





摘要
已经提高了线粒体反应性氧物质(ROS),特别是超氧化物,已提出介绍缺氧肺癌(HPV),慢性缺氧诱导的肺动脉高压和右心室重塑。
我们确定了急性和慢性缺氧的ROS,并研究了线粒体靶向抗氧化丝痘在这些条件下的影响。
MITOQ或其惰性载体物质,甲基苯基膦鎓对急性HPV的影响(1%O2在孤立的无菌灌注小鼠肺中研究了10分钟)。暴露于慢性缺氧的小鼠(10%O24周)或在主要肺动脉的绑扎后用米托克或甲基三苯基鏻处理(50mg·kg-1·天-1)。
肺动脉平滑肌细胞中总细胞超氧化物和线粒体ROS水平增加,但急性缺氧中的肺成纤维细胞中减少。MITOQ显着抑制了超氧化物浓度的HPV和急性缺氧诱导的升高。肺动脉平滑肌细胞中的ROS减少,而慢性缺氧后右心室增加。相应地,MITOQ并未影响慢性缺氧诱导的肺动脉高压的发展,但在慢性缺氧和肺动脉带后衰减右心室重塑。
增加了肺动脉平滑肌细胞的线粒体ros介导急性HPV,但不是慢性缺氧诱导的肺动脉高压。在夸张的急性HPV条件下,MITOQ可能是有益的。
摘要
线粒体靶向抗氧化型米多克衰减急性缺氧肺血管收缩和RV重塑,但不是慢性缺氧诱导的pH值http:///wly/pccu30hsjkk.
介绍
缺氧肺血管收缩(HPV)是毛细血管前肺血管对肺泡缺氧的反应,并用于保持通风灌注匹配,从而优化血液的氧气[1]。相比之下,广义慢性缺氧(例如在慢性阻塞性肺部疾病中,导致肺动脉高压的发育,这是一种以肺血管重塑为特征的渐进疾病,最终导致右心力衰竭[1]。除了血管改造外,广义的HPV还有助于慢性缺氧诱导的肺动脉高压血管抗性增加。
肺动脉平滑肌细胞(PASMC)是这些反应中的关键球员,因为它们对急性缺氧与收缩以及慢性缺氧反应,即使孤立时2那3.]。已经提出线粒体反应性氧物质(ROS)通过与蛋白激酶,磷脂酶和离子通道诱导细胞内钙释放的或通过稳定转录因子的稳定性来发挥至关重要的作用1那4.]。此外,ROS可能参与右心室重塑的发展[5.]。但是,如果肺脉管系统和右心室的急性和慢性缺氧期间ROS增加或减少,则仍然不明确,以及哪种物种(超氧化物或过氧化氢(H.2O.2))触发这些响应[1那3.那6.]。未特异性硫醇化合物的应用N- 减轻许多ROS工艺的乙酰琥珀酰琥珀酰琥珀酰琥珀酸胞嘧啶可抑制缺氧诱导的肺动脉高血压[7.]。线粒体超氧化物歧化酶2(SOD2)的过度表达增加和过表达线粒体靶向过缩酶的过表达降低肺血管重塑,表明线粒体H增加2O.2有助于缺氧诱导的肺动脉高压[8.]。
MitoQ是一种口服线粒体靶向抗氧化剂,由与三苯基膦(TPP)连接的泛醌部分组成+)分子[9.]. 亲脂性TPP+阳离子允许MITOQ通过磷脂双层并积聚由线粒体膜电位驱动的线粒体内膜内[9.]。带正电荷的残留物(TPP+)米托克被吸附到基质表面,而疏水末端(泛醌)插入线粒体内膜的疏水核中[10]。Mitoq,ubiquinol的活性抗氧化形式通过ROS氧化为无活性形式,泛醌,其通过呼吸链的复合II持续再循环到其活性泛醇形式[9.]。MITOQ是一种针对脂质过氧化,过氧化物,水过氧基自由基和超氧化物的有效抗氧化剂,但其与H的反应性2O.2可以忽略不计[10-12]。但是,在特定条件下,特别是在非膜结合形式中,MITOQ也可以增加超氧化物产生[9.那13],这可能只发生体外[9.]. MitoQ已被证明对几种ROS介导的病理具有保护作用,包括心血管疾病[14]和败血症[15]在动物模型和人类中[16]. 作为超氧化物和H2O.2建议调节慢性缺氧诱导的肺动脉高压的发展,MITOQ提供了特异性地靶向线粒体ROS和治疗氧化损伤的可能性。
因此,我们旨在阐明线粒体来源的活性氧在HPV、慢性缺氧诱导的肺动脉高压和右心室重构中的作用。我们假设线粒体靶向抗氧化剂MitoQ有助于1)研究线粒体ROS在这些过程中的作用,2)作为一种可能的治疗工具。
材料和方法
动物实验和试剂
动物实验得到了政府当局的批准。对任意性别的C57BL/6J小鼠进行了研究。除非另有说明,所有试剂均来自Sigma Aldrich(美国密苏里州圣路易斯)。MitoQ和癸基TPP+由Antipodean Pharmaceuticals(新西兰奥克兰)提供。
孤立的无菌灌注和通风的小鼠肺
如前所述分离出小鼠的肺部,通风和无血灌注[17]。
慢性低氧暴露、肺动脉环扎和米托Q治疗
对于慢性缺氧诱导的肺动脉高压,小鼠保存在源极缺氧(10%O2)28天[18]。如前所述进行主要肺动脉(肺动脉带(PAB))[19]. 在慢性缺氧孵育期间和PAB后,小鼠接受50 mg·kg-1·天-1MITOQ或TPP.+溶于H2o通过gavage [9.]。
电子自旋共振光谱和荧光方法测量ROS释放
通过Bruker电子旋转共振(EMXmicro; Bruker Biospin,Rheinstetten,Germany),在PASMC和组织中测定内细胞内和无反应性氮物质(RNS)的ROS和反应性氮物质(RNS),使用旋转探针1-羟基-3-甲氧基羰基-2,2,5,5-四甲基吡咯烷(CMH; 0.5mm)[23那24]. CMH信号的超氧部分通过减去用50%乙醇孵育的样品的ESR信号来确定 U·mL-1聚(乙二醇) - 仅来自CMH样品的信号的缀合的SOD(PSOD)和CMH。
通过荧光染料Mitosox测量线粒体ROS。基于蛋白质的荧光传感器超级细胞用于检测细胞溶质H.2O.2专注 [25]。
肺部和心脏的巨型浓度
通过反相液相色谱和串联质谱法测量MITOQ浓度[26]。
增殖试验
通过测定由5-乙炔脲标记的增殖细胞与Hoechst染色标记的总细胞数进行测定来评价PASMC的增殖。18]。
额外的材料和方法
提供额外的材料和方法补充材料。
统计方法
数值表示为平均值±SEM.。通过在两种实验组的实验中与威尔士策略的T检验和具有两个以上实验组的实验中的一种或双向ANOVA来计算数据的统计学意义。双向ANOVA用于验证治疗因素之间的相互作用(TPP+相对mitoq)和暴露(常氧/缺氧或pab / sham)。p值<0.05被认为是显着的。
结果
MitoQ抑制急性HPV和缺氧诱导的PASMC超氧化物增加
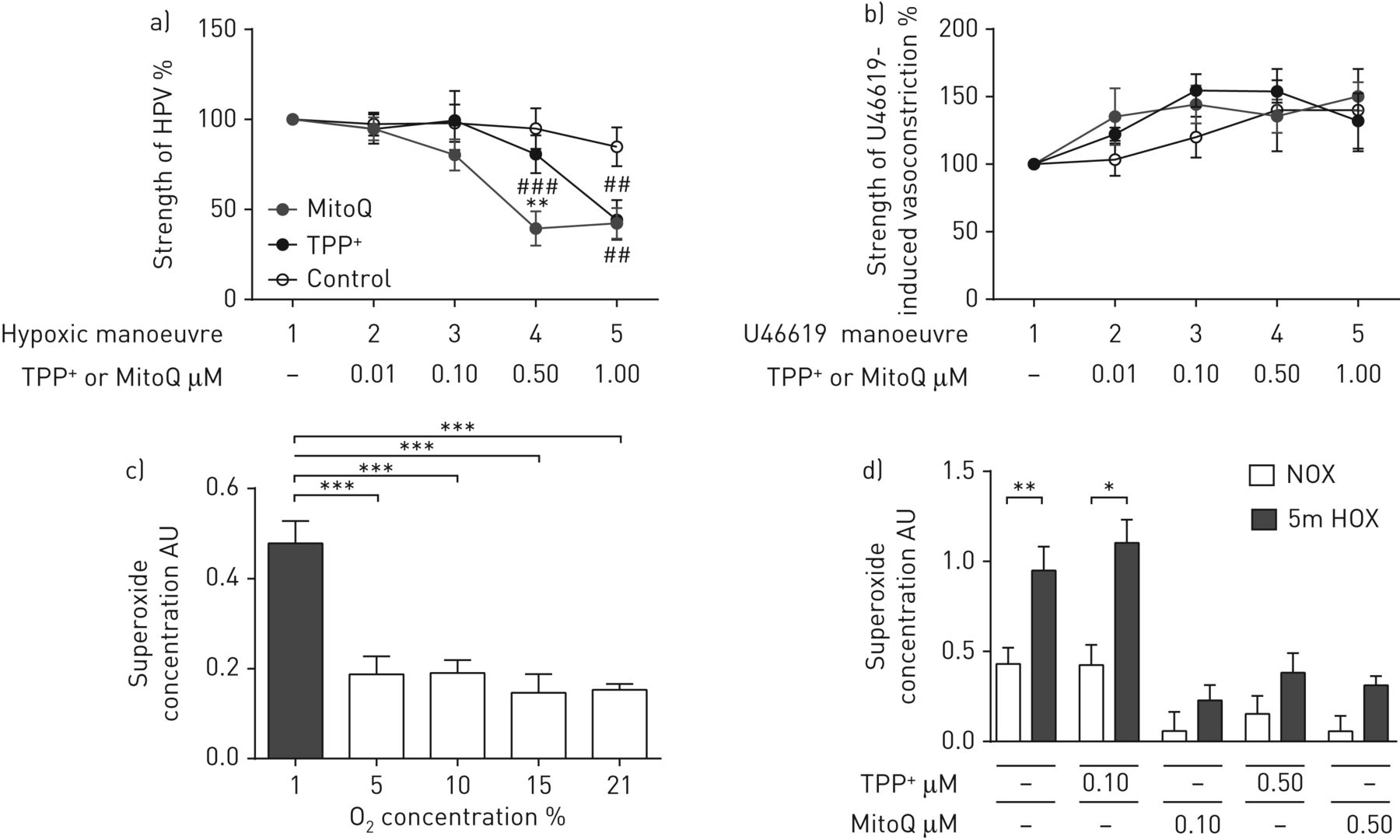
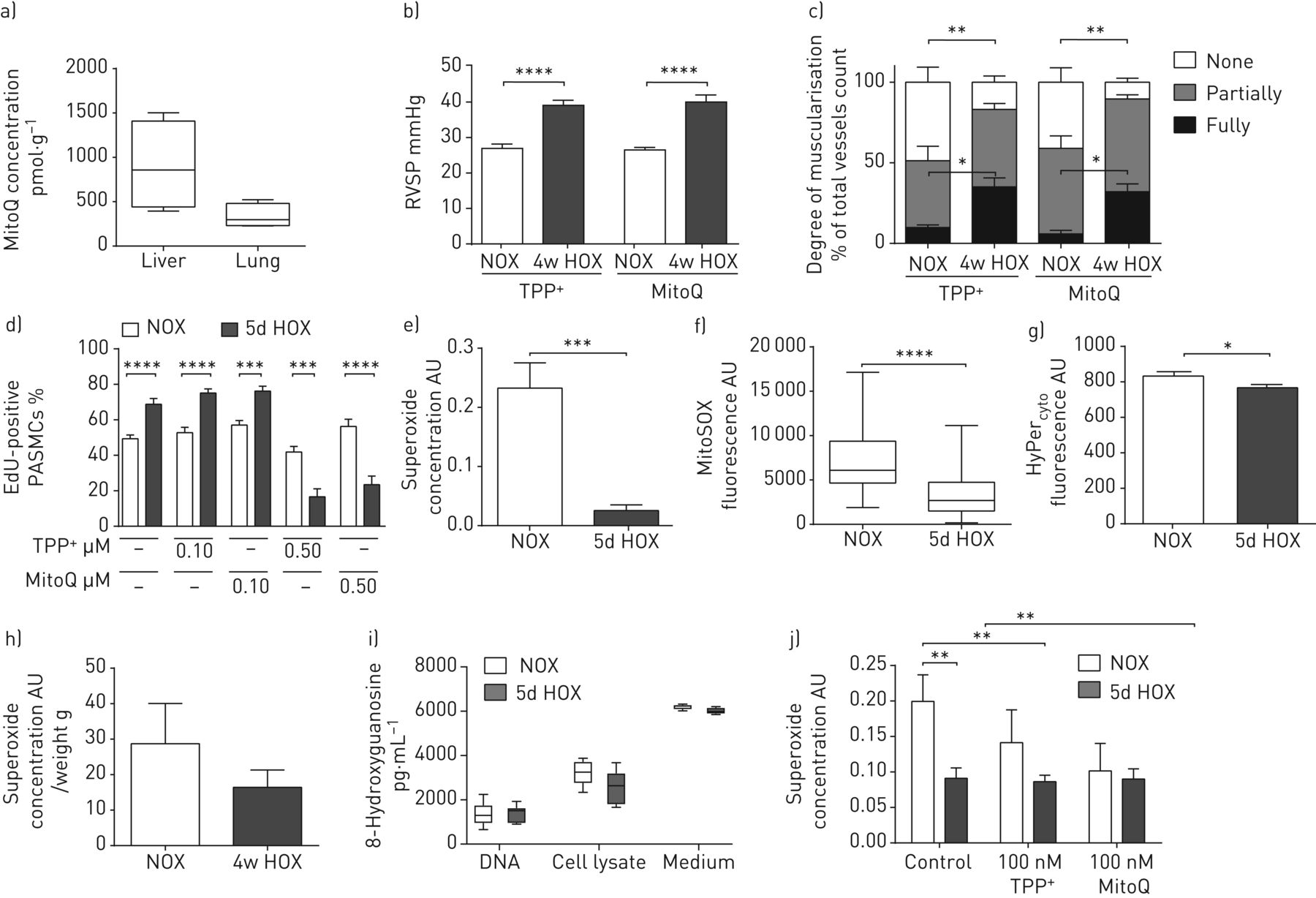
HPV的强度被确定为在离体小鼠肺缺氧通气期间肺动脉压(ΔPAP)的增加。在没有任何物质的情况下进行的第一次低氧运动作为基线(100%)。0.5的应用 与无MitoQ(第一次低氧动作)时的ΔPAP相比,µM MitoQ显著减弱低氧诱导的PAP升高(第四次低氧动作),或在0.5 µM TPP+在第四次演习中(图1A)。相反,较高剂量的mitoq或tpp+(1μm)与对照相比,将急性HPV减少到类似的程度(第五次缺氧机动)(图1A)。米托克和TPP都是+没有改变U46619引起的血管收缩(图1B.),显示HPV的MITOQ效果的特异性。在施用MITOQ或TPP之前,所有实验组中,HPV和U46619诱导的肺动量载体的强度相似+(补充图S1A和B.)。
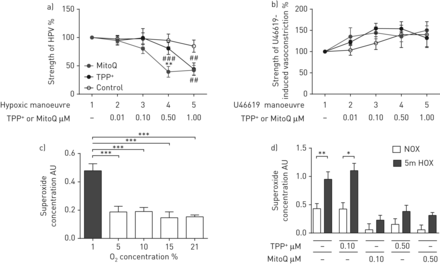
MITOQ对急性缺氧肺动脉平滑肌细胞(PASMCS)中分离出通气和灌注小鼠肺和超氧化物浓度的影响。a,b)急性缺氧通气期间分离出通风和灌注小鼠肺A)中肺动脉压的增加(1%O.2,5.3%的CO2,与N平衡2)或b)在注射血栓素模拟U46619的情况下在不存在(对照)或线粒体靶向抗氧化剂(MITOQ)或非惰性载体物质(TPPP(TPP)的情况下(TPP+)))。n = 5-6各组孤立的肺。**:与TPP相比P <0.01+- 治疗组;##:p<0.01;###:p<0.001,通过Tukey的双向方差分析与各对照组比较后HOC测试。c)5分钟急性暴露于不同o的PASMC中的超氧化物浓度2浓度。通过电子自旋共振光谱法测量超氧化物浓度作为1-羟基-3-甲氧基羰基-2,2,5,5-四甲基吡咯烷信号的差异,具有或不具有聚(乙二醇) - 缀合的超氧化物歧化酶。AU:任意单位。n =每组3-4个单个细胞分离。***:用Tukey单向ANOVA P <0.001后HOC测试。d)急性缺氧5分钟后PASMC中的超氧化物浓度(1%O2,休息n2(“5m HOX”)或常氧(21%O2(“NOx”))在存在或不存在线粒体靶向抗氧化剂(MITOQ)或惰性载体物质(TPP)中孵育(TPP+)。n = 5-6每组单个细胞分离。*:P <0.05;**:P <0.01通过双向ANOVA与TUKEY后HOC测试。
暴露于1%的o2但不到5%,10%和15%o25分钟诱导通过ESR光谱测量的细胞内和细胞外超氧化物浓度的增加(图1C.). 0.1 µM MitoQ特异性地抑制急性缺氧诱导的超氧化物增加,而TPP不起作用+在这种浓度下被检测到(图1D)。在较高的浓度(0.5μm)米托克和TPP+在常氧和缺氧中引起超氧化物浓度的显着降低。
o的值2紧张,公司2在代表性实验期间监测张力和pH(补充图S2A-F.)。
暴露于急性缺氧诱导细胞溶质H的增加2O.2,线粒体ROS和总细胞超氧化物,特别是pasmcs
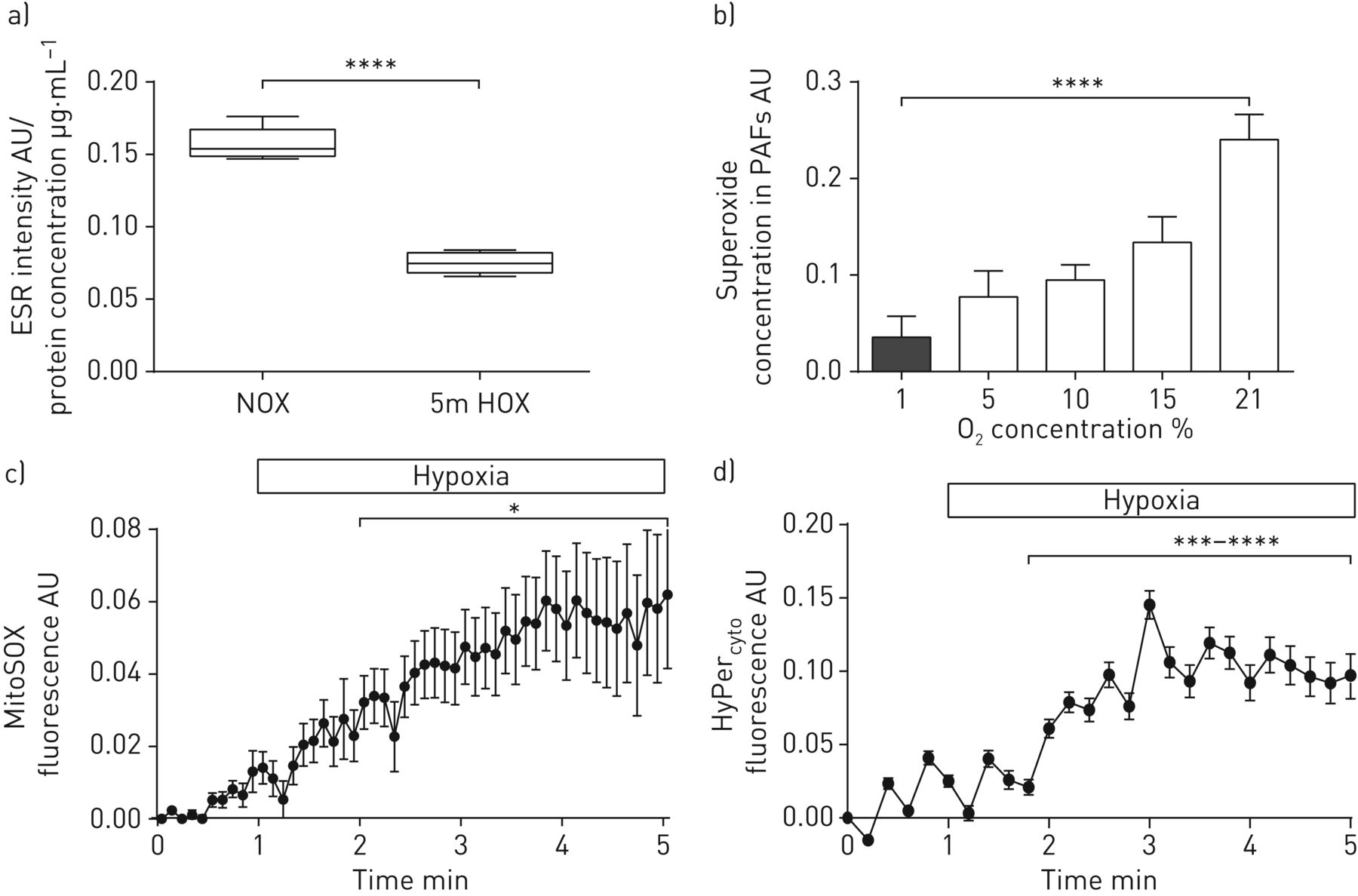
我们进一步专注于完整器官和不同ROS的亚细胞分布中的超氧化物释放。与分离的PASMC相比,从暴露于急性缺氧通气的完整肺部肺匀浆的ROS / RNS水平随CMH探针测定(图2A). 因此,我们检测到缺氧时肺成纤维细胞释放的超氧物减少(图2B.)。关于细胞内分布,急性缺氧增加了Mitosox使用荧光显微镜测量的PASMC中的线粒体ROS(图2C.)。通过共聚焦显微镜证实了弥塞索的线粒体染色模式(补充图S3)。
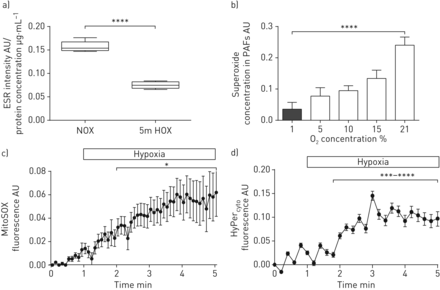
急性缺氧对亚细胞,细胞型特异性活性氧(ROS)浓度的影响。a)在5分钟急性缺氧5分钟后,通过1-羟基-3-甲氧基羰基-2,2,5,5-四甲基吡咯烷-2,2,5,5-四甲基吡咯烷酮(CMH)强度测定的肺匀浆中的肺匀浆(CMH)强度。5分急性缺氧5分钟后(1%o2,5.3%的CO2,与N平衡2(“5M Hox”))或常见通气(21%O2,5.3%的CO2,与N平衡2(“nox”)))。ESR:电子旋转共振;AU:任意单位。n = 4个孤立的肺部在每组中。****:P <0.0001通过威尔士策略的T检验。b)在急性暴露于不同O的5分钟后,原发性小鼠肺成纤维细胞的超氧化物浓度2浓度。通过ESR光谱法测量超氧化物浓度作为具有或不具有聚(乙二醇) - 缀合的超氧化物歧化酶的CMH信号的差异。PAF:肺动脉成纤维细胞。n =每组3-4个单个细胞分离。****:通过单向ANOVA与TUKEY的P <0.0001后HOC测试。c)抗缺氧缓冲液期间肺动脉平滑肌细胞(PASMC)中的线粒体ROS浓度。与基线和常氧值相比,线粒体ROS浓度的水平作为Mitosox荧光信号的变化呈现。n = 4个单独的细胞分离。*:通过威尔士校正的T检验P <0.05。d)细胞内过氧化氢(H2O.2)在灌注期间pasmc浓度与缺氧缓冲液。细胞H水平2O.2呈现为Hyper的变化细胞荧光信号与基线和常氧值相比。n = 4个单独的细胞分离。***:P <0.001;****:P <0.0001通过威尔士策略的T检验。
鉴于H.2O.2已被建议作为急性缺氧期间的下游信使[1],我们接下来量化细胞溶质H.2O.2由Hyper.细胞[25]. 急性缺氧使H2O.2在pasmcs(图2D.)。
米托Q对慢性缺氧性肺动脉高压、PASMC增殖和超氧化物释放的影响
MITOQ浓度4周的治疗后50 mg·kg-1·天-1在此前在展示的范围内,在心脏中具有保护作用(图3A) [26]。小鼠暴露于慢性缺氧(10%O2,4周)诱导MITOQ和TPP中右心室收缩压(RVSP)的右心室收缩压(RVSP)增加+- 治疗群体(图3B.)。根据,缺氧后肺血管重塑程度增加,但不受MITOQ或TPP改变+治疗 (图3C.)。TPP.+与未处理的对照相比,治疗对慢性缺氧期间的RVSP或心脏输出没有任何影响(补充图S4)。
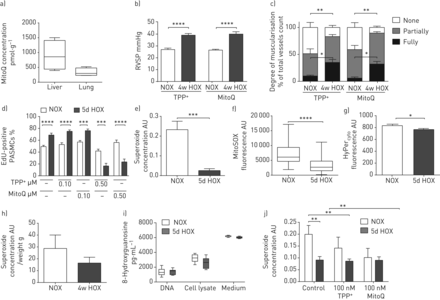
MITOQ对慢性缺氧诱导的肺动脉高压发展的影响(4周,10%O2)。a)在用mitoq治疗4周后,小鼠肺和肝脏中的mitoq浓度(60 mg·kg-1·天-1)通过通过液相色谱法测定饲料和串联质谱法测定。b)右心室收缩压(RVSP)和C)在暴露于慢性缺氧的小鼠中的肺血管系统重塑4周(10%O2(“4W Hox”))或常氧(21%O2(“NOx”))并用线粒体靶向抗氧化剂(MITOQ)或非惰性载体物质(TPHENYLPHSHONOUM)处理(TPP+)). n=每组8只动物。*:p<0.05;**:p<0.01;**:采用Tukey双因素方差分析,p<0.0001后HOC测试。d) 5周后肺动脉平滑肌细胞(PASMC)的增殖 缺氧天数(1%O2(“5d Hox”))或常氧(21%O2)在MitoQ或TPP存在下孵育+。与由Hoechst染色标记的总细胞数相比,增殖呈为5-乙炔氨基吡啶(EDU) - 阳性增殖细胞的百分比。n = 4个单独的细胞分离。***:P <0.001;****:P <0.0001与Tukey双向Anova后HOC测试。e)慢性缺氧5天后PASMCS的超氧化物浓度(1%O2)或常规(21%O2)孵化。通过电子自旋共振光谱法测量超氧化物浓度作为1-羟基-3-甲氧基羰基-2,2,5,5-四甲基吡咯烷信号的差异,具有或不具有聚(乙二醇) - 缀合的超氧化物歧化酶。AU:任意单位。n = 4个单独的细胞分离。***:通过威尔士校正的T检验P <0.001。f)在慢性缺氧5天后通过Mitosox荧光测定的线粒体ROS浓度(1%O.2)或常规(21%O2)孵化。n = 4个单独的细胞分离。****:通过威尔士校正的T检验P <0.001。g)细胞溶质H.2O.2受超级浓度确定的浓度细胞在缺氧孵育5天后PASMC的荧光(1%O2)。n = 4个单独的细胞分离。*:通过威尔士校正的T检验P <0.05。h)从暴露于慢性缺氧的小鼠肺匀浆中每重的超氧化物浓度(10%O2)或常氧(21%O2)4周。n = 3个单独的细胞分离。i)在DNA中的8-羟基核苷酸浓度(在缓冲液中稀释),来自暴露于慢性缺氧的PASMCs的细胞裂解物和生长培养基(1%O2)或常氧(21%O2)5天。j)暴露于慢性缺氧的Pasmc中的超氧化物浓度(1%o2)或常氧(21%O2)在100nm tpp存在下5天+或mitoq。n = 4个单独的细胞分离。**:P <0.01通过双向ANOVA与TUKEY后HOC测试。
此外,低剂量的米托Q不能阻止缺氧诱导的PASMC增殖,而高剂量的米托Q对增殖的影响与TPP一样没有特异性+在高浓度下也减少了增殖(图3d)。
总超氧化物浓度,线粒体ROS水平和细胞H.2O.2在1%o的5天后下降2通过ESR光谱测量的PASMC,MITOSOX荧光和HEAD细胞分别(图3E.-G)。慢性缺氧后小鼠的肺匀浆显示出过量的超氧化物水平降低,但未达到意义(图3H.). 所有ROS测量均在连续缺氧环境中进行,无复氧(补充方法)。此外,8-羟基胍的水平,这是DNA损伤的指标[27[慢性缺氧暴露于常氧值相比,慢性缺氧暴露后,在分离的DNA,PASMC裂解物和培养基中没有改变(图3i.)。在慢性缺氧暴露后MRNA水平上抗氧化酶在慢性缺氧暴露后的mRNA水平上,但在暴露于缺氧的小鼠的肺匀浆中,用细胞外定位的谷胱甘肽过氧化物酶3(GPX3)是唯一在mRNA和PASMC中蛋白质水平上调的酶(补充图S5A-D). 然而,慢性缺氧PASMC的SODs或过氧化氢酶活性和总抗氧化能力没有变化(补充图S5E-G)。线粒体DNA的量减少,而表达水平的表达水平表明线粒体葡萄糖氧化(丙酮酸脱氢酶激酶1(PDK1))和增加的厌氧糖醇(乳酸脱氢酶A(LDHA))(补充图S6a−C)。二氯乙酸酯的PDK1抑制可以增强常氧中的线粒体超氧化物浓度,但不是缺氧(补充图S6D.)。
在低浓度下,米托克没有抑制Pasmc的增殖(图3d),与常氧未处理的对照相比,它降低了常氧中的超氧化物浓度(图3J.). 然而,在未经处理的TPP中,缺氧超氧化物浓度没有差异+-treated或mitoq治疗的pasmcs(图3J.)。
米托Q对慢性缺氧及PAB后右心室重构的影响
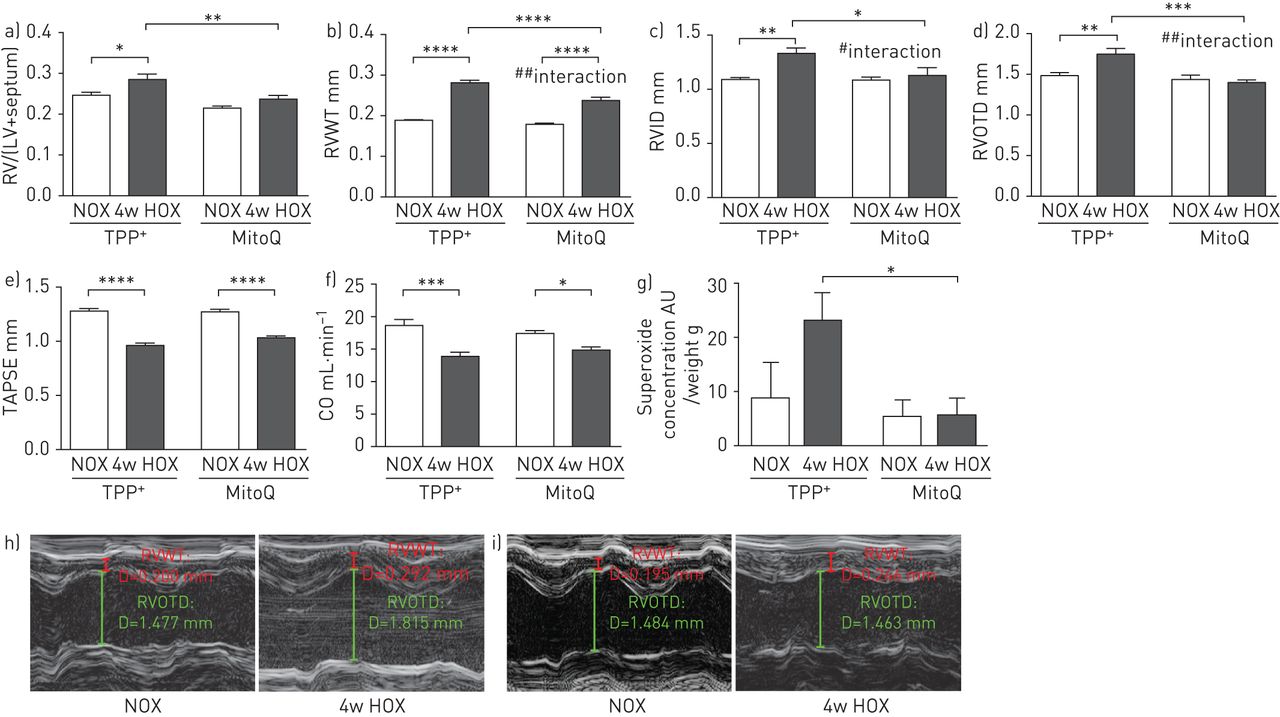
与肺动脉高压/血管重构的发展相反,与TPP相比,在慢性缺氧后,MitoQ组的右心室重构不同+-治疗组(图4G.ydF4y2Ba)。在TPP中缺氧暴露后,右心室质量与左心室加隔膜质量的比例显着增加+- 但不在MitoQ治疗组(图4A)。在两组慢性缺氧暴露后,超声心动图确定的右心室壁厚(RVWT)增加,尽管MITOQ组中的显着较低水平(图4C.)。此外,右心室内径(RVID)和右心室流出道直径(RVOTD)在MITOQ中的慢性缺氧中没有增加 - 与TPP相比+- 在Mitoq存在下,表明慢性缺氧期间的较低右心室扩张(图4C.,d,h和i)。心脏输出和三尖瓣环形平面收缩偏移(Tapse)在慢性缺氧后降低,尽管MITOQ和TPP中的相似程度+- 治疗群体(图4E.和f)。除了MITOQ对右心室的这些特定效果,MITOQ,TPP的载体+,可能对右心室重塑产生一些额外的效果,因为TPP中的RVWT和重量降低+- 与未经治疗的群体相比(补充图S4)。
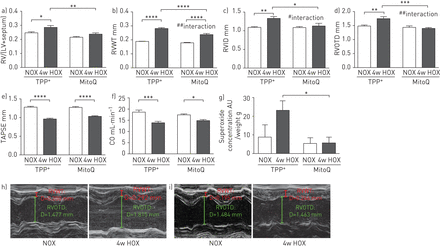
米托Q对慢性缺氧小鼠右心室重构的影响 周。a) 右心室质量与左心室加间隔质量之比(RV/(LV+间隔))和超声心动图参数:b)右心室壁厚度(RVWT),c)右心室内径(RVID),d)右心室流出道直径(RVOTD),e)三尖瓣环平面收缩偏移(TAPSE)作为慢性缺氧(10%O2(“4W Hox”))或常氧(21%O2(“NOx”))4周并用线粒体靶向抗氧化剂(MITOQ)或非惰性载体物质(三苯基鏻(TPP)处理(TPP+)))。n =每组8只动物。*:P <0.05;**:P <0.01;***:P <0.001;****:P <0.0001和#:P <0.05;##:P <0.01与TUKEY双向ANOVA的相互作用后HOC测试。g)暴露于慢性缺氧的小鼠右心室匀浆中的超氧化物浓度(10%O2)或常氧(21%O2)4周并用TPP治疗+或mitoq。通过电子自旋共振光谱法测量超氧化物浓度作为1-羟基-3-甲氧基羰基-2,2,5,5-四甲基吡咯烷信号的差异,具有或不具有聚(乙二醇) - 缀合的超氧化物歧化酶。数据作为与器官质量的比率呈现。AU:任意单位。n = 3-5。*:P <0.05通过双向ANOVA与TUKEY后HOC测试。h,i)右侧胸长轴的RVWT和RVOTD测量的代表性超声心动图:H)TPP+和i)mitoq。D:距离。
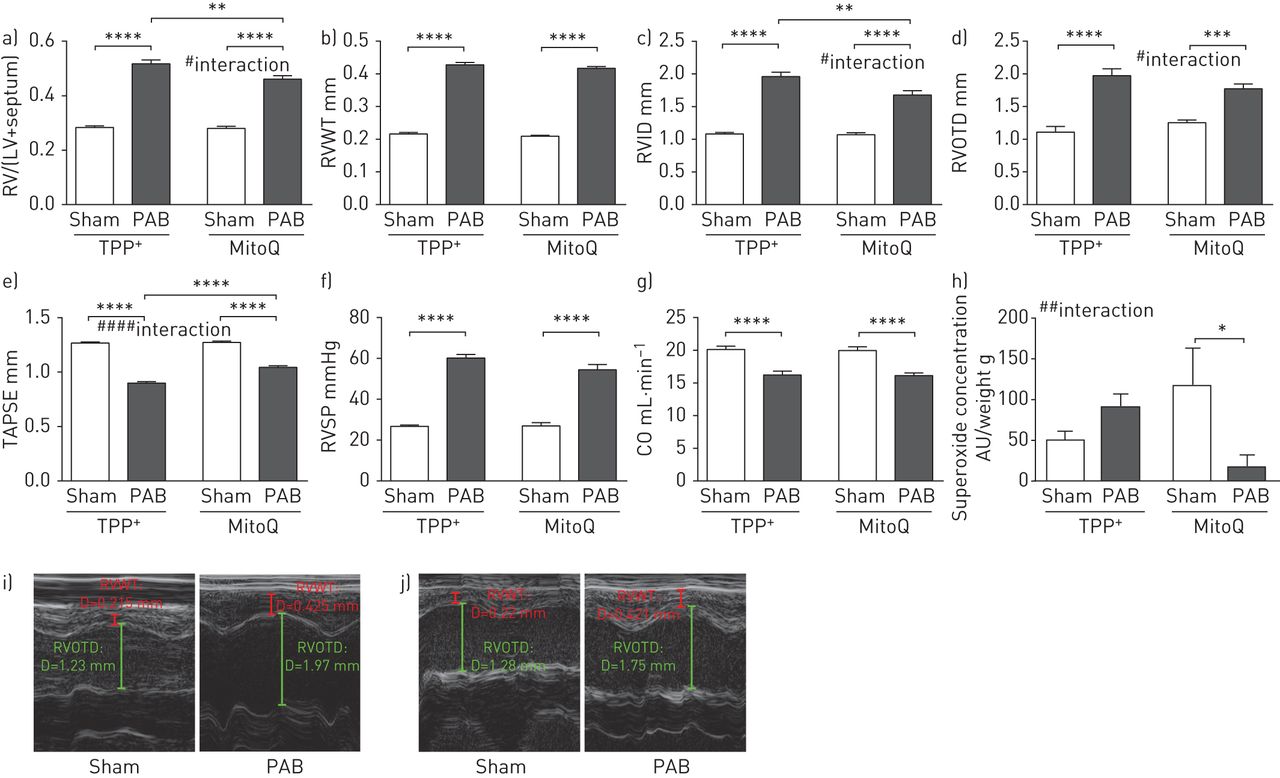
此外,PAB后应用MitoQ,作为右心室肥厚的缺氧非依赖性刺激,也改善了右心室重构(图5.)。RVID显着减少(图5C.),Tapse显着增加(图5E.)与TPP相比,在MitoQ处理的小鼠中使用PAB后+- 治疗的小鼠。
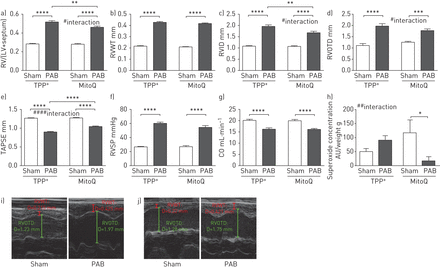
MitoQ对小鼠肺动脉结扎(PAB)后右心室重构的影响。a) 右心室质量与左心室质量加间隔的比率(RV/(LV+间隔)),b)右心室壁厚度(RVWT),c)右心室内径(RVID),d)右心室流出道直径(RVOTD),e)三尖瓣环平面收缩偏移(TAPSE),f)PAB或假手术后小鼠右心室收缩压(RVSP)和g)心输出量(CO)。n=每组8只动物;**:p<0.01;***:p<0.001;**:p<0.0001和#:P <0.05;####:P <0.0001双向ANOVA与TUKEY的交互后HOC测试。h)假手术或用三苯基膦酸铵(TPP)处理右心室匀浆中的超氧化物浓度+)或mitoq。通过电子自旋共振光谱法测量超氧化物浓度作为1-羟基-3-甲氧基羰基-2,2,5,5-四甲基吡咯烷信号的差异,具有或不具有聚(乙二醇) - 缀合的超氧化物歧化酶。数据作为与器官质量的比率呈现。AU:任意单位。n = 5。*:P <0.05和##:P <0.01与TUKEY双向ANOVA的相互作用后HOC测试。i、 j)右胸骨旁长轴RVWT和RVOTD测量的代表性超声心动图图像:i)TPP+和j)mitoq。D:距离。
慢性缺氧暴露后右心室匀浆的超氧化物浓度显着较高(补充图S4H.)。与TPP相比,MITOQ治疗减少了慢性缺氧暴露后的超氧化物浓度+-治疗组(图4G.)与假群相比,PAB(图5H.)。
讨论
该研究表明,超氧化物增加,最有可能源于线粒体,调节急性,但不是慢性缺氧诱导的肺动脉高压。这一结论是基于1)细胞超氧化物和线粒体ROS生产在缺氧暴露5分钟后的缺氧暴露后增加,并且在缺氧暴露5天后下降,2)线粒体靶向抗氧化型MITOQ可以抑制急性缺氧 -诱导超氧化物的增加,但不是慢性缺氧诱导的肺动脉高压。适应线粒体代谢/量可能使ROS在急性和慢性缺氧中的差异作用。
据我们所知,这是第一次调查特异性线粒体靶向抗氧化的抗氧化,关于其对肺脉管系统对急性和慢性缺氧的反应的影响。
虽然最近有证据表明线粒体衍生的超氧化物在肺脉管系统中对急性和慢性缺氧信号传导起着重要作用[3.那6.那28-30]存在两个相反的概念,表明缺氧时ROS增加或减少[3.那6.那28那29]。这一争议主要基于1)缺氧期间ROS测量的事实,易于遗体,2)局部化,ROS的定位,时间和种类必须考虑到,3)没有用于线粒体的特定药理学抗氧化剂。在这里,我们使用了三种方法来测量ROS:ESR技术,荧光染料和基于蛋白质的传感器。此外,在连续缺氧下进行该研究细胞和组织匀浆的实验,从而排除了由于测量样品的重新氧化引起的人工制品。我们发现线粒体ROS的浓度以及细胞溶质H.2O.2,其作为超氧化物的可能下游信号介体作用[1],在急性缺氧期间的PASMC中增加。线粒体生产的超氧化物可以转化为H.2O.2通过线粒体SOD,其可以将其扩散到细胞溶溶胶中或通过线粒体定位的GPX转化为水,或者从线粒体释放到细胞溶胶中通过电压依赖的阴离子通道[31]哪里可以转换为h2O.2通过SOD1。因此,H2O.2或局部超氧化物(例如通过线粒体和离子通道的分层化)或两者都可以充当急性缺氧中的急性缺氧中的下游介质。最近,我们提供了H.2O.2源自线粒体超氧化物对于PASMCS中的缺氧信号传导和HPV至关重要[30]。虽然急性缺氧期间超氧化物释放的机制仍然没有完全阐明,但我们可以表明它取决于线粒体细胞色素的特异性同种型的存在C导致急性缺氧中的线粒体超极化的氧化酶亚基4(COX4I2),众所周知的条件是从线粒体呼吸链的复合I或III促进超氧化物释放的条件[30]。h的下游目标2O.2是电压门控钾通道[30],但可能还有其他几个离子通道[3.那32]。与PASMC相比,急性缺氧降低了肺成纤维细胞中的超氧化物浓度。这种发现可以解释为什么肺匀浆中的ROS浓度在急性缺氧期间减少,尽管我们的测量无法区分整肺的不同ROS种类。尽管如此,我们的结果可以通过ROS的细胞类型特异性调节来解释整个肺和PASMC中ROS浓度的测量之间的差异。我们的研究结果与其他研究一致,显示急性缺氧中的线粒体ROS浓度增加[33-37]。随着MITOQ抑制我们研究中的超氧化物升高和HPV,显示了增加的超氧化物中的超氧化物中的增加的超氧化物。
抑制HPV的效果达到略微较高剂量的MITOQ的显着水平,而不是缺氧诱导的超氧化物释放的抑制剂可以通过分离的PASMCs对MITOQ更敏感而不是孤立的器官,其中该物质具有较高的分布体积和扩散距离。
MITOQ和TPP.+已经显示高剂量以抑制氧化磷酸化[38]并与线粒体钠/钙交换器相互作用[39[从而可能降低线粒体膜电位。但是,低剂量的TPP+,在本前研究中使用体外,没有改变线粒体呼吸[40]。因此,TPP的影响+在急性HPV和浓度下缺氧诱导的超氧化血氧诱导的升高>0.5μm可能与其性质有关,以降低线粒体膜电位,因为在急性HPV期间已经观察到线粒体高苯化术[37]。然而,由于米托克抑制较低浓度范围内的急性缺氧反应,而TPP+没有,这种效果可以被认为具体对米托克的抗氧化特性。
除HPV外,慢性缺氧由于病理肺血管重塑导致肺动脉高血压,其特征在于,部分地,通过肺血管的PASMC的增殖[18]。与HPV的情况一样,减少的证据[41]细胞ROS浓度触发肺动脉高压发育的增加[8.已经提供了。最近,一个优雅的研究表明,特别是线粒体的增加2O.2可能促进慢性缺氧性肺动脉高压的发展[8.]。在我们的研究中,超氧化物,线粒体ROS和细胞源性H水平2O.2通过ESR光谱法测量,Mitosox和Hyper细胞分别在1%o温育5天后,荧光分别在吡螺溶胶和线粒体中显着降低了PASMCs的培养5天后2以及慢性缺氧小鼠的肺匀浆,支持慢性缺氧诱导总细胞ROS的假设。另外,8-羟基胍的水平(氧化诱导的DNA损伤的指示剂[27])在PASMCS的慢性缺氧期间没有改变。此外,用米托克达到肺组织水平的口服治疗,证明了先前研究中的保护作用[26]没有抑制慢性缺氧诱导的肺动脉高压或脂肪增殖。有趣的是,我们发现在慢性缺氧PASMC中的细胞内抗氧化酶但不变的抗氧化能力下降。随着线粒体葡萄糖氧化的降低,通过降低的线粒体DNA量表明,PDK1的表达增加(抑制线粒体丙酮酸氧化)的表达增加和LDHA的表达增加(介导厌氧丙酮酸代谢),我们表明细胞新陈代谢的适应导致与急性缺氧相比,慢性缺氧中的ROS浓度降低。在这方面,提出了以前认为线粒体代谢可以在慢性缺氧中下调,以避免过量的线粒体ROS生产[42]。有趣的是,二氯乙酸乙酸酯的线粒体葡萄糖氧化激活,可抑制缺氧诱导的肺动脉高压和人肺动脉高血压[43],可增加常氧条件下的超氧物释放,但不增加缺氧条件下的超氧物释放,表明线粒体对缺氧诱导的活性氧水平的适应是多因素的,二氯乙酸通过非超氧物独立的机制抑制肺动脉高压。因此,我们的数据表明,线粒体ROS的上调在慢性缺氧诱导的肺血管重塑中不起作用,这是由于降低超氧化物浓度的多种适应性机制所致。
此外,PASMC中线粒体ROS的减少似乎与PASMC中的增殖途径没有相互作用,因为在MitoQ治疗期间,常氧条件下的超氧化物水平降低也不会改变肺血管重塑。
然而,不能排除的是,细胞类型特异性和特异性ROS的局部亚细胞释放可能激活增殖途径。其他活性氧源的上调可能有助于这一发现,因为缺氧诱导的活性氧释放也可能源于NADPH氧化酶的特定亚型[4.]。
有趣的是,在慢性缺氧条件下,应用米托Q可减少右心室肥厚和右心室扩张的发生。TPP+与未处理的对照动物相比,右心室肥厚也降低,尽管略低于米托克,但对右心室扩张几乎没有影响,表明线粒体ROS特异性可以影响右心室重塑。此外,MITOQ治疗衰减了右心室的肥大和扩张,并在PAB后防止右心室功能障碍的发育。因此,MITOQ治疗在右心室中降低过氧化物浓度。
之前已被证明右心室重塑可以依赖于依赖ros依赖的信号传导途径[5.那44]从有益的适应性向心性右心室肥大到右心室扩张和衰竭的转变可能取决于ROS释放的数量[45]. 我们的研究没有回答右心室线粒体ROS释放激活机制的问题,但表明右心室后负荷增加而非缺氧导致ROS增加。此外,米托Q治疗肺动脉高压右心室功能障碍的治疗潜力和潜在机制有待进一步评估。
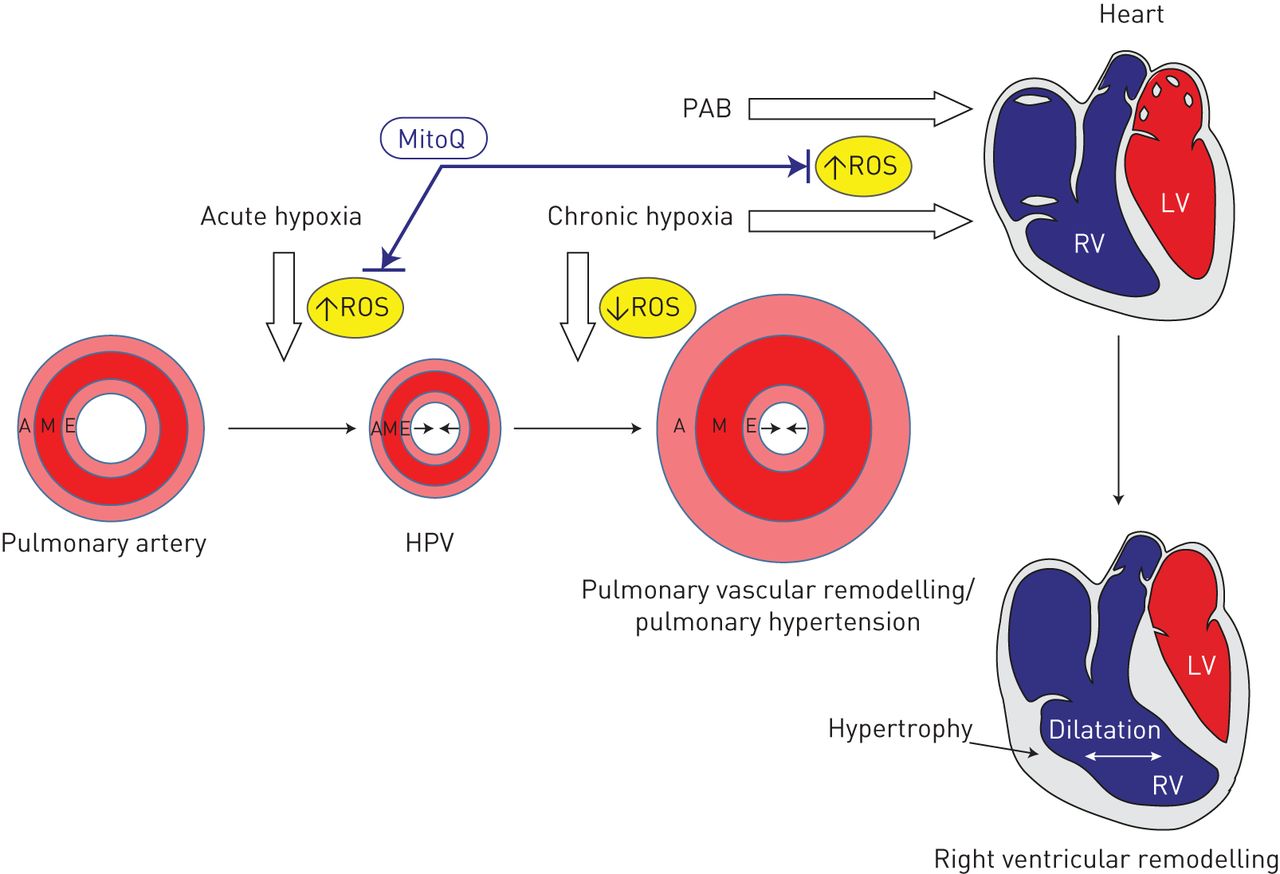
总之,我们的研究表明,急性缺氧暴露在调节HPV调节的PASMC中提高了线粒体ROS,最多的超氧化物,而慢性缺氧暴露与PASMC中不同RO的浓度降低有关。施用线粒体靶向抗氧化米科克的应用特异性衰减HPV,但没有抑制肺血管重塑和慢性缺氧诱导的肺动脉高压的发育,而其衰减右心室扩张(图6.)。
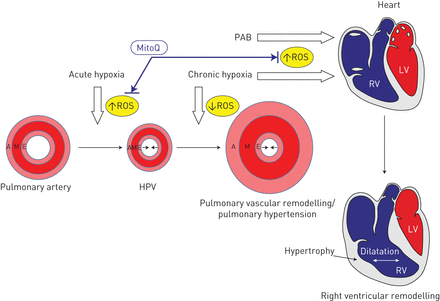
MITOQ在缺氧中的影响。急性缺氧增加反应性氧物种(ROS),而慢性缺氧降低肺脉管系统中的RO,但右心室(RV)中的ROS浓度增加。线粒体靶向抗氧化型MITOQ抑制急性缺氧诱导的肺血管收缩和慢性缺氧,以及肺动脉带(PAB) - 诱导的右心室扩张。LV:左心室;答:冒险;M:媒体;E:内皮;HPV:缺氧肺血管收缩。
补充材料
致谢
作者感谢K。霍姆伯格,I。布雷滕伯恩·穆勒,N。舒普,M。韦斯森多夫和E。Kappes(卓越集群心肺系统,德国吉森)提供技术援助。
脚注
本文提供了补充材料www.qdcxjkg.com.
利益冲突:没有宣布。
支持声明:由Deutsche Forschungsgemeinschaft AZ支持我们1978/4-2和SFB 1213,项目A06和CP02,由医学研究委员会(MC_U105663142)和Wellcome Trust调查员奖(110159 / z / 15 / z)进行m。P.。墨菲。
- 已收到2017年5月19日。
- 公认2018年1月10日。
- 版权所有©ers 2018











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}