





文摘
我们评估治疗的安全性和耐受性pirfenidone (1602 - 2403 mg·−1)和nintedanib (200 - 300 mg·−1)患者的特发性肺纤维化(IPF)。
这24周,随访时间、非盲、第四阶段研究(ClinicalTrials.gov标识符NCT02598193)注册IPF患者用力肺活量% pred≥50%,肺的一氧化碳扩散能力% pred≥30%。启动nintedanib之前,病人收到pirfenidone≥16周和容忍一个稳定剂≥1602 mg·−1≥28天。主要终点是完成了24周的患者的比例组合治疗pirfenidone (1602 - 2403 mg·−1)和nintedanib (200 - 300 mg·−1)。调查人员记录治疗诱发不良事件(流泪),将他们pirfenidone, nintedanib,或没有。
89患者登记;73完成了24周的治疗(69会议的主要终点)和16过早停止治疗由于流泪(13)。74名患者有418治疗相关的流泪,腹泻,恶心和呕吐是最常见的。两名患者有严重的治疗相关的流泪。
pirfenidone和nintedanib用于24周被大多数IPF患者和容忍的流泪的预期关联到一个类似的模式要么单独治疗。这些结果与pirfenidone鼓励联合治疗的进一步研究和nintedanib IPF患者。
文摘
结合pirfenidone和nintedanib容忍了大多数IPF患者,鼓励进一步的研究http://ow.ly/1Iq030kaZuD
介绍
特发性肺纤维化(IPF)是一种进步的,不可逆转的,致命的,fibrosing肺病的原因不明1,2],存活率低于报道对于许多常见的癌症类型(1,3- - - - - -5]。Pirfenidone和nintedanib被批准为单方治疗IPF (6,7),都收到了有条件的推荐使用在2015年更新美国胸科学会(ATS) /欧洲呼吸学会(人)/日本呼吸协会(青年队)/拉丁美洲胸协会(ALAT)临床实践指南188bet官网地址8]。虽然pirfenidone和nintedanib都证明疗效在降低疾病进展与安慰剂相比,这种疾病是停止和逆转,继续体验肺功能下降,而治疗的患者(9- - - - - -11]。
不同的假定的机制的作用pirfenidone和nintedanib12- - - - - -14)提供一个生理原因结合这两个特工为了进一步减少IPF患者肺功能下降(15]。即使目标纤维级联两个代理,减少纤维母细胞和myofibroblast生产,积累的细胞外基质(12- - - - - -14,16,17),证据表明,他们可能目标纤维级联的不同方面。尽管它的作用机制尚未完全建立,pirfenidone已被证明对多个目标采取行动在体外,包括转变增长factor-β-triggered事件,通过glioma-associated致癌基因介导的同族体2 (12,13]。Nintedanib是一种酪氨酸激酶抑制剂,街区血小板源生长因子受体的胞内信号,纤维母细胞生长因子受体和血管内皮生长因子受体(14]。
Pirfenidone和nintedanib都与胃肠道不良事件(AEs),与Pirfenidone主要与恶心和nintedanib主要与腹泻相关(9,10,18]。一个小日本的随机、双盲、二期的剂量递增试验nintedanib (100 - 300 mg·−1)添加到正在进行的pirfenidone治疗(≤1800 mg·天−1为≤28天),其1年,非盲扩展,表明联合治疗的安全性和耐受性IPF患者符合AE概要文件为每个单独药物(19,20.]。同样,最近INJOURNEY试验,一个非盲、随机试验的pirfenidone (≤2403 mg·−1)添加到nintedanib治疗(≤300 mg·天−112周),还发现,联合治疗的安全性和耐受性方面更好可比与个人药品(21]。然而,有限的数据联合治疗的长期安全。在这里,我们从一个24周的研究调查报告结果的安全性和耐受性增加nintedanib稳定pirfenidone IPF患者治疗。
方法
病人
符合条件的患者年龄在40 - 80岁开始检查,与IPF基于ATS /人/青年队/ ALAT 2011指南(2]。病人收到pirfenidone≥16周和容忍一个稳定剂1602 - 2403 mg·天−1≥28天没有经历任何中度或重度不良反应被研究者认为是pirfenidone有关,或pirfenidone治疗中断> 7天任何理由。病人也有用力肺活量(FVC) % pred≥50%,一氧化碳(肺的扩散能力DLCO在筛选)% pred≥30%。从每个病人知情同意了之前研究活动或进行治疗。其他包含和排除标准中列出补充表S1。
研究设计
这是一个国际,随访时间、非盲、第四阶段研究(ClinicalTrials.gov标识符NCT02598193),评估24周的治疗的安全性和耐受性与pirfenidone (1602 - 2403 mg·−1)和nintedanib (200 - 300 mg·−1)IPF患者(图1)。病人开始nintedanib治疗添加到稳定pirfenidone治疗研究的第一天。联合治疗政府进一步描述补充的方法。
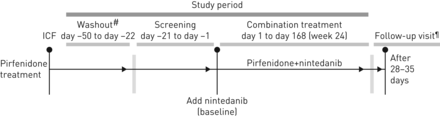
研究设计。ICF:知情同意的形式。#:接受禁止药物停用药物的病人在冲刷(其他病人直接进入到筛查);¶:发生28-35天结束后综合治疗患者完成了24周治疗期组合,或决定停止nintedanib 28-35天后患者过早停止或中断治疗≥连续28天。
在描述的研究药物禁止补充表S2。
评估
的主要目标是调查的安全性和耐受性增加nintedanib稳定pirfenidone IPF患者治疗。这是评估与下面的端点。1)主要终点:完成了24周的联合治疗的患者的比例在pirfenidone (1602 - 2403 mg·−1)和nintedanib (200 - 300 mg·−1)。2)二级终端:1)的比例的患者停止pirfenidone, nintedanib或两者治疗24周之前由于治疗诱发的AEs(流泪;联合治疗,直到开始后的AEs发生后28天的最后剂量pirfenidone nintedanib), b)总病人天的联合治疗,c)总持续时间天发起联合治疗中止pirfenidone, nintedanib或治疗,和d)的频率和时间流泪和严重的流泪。改变从基线FVC,DLCO和王短暂的间质性肺病(K-BILD)问卷(22分数被评估为探索性的端点。进一步研究评估中所描述的细节补充的方法。
统计分析
本研究计划招收80名患者基于描述的样本容量预测补充的方法。
人口安全被定义为所有病人的人口登记接受至少一个剂量的pirfenidone或nintedanib基线。比例常数计算基于数量的患者数据。置信区间是基于二项分布。这是一个安全性和耐受性研究中,只有一个治疗组,用来评估联合治疗的效果。因此,没有正式的统计假设被评估和分析局限于描述性统计对多样性的端点或没有调整within-subgroup比较。
结果
总共89名患者来自36个中心在加拿大、丹麦、法国、德国、意大利、荷兰、西班牙和美国在2016年1月至11月之间。基线病人的人口统计数据和特点所示表1。患者平均±seFVC % pred 71.8±1.7%DLCO在基线% pred 48.4±1.3% (表1)。测量FVC和DLCO最近6个月前开始筛选意味着±se72.6±1.7%和50.0±1.4% (表1)。
最常报道的病史首选项(> 10%的患者):gastro-oesophageal返流性疾病(49例(55%))、高血压(44例(49%)),hyperlipidaemia(20例(23%)),咳嗽(18例(20%)),季节性过敏(17例(19%)),IPF(16个病人(18%);进入IPF病史是可选的),睡眠呼吸暂停综合征(16例(18%)),鸡蛋(14例(16%))、糖尿病(13例(15%)),骨关节炎(12例(14%)),过敏性鼻炎(11例(12%),甲状腺功能减退(11例(12%))、抑郁(10例(11%)),焦虑(9例(10%))、失眠(9个病人(10%))、良性前列腺增生(9例(10%))和肥胖(9例(10%))。
最常报道的药物的患者(> 25%):质子泵抑制剂(66名患者(74%),支气管扩张剂和平喘药(如。舒喘灵、ipratropium和布地奈德;52例(58%),他汀类药物(41例(46%)),维生素和矿物质(41例(46%)),类固醇(38例(43%))、水杨酸盐(35例(39%)),antidiarrhoeals(28例(32%)),抗组胺药(26例(29%))、草药、顺势疗法和膳食补充剂(24例(27%),及非甾体类抗炎药(23例(26%))。
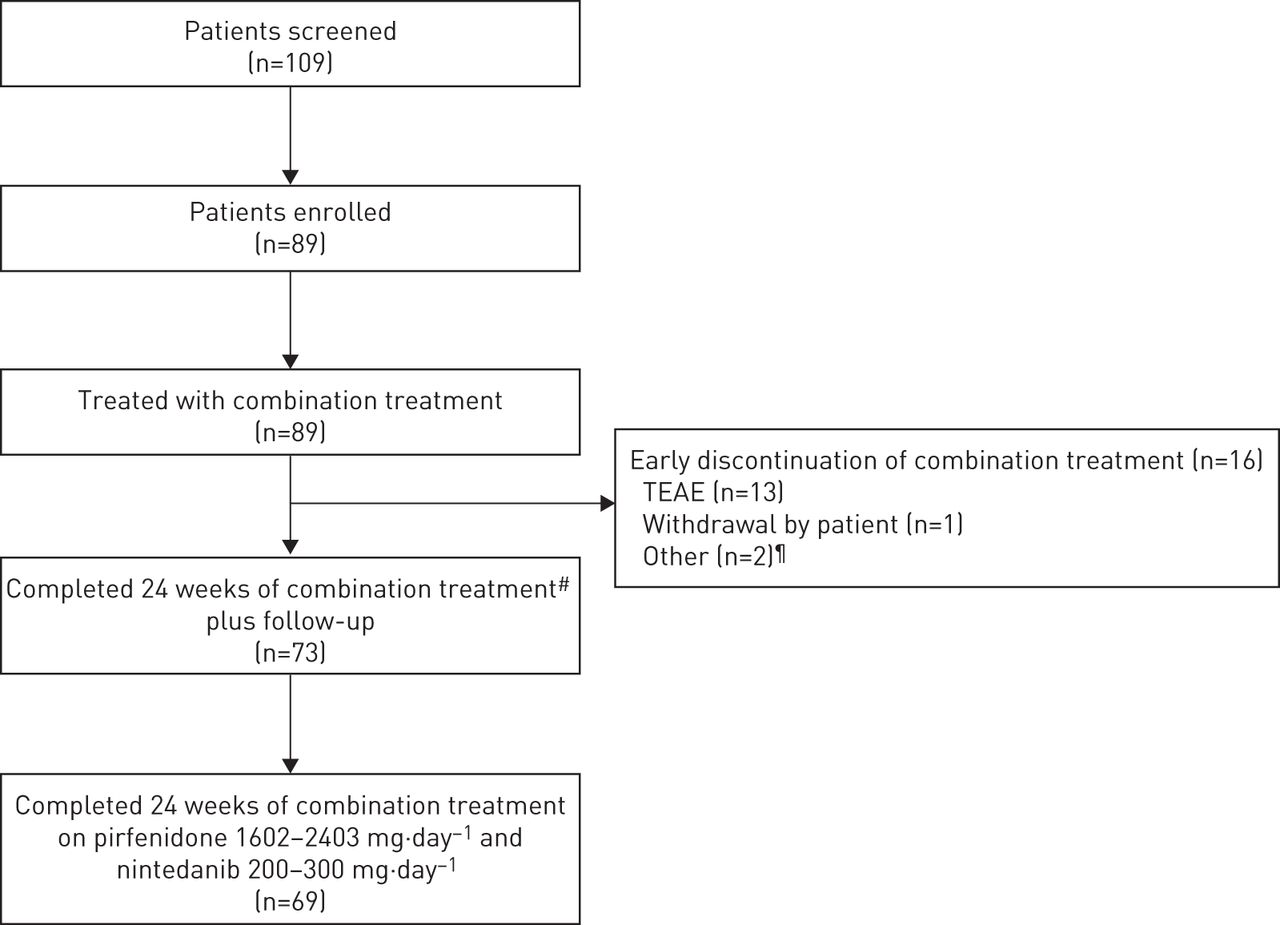
73 89(82%,95%置信区间72.5 - -89.4%)患者完成了24周的治疗以及随访和69年的89名患者(78%,95%置信区间67.4 - -85.7%)会见了主要终点(完成24周的治疗剂量的组合pirfenidone 1602 - 2403 mg·天−1和nintedanib 200 - 300 mg·−1)(图2)。16个病人过早停止治疗,13个病人停止流泪,一个病人撤回了自己的研究和两个病人停止其他原因(一个病人不想继续nintedanib,另一个是肺移植)上市(图2)。
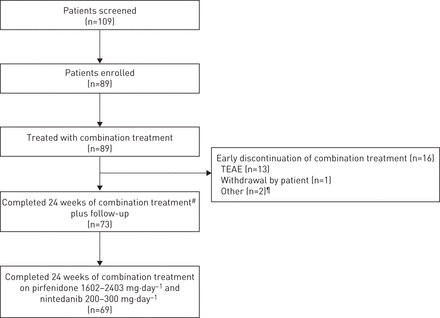
耐心的性格。TEAE:治疗诱发的不良事件。#:结合pirfenidone和nintedanib在任何剂量;¶:一个活跃的肺移植名单中列出的病人,一个病人不愿采取nintedanib。
均值±sd每日剂量联合治疗期间采取pirfenidone 2339.3±183.7 mg·天−1和nintedanib 255.4±43.7 mg·−1(包括nintedanib滴定时间剂量)。虽然78个病人(88%)接受>的目标剂量1602毫克·−1pirfenidone, > 200 mg·天−1nintedanib, 61名患者(69%)接受不到pirfenidone的剂量(2403毫克·天−1)和/或nintedanib (287 mg·−1当包括nintedanib滴定剂量)整体在研究;大多数病人因为临时减少剂量和/或中断后流泪。病人的总数与pirfenidone天的联合治疗,nintedanib 13岁304天,不包括剂量中断(预期的治疗病人天14岁952天)。均值±sd治疗时间在研究期间为21.3±6.3,20.8±6.2,21.4±6.3周,pirfenidone nintedanib和联合治疗,分别包括剂量中断。接触分析认为治疗管理从药物联合治疗的开始,直到研究完成/中止从联合治疗早期(按电子病例报告形式),中断完成之前的药物组合治疗期导致个体治疗的平均持续时间短于,为联合治疗记录。
23个患者(26%)治疗中断:一个病人(1%)有一个中断pirfenidone仅16个病人(18%)有28个中断nintedanib孤独和六名病人(7%)有一个中断的联合治疗(同时pirfenidone和nintedanib中断);中断的联合治疗持续了平均时间是4.3天。意味着所有pirfenidone和nintedanib中断的长度是7.6和10.3天,分别。
88名患者(99%)有经验670流泪;这些被认为是相关治疗74个病人(83%;418年治疗相关的流泪)(表2)。中位数(四分位范围)期间,流泪是1.9(0.3 - -11.3),0.4(0.1 - -7.1)和0.5(0.1 - -6.4)周有关pirfenidone流泪认为,nintedanib pirfenidone和nintedanib分别。67名患者(75%)有经验的胃肠治疗相关的流泪;胃肠道事件的大多数被调查者认为独自nintedanib(61名患者)。7例(8%)有经验治疗相关的光敏性或皮疹流泪(表2)。
13例(15%)停止治疗由于流泪;这些流泪是归因于nintedanib独自在10位病人(11%)、pirfenidone和nintedanib一个病人(1%),和治疗被认为是不相关的两个病人(2%),由调查员评估(表2)。停药的联合治疗发生在整个试验和没有明确的模式在早期停用的时间治疗期(图3)。
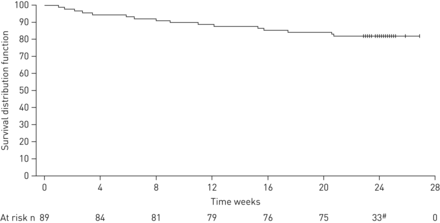
时间停止人口(安全):包括早期停用或研究完成时间。#:40例完成了计划研究前的联合治疗24周一天168,因此审查kaplan meier曲线(由竖线)。
18例(20%)遭遇了严重不良事件(常见的术语标准等级≥3)流泪;这些被认为是治疗相关的六个患者(7%)(表3)。16个病人(18%)有经验的18严重流泪(表3和补充表S3);两个病人(2%)有经验的一个严重的治疗相关的TEAE,两者都是归因于nintedanib孤单:一个病人经历了短暂性缺血性发作但继续联合治疗没有修改,和一个病人经历了深静脉血栓形成和停止nintedanib但pirfenidone继续治疗。没有致命的流泪是在研究报道。
治疗相关的肝流泪是6个病人(7%);这是归因于nintedanib独自在5例(表3)。所有事件都是1级(n = 4)或2级(n = 1)海拔在肝酶,或1级肝脏功能异常(n = 1)。
在一个探索性的分析效果,意味着±seFVC % pred及DLCO% pred下降了0.8±0.6%和1.4±0.8%,分别从历史值基线和0.4±0.5%和1.9±0.8%,分别从基线到24周的患者可用数据(补充图S1)。探索性分析K-BILD从基线到24周发现意味着±sd总分下降了1.3±8.7,意味着±sd个人部分的得分下降了2.4±14.3“心理”域,0.7±15.9“呼吸困难和活动”的领域,和3.1±17.0“胸部症状”域联合治疗的患者完成24周;这些变化可能是由于机会的标准差比降低(补充图S2)。
讨论
本研究发现,联合治疗pirfenidone (1602 - 2403 mg·−1)和nintedanib (200 - 300 mg·−1)在89年为24周IPF患者完成78%的病人。联合治疗的安全性并没有揭示不同模式的流泪独自预期与治疗。大多数流泪影响胃肠道系统和轻度至中度的严重性。此外,联合治疗24周与肝毒性的风险增加无关或光敏性。然而,流泪的频率(99%)高为6个月的研究中,比较和频率在12个月pirfenidone nintedanib单一疗法试验(18,24];虽然常见胃肠道流泪的6个月后较少发生pirfenidone或nintedanib单一疗法在这些研究[25,26]。
24周的研究结果与最近的12周INJOURNEY试验的总体TEAE频率(21]。两项研究发现,联合治疗与nintedanib pirfenidone有类似的安全性pirfenidone或nintedanib单一疗法的病人经历流泪比例和类型的流泪。在当前的研究中,病人已经容忍一个稳定剂pirfenidone nintedanib启动之前,这或许可以解释流泪归因于nintedanib发病率就越高与pirfenidone调查人员。同样,INJOURNEY试验(启动前病人已经耐受性nintedanib pirfenidone)联合治疗组中发现更多的病人停止pirfenidone nintedanib相比,尽管它应该注意的是,这项研究协议之前推荐的剂量减少pirfenidone nintedanib剂量管理以外的AEs腹泻(21]。
在这项研究中,治疗相关的腹泻(49%的患者)报告比任何更频繁腹泻流泪的先前的研究pirfenidone单一疗法(25%)(24)和联合治疗的INJOURNEY试验(38%)(21),但比任何腹泻次数少流泪的先前的研究nintedanib单一疗法(62%)(18]。此外,治疗相关的恶心(46%的患者)报告了更多的病人比先前的研究中任何恶心流泪pirfenidone或nintedanib单一疗法(分别为36%和24%)18,24]和相当恶心流泪在INJOURNEY试验报告(42%)21];然而,这两项研究的持续时间是不同的,这样比较困难。
虽然有些流泪的频率是在这个24周研究高于仅在每个治疗的研究,停药率(18%和15%由于流泪)数值低于12周以上INJOURNEY试验和在52周INPULSIS试验,和类似的数值超过52周或更多的提升和能力试验9- - - - - -11,21]。现实世界的数据来自美国建议停药率pirfenidone和nintedanib较高临床实践(nintedanib pirfenidone为24%和34%;平均随访8 - 9个月)比在临床试验中6,7,27]。因此,这将是重要的进一步调查联合治疗的耐受性和发展战略来帮助减轻胃肠道的负担流泪。目前的策略来减少恶心与pirfenidone包括在每个剂量与食物;减少腹泻或恶心与nintedanib,确保足够的水合作用和使用antidiarrhoea或止吐剂药物治疗建议(6,7,28,29日]。剂量调整也推荐管理AEs pirfenidone和nintedanib [6,7),和可用数据表明,剂量调整治疗的持久性(产生积极的影响30.)在维持治疗的好处FVC下降(31日]。在这项研究中,26%的患者进行了联合治疗的中断,但只有7%都同时治疗中断(中断的长度平均∼4天)。这种整体速度类似于其他临床试验数据pirfenidone单一疗法(25%)(31日]。
探索性功效分析发现,几乎没有肺功能下降与pirfenidone和nintedanib联合治疗期间,这是类似于INJOURNEY试验结果(21]。然而,由于缺乏一个对照组和更大的样本量,没有确定的结论可以得出关于IPF患者联合治疗的效果。生活质量,使用K-BILD问卷测量,没有恶化的患者完成了24周的治疗,这是符合生活质量调查结果从EuroQoL-5D INJOURNEY试验中使用的调查问卷(21]。
结合疗法的好处与不同机制的行动证实了多种慢性疾病,包括慢性阻塞性肺疾病、哮喘和肺动脉高血压(PAH) [32,33]。PAH长期研究的证据表明联合治疗靶向内皮素、一氧化氮和/或环前列腺素途径有可能延缓疾病进展(33]。事实上,PAH的治疗策略是转向使用一线联合治疗(32]。目前的研究结果表明,联合治疗与pirfenidone nintedanib IPF患者可以提供一个可行的未来选择。然而,联合治疗可能不是适合所有患者,并且需要进一步的研究来研究长期联合治疗的益处和风险控制研究与治疗持续时间> 6个月。
本研究的局限性包括缺乏控制或比较器组和小样本大小,这是由于探索性研究的性质。此外,只有病人容忍pirfenidone被包括在内,这可能引入一个偏向患者不太可能经历流泪,和病人可以治疗中断连续< 28天,仍然被认为已经完成了24周的联合治疗。因此,宽容在实际治疗可能会反映在完成率不到。鉴于pirfenidone nintedanib治疗会导致类似的AEs (9,10,18),可能是不确定性关于是否有专门pirfenidone或nintedanib流泪有关。nintedanib加入稳定pirfenidone治疗在这项研究中,结论不能pirfenidone添加到稳定nintedanib治疗或治疗都同时开始。虽然更高级的IPF患者(FVC % pred < 50%DLCO% pred < 30%)也可能会受益于联合治疗,这些患者被排除在审判。然而,合格标准的关键试验(9- - - - - -11),允许之间的安全数据比较pirfenidone单一疗法的研究和临床试验。最后,FVC的变化,DLCO作为探索性研究和K-BILD测试端点,这项研究目的不是正式评估联合治疗的疗效;历史价值被用来比较预处理FVC和变化DLCO。
结论
结合使用pirfenidone和nintedanib 24周被大多数IPF患者,容忍和与类似的流泪和停药率预期与单独治疗。这个试验的结果添加到有限的安全数据的联合治疗> 3个月,并鼓励进一步的研究来确定pirfenidone的疗效和安全性,nintedanib作为联合治疗与单一疗法在IPF患者。
补充材料
补充材料
请注意:补充材料并不是由编辑部,编辑和上传已由作者提供。
补充方法。erj - 00230 - 2018 - _supplement
补充表S1。额外的进入标准文本中没有描述。erj - 00230 - 2018 - _supplementary_table_s1
补充表S2。药物禁止在28天前筛查和在研究的开始。erj - 00230 - 2018 - _supplementary_table_s2
补充表S3。人口治疗诱发的严重不良事件(安全)。erj - 00230 - 2018 - _supplementary_table_s3
补充图S1。探索性的分析预测百分比变化)FVC和b)DLCO人口(安全)。*:肺功能值最接近6个月前开始筛选。DLCO:一氧化碳扩散能力;FVC:用力肺活量;SE:标准误差。erj - 00230 - 2018 - _supplementary_figure_s1
补充图S2。探索性分析K-BILD人口(安全)。*:所有患者non-missing数据。芭:呼吸困难和活动;胸部:症状;K-BILD:王短暂的间质性肺疾病问卷;心理学:心理域;SD:标准差。erj - 00230 - 2018 - _supplementary_figure_s2
确认
医学写作提供的支持是丽贝卡水域代表CMC亲和力,完成医疗通信有限公司的一个部门,曼彻斯特,英国由f .罗氏公司,作者要感谢病人参与试验和研究团队,促进了审判。这项研究的独立的数据监测委员会成员是玛丽莲·k·Glassberg(椅子;美国佛罗里达州迈阿密大学、迈阿密、),乌尔里希Costabel (Ruhrlandklinik、埃森、德国)和Diethelm梅辛杰卖力地(PROMETRIS GmbH,曼海姆,德国)。
脚注
本文根据修正修正发表在2018年10月出版的欧洲呼吸杂志。
这项研究是在注册ClinicalTrials.gov标识号NCT02598193。
调查人员名单:Charlene d(卡尔加里,加拿大),Shane Shapera(加拿大多伦多),sah b瓶(Hellerup、丹麦),文森特Cottin(布朗、法国),史蒂芬Jouneau(雷恩、法国),希拉里奥Nunes(博比尼、法国),弗朗西斯科·托马索Bonella(德国埃森),德克Koschel (Coswig、德国),菲利普Markart(富尔达,德国),卡洛Albera(当过,意大利),阿尔贝托Pesci(意大利蒙扎),Paola Rottoli(意大利锡耶纳),劳拉Tavanti (Cisanello、意大利),马塞尔Veltkamp (Nieuwegein、荷兰),土地肥沃的Wijsenbeek(鹿特丹,荷兰),奥兰多Acosta (La拉古纳、西班牙),埃琳娜豌豆粉油煎饼(Leon,西班牙),Casanova阿尔瓦罗·埃斯皮诺萨(马德里,西班牙),胡安Gustavo(瓦伦西亚,西班牙),玛丽亚Molina-Molina(西班牙巴塞罗那),何塞·a . Rodriguez-Portal(西班牙塞维利亚),克劳迪娅Valenzuela(马德里,西班牙),约翰Belperio(美国洛杉矶CA),约翰·巴特勒(波特兰,或美国),Sakshi Dua(美国纽约)、凯文·r·费海提(美国安阿伯市MI),托德Horiuchi(美国佛罗里达州萨拉索塔),j . Terrill哈金斯(美国查尔斯顿,SC) Gaurav卡纳(美国辛辛那提,哦),克里斯托弗·王(美国弗吉尼亚州瀑布教堂),安德鲁Labelle(切斯特菲尔德,密苏里州,美国),丽莎·h·兰开斯特(美国纳什维尔,TN),约书亚穆尼(美国斯坦福大学,CA),李明日(美国奥马哈,NE),尼娜·帕特尔(美国纽约),Murali Ramaswamy(格林斯博罗、数控、美国),罗伯特•苏斯曼(美国新泽西的斯普林菲尔德,多),约瑟夫Zibrak(美国波士顿)。
可以从本文的补充材料www.qdcxjkg.com
作者的贡献:所有作者都参与这项研究的设计和研究结果的解释,从一开始就导致了手稿,阅读和批准了最终稿。所有作者保证内容的准确性包括在最后的手稿。
利益冲突:投资者Flaherty收到f .罗氏公司的顾问和研究支持费用/基因泰克和勃林格殷格翰的发言,从FibroGen顾问费用,Veracyte,埃俄罗斯,PharmaKea Sanofi-Genzyme Celgene公司,研究支持从传入医药费用。C.D.下跌已收到顾问和研究支持费用从f .罗氏公司加拿大,从勃林格殷格翰集团顾问费用。j.t哈金斯得到了顾问和研究支持费用从f .罗氏公司/基因泰克和勃林格殷格翰的发言,并从基列和iBIOS顾问费用。h . Nunes已经收到f .罗氏公司的顾问和研究支持费用/基因泰克和勃林格殷格翰的发言,并临床试验的研究者赛诺菲和基列地。r·苏斯曼已经收到了研究支持和演讲者和咨询费用从f .罗氏公司/基因泰克和勃林格殷格翰集团,和研究支持在基列山旁。c . Valenzuela已经收到了演讲者和咨询费用从f .罗氏公司和勃林格殷格翰的发言。J.L. Stauffer基因泰克公司的一名雇员和前雇员FibroGen,来自并持有股票。f . Gilberg f .罗氏公司的员工。m . Bengus f .罗氏公司的员工。m . Wijsenbeek已经收到了演讲者和咨询费用和不受限制的研究经费从f .罗氏公司和勃林格殷格翰的发言,从加拉帕戈斯群岛和咨询费用; all fees and grants were paid to her institution.
支持声明:这个试验是由罗氏公司,有限公司融资信息本文已沉积的Crossref资助者注册表。
- 收到了2018年2月1日。
- 接受2018年5月16日。
- 版权©2018人队
这收获打开文章是开放和分布式根据创作共用署名非商业188滚球软件性4.0许可证。











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}