





抽象的
两项新研究证实了作用BMP9.突变是遗传PAH的一个原因,并提供了第一个证据BMP10也可能是因果的;这些结果对PAH的新治疗的发展具有相关性http://our.ly/uhmg30ncl3x.
血管保护的循环轴的循环轴产生了证据,其中分别从肝脏和右心房释放骨形态发生蛋白(BMPS)9和10,为血管内皮术提供滋补静态信号[1].这些配体以生理活性浓度在血浆中循环,刺激内皮细胞中的BMP信号通过包含活素受体样激酶1(ALK1;也称为ACVRL1)和骨形态发生蛋白受体类型2(BMPR2)的受体络合物(BMPR2)[2].我们现在知道,几乎只在内皮细胞上表达的ALK1/BMPR2受体复合物对BMP9和BMP10具有高亲和力(半最大有效浓度(EC50)50-100 pg·ml−1)但不绑定其他BMP [3.,4].很好地确定,编码BMPR2基因中的杂合酶突变是肺动脉高压(PAH)的主要遗传原因[5].此外,已知ALK1中的杂合酶突变在罕见的PAH病例中是因果的,尽管更常见的是遗传性出血性毛细诊炎[6].携带,人类遗传证据表明,负责维持内皮完整性和功能的关键BMP9-BMP10 / ALK1 / BMPR2轴(图1)。该轴在肺循环中似乎特别重要,其中Alk1和BMPR2的mRNA水平高于任何其他器官[7,这可能解释了肺动脉高压患者孤立性肺血管受累的原因。减少信号通过这种途径发生在PAH的遗传和非遗传原因中[8,9],促进内皮功能障碍,增加血管渗透性和凋亡,无序的血管生成,最终,PAH的病理变化[10.].
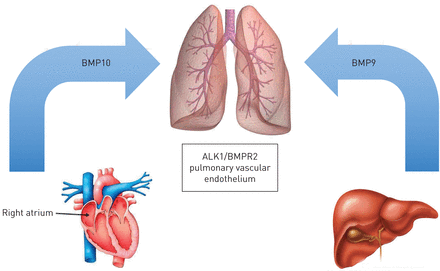
骨形态发生蛋白(BMP)9和骨形态发生蛋白10在血浆中以生理活性水平循环,并选择性地结合内皮受体复合物激活素受体样激酶1 (ALK1)/骨形态发生蛋白受体2型(BMPR2),以促进肺血管健康。突变、配体循环水平的缺失或受体的下调导致这条轴的破坏,促进了肺动脉高压的发展。
最近,在一个大型欧洲队列中发现了导致PAH的新基因,包括编码BMP9的杂合突变(误义和过早截断变异)(GDF2)作为PAH的重要原因[11.]加强BMP9信号通路的核心作用。最近的一份报告还证明,具有促进尿液高血压的患者具有非常低的BMP9血浆水平,这促进了肝硬化患者的PAH [12.].此外,我们之前已经证明了该途径的治疗潜力,因为给药重组BMP9逆转了几个PAH临床前模型的疾病[10.].
至关重要的是,BMP9.敲除小鼠没有表现出明显的血管表型(除了淋巴),这表明BMP9.在开发期间,损失在很大程度上得到了补偿。虽然循环BMP10水平不变BMP9.敲除小鼠,BMP10可以作为非活性蛋白循环[13.].最近的一项研究报道BMP9和BMP10作为生物活性异源二聚体循环[14.],乍一看与他们独特的组织表达模式发生冲突。对肝脏BMP9细胞来源的研究将肝星状细胞均为主要生产部位[15.],这也表达了低水平的BMP10 [14.,所以异二聚体可能确实有助于循环活性。
除了血管保护外,新的证据表明BMP9和BMP10也能保护成人心肌。BMP10在胚胎心脏中高度表达,成人的表达仅限于右心房[16.].BMP10基因敲除小鼠在胚胎第10天因心脏发育失败而死亡[17.].反过来,BMP10发育过程中的过度表达增加了心室肥大和三张分枝杆菌[17.].虽然BMP9.敲除小鼠表现出正常的心脏发育,最近的一项研究报道BMP9对心衰患者的心脏纤维化和心功能有有益的影响[18.].这些数据表明,成人心脏中BMP9和BMP10信号传导的丧失可能易于易于心肌功能障碍和纤维化,尽管需要在PAH的背景下的右心室研究。
在这一期的欧洲呼吸杂志,来自法国的两个基因研究[19.]和中国[20.,进一步了解BMP9和BMP10在PAH病理生物学中的作用。两项研究均独立证实了特发性PAH患者队列中BMP9可能致病突变的发生。两项研究均未报道BMP9突变携带者的家族史,表明两项研究都发生了突变新创或者,类似于BMPR2.突变,疾病基因渗透降低。法国学习eyrieset al。[19.],其中包括肺静脉闭塞性疾病(PVOD)患者,使用了一个定制的诊断基因面板,用于以前报道的PAH/PVOD基因,但也包括BMP10,因为它与BMP9相似(65%的氨基酸序列相同)。Eyrieset al。[19.]证实了突变BMPR2.是PAH最常见的遗传原因,其次是TBX4.,ALK1(也称为ACVRL1),BMP9.和Smad9。PAH的遗传和基因组学最近已审查[21].重要的是,突变TBX4.和BMP9.是高度有害的截断突变,与涉及单一氨基酸替代的错义突变相比,它更有可能被确定为致病状态。Eyrieset al。[19.进一步证实了双胞胎突变EIF2AK4[22]是散发性PVOD最常见的遗传原因(本系列中的13.3%)。发现一名患者被发现通过参考临床医生有PAH患者有双倍EIF2AK4突变,使它们被重新分类为PVOD,证实了这一方法在以前的研究中的效用[23].在基因面板中包含BMP10允许鉴定BMP10在两名患有严重PAH的年轻患者(11岁和28岁)中发现突变。其中一个突变被预测在生长因子结构域之前截断蛋白质,而另一个突变被预测破坏生长因子结构域的错义突变。尽管这些突变看起来具有令人信服的致病性,但仍有必要更全面地确定其功能效应,例如通过测量突变的BMP10水平和活性。11岁的病人,截骨BMP10突变还具有先天性心脏病的特征,与BMP10在心脏发育过程中的已知重要性保持一致。来自这个法国队列的另一个有趣的点是,在11个有家族史的PAH患者中,只有6个从基因面板测试中得到了基因诊断(这6个都是)BMPR2.突变)。虽然小组没有包含所有最近报告的PAH基因,但这种发现意味着大量患者尚未识别遗传诊断。这些患者可以通过全外销或全基因组测序提供进一步的无偏遗传检测,以寻找新的因果PAH基因。
中国的研究来自中国anget al。[20.]对331例成人和儿童发病的特发性PAH病例进行了全外显子组或全基因组DNA测序,对比对照组的10 508例。根据已建立的PAH基因进行遗传诊断的PAH患者所占比例[21在群组中较高(36.3%)比一般报道。类似于法国的研究yrieset al。[19.],四个床位EIF2AK4突变在PAH患者中被发现,允许将这些患者重新分类为PVOD。在PAH患者中,基因突变BMPR2.(19.5%)是最常见的,其次是ALK1(6%)和TBX4.(4%),其他基因在整个队列中所占比例均小于1%。在确定了这些已确定基因的突变频率后,Wanget al。[20.[进行了发现和复制队列的病例对照分析,以寻找其他基因中在全基因组水平上具有显著意义的罕见变异的过度代表。这导致了预测有害杂合突变的鉴定BMP9.在6.7%的病例(21例不同的突变中),使突变成为BMP9.中国队列中PAH最常见的原因。突变包括三个无意义的突变,两个框架和破坏翻译起始网站的一个框架。剩下的15个突变是畸形的。大多数(14个中的15个)的小姐突变完全没有来自中国血统的6512个人,并且预计常用算法都预计是有害的。为了克服呼叫罕见的畸形变体的潜在缺陷,没有功能实验数据,Wanget al。[20.]显示了一组错义突变导致转染细胞中成熟BMP9蛋白分泌减少,这与之前的报告相似[11.,或在报告细胞系中显示信号活性降低。最后,Wanget al。[20.通过ELISA测量的血浆BMP9 19BMP9.与年龄和性别匹配的特发性PAH患者和健康对照者进行比较。特发性肺动脉高压患者的中位血浆BMP9水平显著降低,携带肺动脉高压患者的中位血浆BMP9水平甚至更低BMP9.突变,虽然具有各种级别和群体之间的重叠值。这些测量提供了证据表明畸形突变BMP9.实际上影响配体的血浆水平,进一步增加了它们是致病性的证据负担。血浆BMP9活性的测量将提供进一步的确认。
携带,这些报告提供了重要的确认BMP9.突变是遗传PAH的一个原因,并提供了第一个证据BMP10也可能是因果关系。需要进一步确认BMP10的发现,同时进行功能研究。这些发现对开发PAH的新疗法有意义吗?当然,在BMP9突变患者中发现的BMP9水平降低,表明在这些患者中补充BMP9将是一种有吸引力的精准治疗方法。事实上,外源性BMP9可以提供一种直接、靶向的方法来恢复遗传和非遗传形式PAH中的生理ALK1/BMPR2内皮信号[10.],如果它可以安全地管理。这种方法现在正在积极发展。
脚注
利益冲突:N.W.Morrell在提交的工作之外向Morphogen-IX,GSK和Actelion报告个人费用;此外,他持有使用BMP9和BMP10作为疗法的专利,既挂起和许可的Myphogen-IX。
支持声明:N.W.Morrell由英国心脏基金会个人主席奖资助。本文的资助信息已被存入Crossref资助者注册表.
- 已收到2019年1月11日。
- 接受2019年1月13日。
- 版权所有©2019年











{kind=link}
{kind=link}