量表达能力相关联。其他更多不寻常的特性存在于丹家庭:缩短端粒导致预期的继承,有时发生的肺拟表型。虽然存在突变associates高危疾病,没有突变的丹家庭不排除疾病由于其他遗传风险增加端粒异常(93年]。进一步研究基因测序的预测能力有致病性丹突变的家庭是必要的。
叙事问题8:临床筛查和随访参数可能被视为家庭成员?
声明
工作组成员的多数提供所有无症状患者的一级亲属基维辛迪周期临床评估(早期)ILD识别。频率是目前未知。
在临床实践中,工作组成员的多数建议所有患者的直系亲属基维辛迪有胸部CT扫描和肺功能测试至少在经历持续的呼吸道症状,如呼吸困难、咳嗽。
大多数的工作组成员包括一个完整的血细胞计数和肝酶评价短端粒综合症患者的一级亲属。
肺纤维化的所有已知的危险因素如吸烟或其他应避免吸入的曝光基维辛迪患者的一级亲属。
总结的证据
疾病过程可以分为临床(症状)和临床前阶段(无症状)。考虑到许多成年患者在诊断不可逆转地失去了多达50%的扩散能力,早期发现ILD是十分必要的。比较年龄出现症状随着年龄的增长在诊断表明,有一个诊断延迟1 - 3年基维辛迪[27,28,72年]。此外,在出现症状筛查可以发现疾病。在家族在一项研究中,患者在检查后诊断为58.5岁,而病人在出现症状后诊断的年龄为61.4岁(72年]。
疾病筛查临床前向一级亲属在几个国家在临床或研究设置。没有指导方针,标准化,或更少,循证实践包括哪些测试筛选,年龄应该开始和它的频率。疾病的风险与年龄和特定的基因。最常见的高外显率的常染色体显性遗传模式,导致疾病的风险高达50%的一级亲属。
在年龄中位数91成人患者的诊断分析突变是45岁(范围18 - 72年)(58]。在分析航空公司中,年龄中位数SFTPC突变携带者显著低于SFTPA1 / SFTPA2突变携带者(37与48年)[58]。只有一个儿科患者SFTPA基因突变(变异)过去一直是报道与51成人病例,这表明疾病的风险SFTPA1/SFTPA2通常局限于成年人。相比之下,SFTPC -和ABCA3有关的疾病可能会出现在所有年龄段(58]。
在丹突变携带者,在诊断ILD的年龄中位数是62年(35 - 79年)(58]。内丹突变携带者,病人携带,TERC突变确诊年龄比早些时候明显PARN在一个队列(51±11突变与64±8年)(10]。而surfactant-related疾病的性别比例是50/50所有年龄(58),男性的优势已经报道了telomere-related疾病。134年的一项研究主题与杂合的叔突变,肺纤维化被发现在38%的男性和14%的女性年龄< 60年,60%的男性和50%的女性年龄≥60岁(31日]。然而,多个亲戚基维辛迪的患者的研究显示,只有年龄,而不是性,同事与HRCT异常筛选[21,110年,125年,126年]。
预期的现象在家庭与丹突变使问题进一步复杂化。丹突变患者独立传播他们的短端粒突变和染色体端粒缩短的传播在后代的一个年轻的年龄(34,127年]。遗传基因预测的现象解释道,这被定义为与每一代更早和更严重的疾病(图1)[34,93年,127年,128年]。诊断年龄是影响家庭成员之间高度相关的相同的一代(2,26)和几代人降低了平均18年基维辛迪[10,26,28]。< 30岁,肺纤维化的病人作为单一短端粒综合征是常见的表现。当疾病发展的风险随着年龄的增加,依赖于基因或类型的突变和基因预测,没有足够的证据证明基因——或者筛查年龄的建议。几个SRGs引起肺部疾病在童年,而肺疾病作为唯一的表现telomere-related疾病在儿童时期没有被报道。
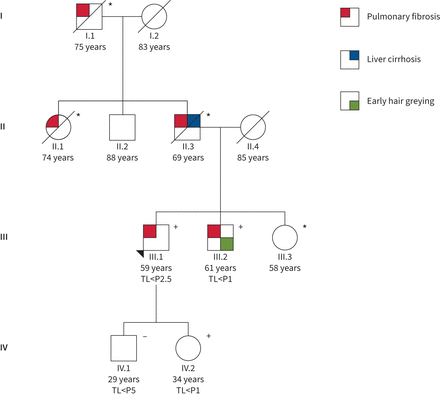
代表血统的家庭telomere-related与肺纤维化相关基因突变,肝硬化和头发灰白,常染色体遗传预期和端粒缩短传输。渊源者是由一个箭头表示。亲属IV.1 IV.2接受pre-symptomatic筛选。年龄是表示。+:突变的载体;−:非承运人的突变;*:未知基因状态。TL:端粒长度;P:百分位。
鲜为人知的发病机理,临床前疾病,定义为在起始的致病过程和疾病的发病症状。在基维辛迪,亲属患疾病。一些证据表明额外的遗传等因素MUC5B运输风险等位基因和环境因素,包括一个强大的吸烟的影响,在肺纤维化的发展发挥作用2,21,110年]。
先前的研究使用HRCT作为筛查工具非常一致的患病率间质变化在HRCT上无症状高危病人的亲属家族。这些研究包括亲戚> 18岁、40 - 65 (21],> 40 [110年,126年),48 (>129年)或在10年内最年轻的家庭成员的年龄诊断和报告14 - 25%的患病率间质变化在HRCT上首次筛选访问(21,110年,125年,126年,129年),
在无症状的亲戚间质HRCT异常的危险因素包括年龄、外围端粒长度短,肺泡上皮细胞端粒长度,SP-D血浆浓度升高和矩阵metalloproteinase-7 [2,21,110年,126年]。
间质HRCT的发展速度异常对临床疾病基维辛迪已经替代。一项研究在129年描述了放射性异常的变化自我报告的影响患者的直系亲属基维辛迪没有广泛的肺间质异常在第一次访问,其中25例(19.4%)开发广泛的HRCT异常或临床ILD 5年后报名(126年]。值得注意的是,63.3%的患者肺间质异常在有限的招生经验的发展相比,只有6.1%的病人与正常HRCT在基线126年]。
尽管在亲戚稀疏评估风险,初步研究表明,在HRCT上可见的变化可能与肺功能的减少。无症状的筛选与HRCT亲戚发现符合早期ILD的意思DLCO预测的97%,远远低于平均值DLCO105%的预测在亲戚正常HRCT扫描(125年]。同样,年代alisburyet al。(126年]发现高危亲属与肺间质异常了DLCO89%的与预计93%的亲戚没有肺间质异常。在无症状的患者家庭的亲属或零星ILD,间质异常有显著降低DLCO而预期的73%DLCO在那些没有间隙异常(预期的87%129年]。FVC与无症状通常是在正常价值在亲戚间质异常(21]。肺功能在正常范围内,因此不排除临床前疾病;然而,肺功能应该初步评估和跟踪的一部分。肺功能测量是相对非侵入性程序和年度重复检测显著减少或低于正常的值表明ILD的存在。
在家庭丹突变,无症状携带者的突变在ILD变化中能HRCT和略有减少DLCO相比同龄非承运人。与无症状的主题从家庭与肺纤维化,叔突变携带者明显减少DLCO77%的与88%的预测非承运人(36]。此外,短端粒syndrome-associated差异异常,如降低红细胞和血小板计数,和更高的平均微粒体积和平均微粒血红蛋白浓度,更频繁的早些时候头发老龄化和较短的端粒长度,在无症状的出现叔突变携带者(36]。
疾病外显率分析突变携带者和记录无症状高突变携带者是罕见的58]。这样的亲戚发现亚临床疾病的筛查与肺功能低于正常或ILD的成年人(HRCT的变化62年,130年- - - - - -132年]。在无症状的儿科运营商所知甚少。然而,一些儿童surfactant-related疾病显化在新生儿期疾病症状的消失(恢复56,133年]。他们的长期预后尚不得而知。
从研究结果筛选亲戚归纳了临床前ILD的风险表7。筛选标记包括HRCT、肺功能在一个家庭中带有未知疾病的遗传原因,和血液学的肝评估病人的亲属丹突变。
筛查是国家特定的可能性。亲戚的风险包括所有患者的直系家庭成员单基因或家庭ILD定义。适当的年龄开始筛查是未知的,而且可能取决于基因和疾病发病年龄的家庭成员,但这需要进一步的研究(2,10,26,128年]。
然而,在家庭与肺纤维化的发病年龄> 60岁,没有一个致病突变,也没有证据疾病预期,工作组成员通常提供筛选所有基维辛迪患者的直系亲属,与胸部HRCT扫描至少从50岁开始,或更早如果经历持续呼吸道症状或者听诊异常识别(129年]。
在儿童,病史的细节通常包括新生儿呼吸适应(呼吸支持必要的,多长时间),长期肺感染(> 2周),tachypnoea /呼吸道感染之间的呼吸困难,任何慢性呼吸道疾病,夜总会和肺功能的变化。总体而言,在HRCT上间隙变化可能表明临床前的疾病,但HRCT不是通常在每年重复。血液学的异常值和肺外症状可能通知短端粒综合症的发展。
在影响儿童的兄弟姐妹,工作组成员通常提供定期完成评估。然而,最佳的年龄进行检查和监测的频率将会在未来指定。
结论
这个声明的目的是协助卫生专业人员,基维辛迪病人和他们的家庭成员与单基因的治疗肺纤维化和基维辛迪。目前可用的数据和设备有限,我们希望这句话能帮助基因排序的实现在日常实践在世界范围内,尽管我们强调的重要性进行基因测序在高度专业化的中心,所有能力是可用的。此外,这句话总结了最相关信息分析/丹遗传学ILD和倡导未来临床试验以增加强有力的国际准则的证据基础。
补充材料
可共享的PDF
脚注
这个文档被人认可执行委员会2022年11月29日。
利益冲突:r . Borie报告从勃林格殷格翰集团资助,从赛诺菲咨询费,从勃林格殷格翰的发言演讲酬金,罗氏公司和赛诺菲安万特,和旅行勃林格殷格翰集团的支持,在提交工作。k·安东尼奥由于从勃林格殷格翰集团报告咨询费,罗氏公司和葛兰素史克公司,从勃林格殷格翰的发言演讲酬金,罗氏,基耶西,Menarini,葛兰素史克和阿斯利康,在提交工作。f . Bonella报告讲座从勃林格殷格翰集团谢礼,Fujirebio,加拉帕戈斯群岛NV罗氏,旅行勃林格殷格翰的发言和罗氏公司的支持,参与与勃林格殷格翰集团顾问委员会,BMS, Fujirebio,加拉帕戈斯群岛NV,葛兰素史克,罗氏,武田在提交工作。报告赠款BMS伽马安基丁酸,勃林格殷格翰集团和罗氏公司,从Apellis咨询费,从勃林格殷格翰的发言演讲酬金,BMS,罗氏公司,赛诺菲,诺华、阿斯利康和基耶西,从翻译简历,收到设备,在提交工作。a . Froidure报告从勃林格殷格翰集团拨款和咨询费,演讲酬金勃林格殷格翰集团和葛兰素史克和旅行赛诺菲的支持,在提交工作。l .加尔文报告旅行欧洲肺基金会的支持下,欧洲呼吸学会和欧洲的肺纤维化联合会领导角色与欧洲联合肺纤维化,爱尔兰肺纤维化协会,欧洲肺基金会188bet官网地址和欧洲罕见的呼吸道疾病,参考网络外的提交工作。m . Griese报告赠款和旅行勃林格殷格翰集团的支持,在提交工作。m . Molina-Molina报告赠款从罗氏公司和勃林格殷格翰的发言,从Esteve-Teijin咨询费,费雷尔,罗氏,勃林格殷格翰的发言,并从基耶西演讲酬金,罗氏,勃林格殷格翰的发言,在提交工作。诉Poletti报告讲座从AMBU谢礼,勃林格殷格翰的发言,ERBE和罗氏公司,在提交工作。大肠Renzoni报告从勃林格殷格翰集团资助,从勃林格殷格翰的发言和演讲酬金,罗氏、诺华,在提交工作。 C.H.M. van Moorsel reports grants from the European Respiratory Society supporting the present manuscript, grants and lecture honoraria from Boehringer Ingelheim, and a leadership role as co-chair of the European Respiratory Society Task Force for genetics in pulmonary fibrosis, outside the submitted work. All other authors have nothing to disclose.
支持声明:这项工作得到了欧洲呼吸学会。188bet官网地址资金信息,本文已沉积的Crossref资助者注册表。
- 收到了2022年7月9日。
- 接受2022年10月26日。
- 版权©2023年作者。生殖权利和权限接触权限在}{ersnet.org







{kind=link}
{kind=link}
{kind=link}