





摘要
我们之前报道过上皮来源的白细胞介素(IL)-1α在肺上皮-间充质营养单位中驱动成纤维细胞来源的炎症。由于miR-146a-5p已被证明负向调节IL-1信号,我们研究了miR-146a-5p在慢性阻塞性肺疾病(COPD)中IL-1α驱动炎症调节中的作用。
人支气管上皮细胞(16HBE14o-)与对照和copd来源的原代人肺成纤维细胞(PHLFs)共培养,使用和不使用IL-1α中和抗体评估miR-146a-5p的表达。基因组DNA评估单核苷酸多态性(SNP) rs2910164的存在。miR-146a-5p模拟物用于过表达研究,以评估il -1α诱导的信号通路和PHLFs产生IL-8。
PHLFs与气道上皮细胞共培养显著增加了miR-146a-5p的表达,这种诱导依赖于上皮来源的IL-1α。miR-146a-5p过表达降低了il -1α诱导的phlf中IL-8的分泌通过IL-1受体相关激酶-1下调。在COPD PHLFs中,与对照组相比,miR-146a-5p的诱导显著减少,并与miR-146a-5p基因中的SNP rs2910164 (GG等位基因)相关。
我们的结果表明,miR-146a-5p的诱导参与了肺中上皮细胞-成纤维细胞的通信,并负调控成纤维细胞上皮来源的IL-1α对IL-8的诱导。COPD成纤维细胞中miR-146a-5p水平的降低可能诱导更促炎症表型,有助于COPD的慢性炎症。
摘要
肺成纤维细胞中miR-146a-5p的缺陷上调导致COPD成纤维细胞介导的炎症http://ow.ly/3awN308JqRH
简介
慢性阻塞性肺疾病(COPD)是一种进行性疾病,慢性中性粒细胞性炎症与肺实质破坏(肺气肿)和小气道疾病相关,这两种疾病都导致气流受限和肺功能下降[1].吸入有害颗粒,特别是来自香烟烟雾的有害颗粒,是慢性阻塞性肺病最常见的危险因素,已知吸烟会诱导中性粒细胞的招募[1,2].然而,并不是所有吸烟者都会患慢性阻塞性肺病,这表明疾病过程的遗传易感性[3.].不同研究人群中COPD相关靶基因复制的不一致性表明表观遗传调控的重要作用[4].表观遗传介质如microRNAs (miRNAs)可以调节各种参与肺功能和炎症的基因的转录活性,这些基因被认为与COPD的发病机制有关[5].
miRNAs是由大约19-25个核苷酸组成的小的非编码rna,通过增加mRNA降解或抑制特定mRNA靶标的蛋白质翻译来引起转录后基因抑制[6].它们参与各种生物过程,其表达的改变可导致病理状况,包括肺部疾病[7].我们最近提供了一份最新的研究综述,显示miRNA失调在COPD中的作用,以及它如何与COPD的各种特征相关[5].特别是,研究表明,仅暴露在香烟烟雾中就可以改变肺部miRNA的表达[8].多项研究表明,吸烟的COPD患者与不吸烟的COPD患者相比,全肺组织、血清和/或痰中各种miRNAs的表达存在差异[9- - - - - -12].其中,miR-146a-5p已被证明可调节肺上皮细胞释放白细胞介素(IL)-1诱导的炎症介质,包括中性粒细胞趋化剂IL-8 [13].我们最近在共培养模型中发现,香烟烟雾提取物(CSE)诱导的IL-1α在COPD患者气道上皮细胞(AECs)中表达高于对照来源的上皮细胞[14].此外,较高水平的上皮细胞IL-1α诱导肺成纤维细胞释放更强的前炎症细胞因子,包括IL-8 [14].有趣的是,其他研究表明,当肺成纤维细胞被IL-1β和肿瘤坏死因子(TNF)-α刺激时,与对照成纤维细胞相比,COPD患者肺成纤维细胞中miR-146a-5p的表达被诱导程度较低[12].
在本研究中,我们假设肺成纤维细胞中miR-146a-5p的上调失败导致COPD中上皮-间充质营养单元内的通信紊乱,导致疾病炎症增加。因此,我们在共培养模型中研究了miR-146a-5p在COPD和非COPD对照来源的原代人肺成纤维细胞(PHLFs)中的表达,并进行功能检测,以研究miR-146a-5p在肺成纤维细胞中调节促炎活性的机制。
材料与方法
实验对象和细胞培养条件
人支气管上皮细胞系16HBE14o-由dc . Gruenert博士(加州大学旧金山分校,CA, USA)捐赠,并在Eagle's最低必需培养基(EMEM)/10%胎牛血清(FCS;Lonza/BioWhittaker, Verviers,比利时)的胶原蛋白/牛血清白蛋白涂层烧瓶,如前所述[15].
胎儿肺成纤维细胞(MRC-5;BioWhittaker, Walkersville, MD, USA)在实验前在24孔板上EMEM/10% FCS中培养。从8例接受肿瘤切除手术的非COPD对照组供体的外周肺实质组织中分离出phlf,以及12例采用前文所述的外植体技术进行肺移植的严重疾病COPD患者[16,17].对照组的PHLFs从组织学正常的组织中分离出来,尽可能远离肿瘤。由一位经验丰富的病理学家检查该组织是否有癌症或其他病理,结果发现该组织没有癌症,也没有其他病理。COPD患者和非COPD对照组的临床信息在表1.本项目的研究方案与格罗宁根大学医学中心(www.rug.nl umcg / onderzoek / researchcode /索引)及国家道德及专业指引(www.federa.org).实验前,PHLFs以Ham’s- f12培养基/10% FCS (Lonza)在24孔培养板上培养。
共培养模型
我们使用16HBE14o-和MRC-5共培养进行机制研究。16HBE14o-细胞与COPD和对照来源的PHLFs共同培养,以确定疾病特异性效应。
简单地说,16HBE14o-细胞被镀在涂层的0.4-µM孔6.5 mm跨孔膜上(Costar;Corning, New York, NY, USA),而成纤维细胞分别在24孔板上培养。在获得两种细胞类型的融合层后,将含有16HBE14o-细胞(上隔室)的transwell与肺成纤维细胞放在24孔板(下隔室)中共培养,并在EMEM/10% FCS或Ham's-F12培养基/10% FCS (Lonza)中分别与MRC-5细胞或PHLFs共培养72小时[14].在实验之前,细胞被整夜剥夺血清。
卷烟烟气提取物制剂
CSE实验在无血清、添加激素的支气管上皮基础培养基(Lonza, Basel, Switzerland)中进行。在每次实验前,将两支无过滤嘴香烟在25 mL介质中起泡以制备100% CSE,然后如前所述将其稀释至20% CSE [18].
条件培养基和中和抗体实验
16HBE14o-细胞被剥夺血清过夜,并在有或没有20% CSE的情况下刺激6小时,我们之前报道过,这不会影响细胞活力[14].刺激后,彻底洗掉CSE,在进一步孵育24小时后收集无CSE条件培养基。无cse条件培养基加或不加4µg·mL预孵育1 h−1IL-1α中和抗体(AB-200-NA, MAB601;R&D Systems, Abingdon, UK),并用于刺激无血清成纤维细胞24小时。对于重组人(rh) IL-1α (R&D Systems)的实验,a为1 ng·mL−1之所以选择浓度,是因为这与之前报道的COPD患者痰液中的浓度相当[19].采用酶联免疫吸附法(ELISA)收集无细胞上清液,用TRI试剂(Molecular Research Center, Cincinnati, OH, USA)收集细胞裂解液进行RNA分离。
miR-146a-5p模拟转染
用miR-146a-5p模拟物在25 nM处转染MRC-5成纤维细胞(MirVana miRNA模拟物,分析ID: MC10722;应用生物系统公司(Applied Biosystems, Carlsbad, CA, USA),并在25 nM条件下进行小RNA(全明星阴性对照siRNA;Qiagen, Hilden, Germany)作为非靶向对照,以评估miR-146a-5p诱导对肺成纤维细胞IL-1信号通路的影响。如补充材料所述,采用定量PCR方法分析miR-146a-5p的表达,Western blot检测蛋白裂解物,ELISA (R&D系统)检测无细胞上清中IL-8的浓度。
单核苷酸多态性基因分型
从PHLF捐赠者的肺组织中提取用于基因分型的基因组DNA,并评估单核苷酸多态性(SNP) rs2910164的存在。在补充材料中有详细的说明。
统计分析
使用SPSS统计版本23 (IBM, Armonk, NY, USA)进行数据分析。用Mann-Whitney u检验分析COPD患者与非COPD对照组的差异。用细胞系的配对t检验和原代细胞的Wilcoxon符号秩检验分析了组内处理之间的差异。P <0.05为差异有统计学意义。
结果
上皮细胞来源的IL-1α是肺成纤维细胞miR-146a-5p表达增加的原因
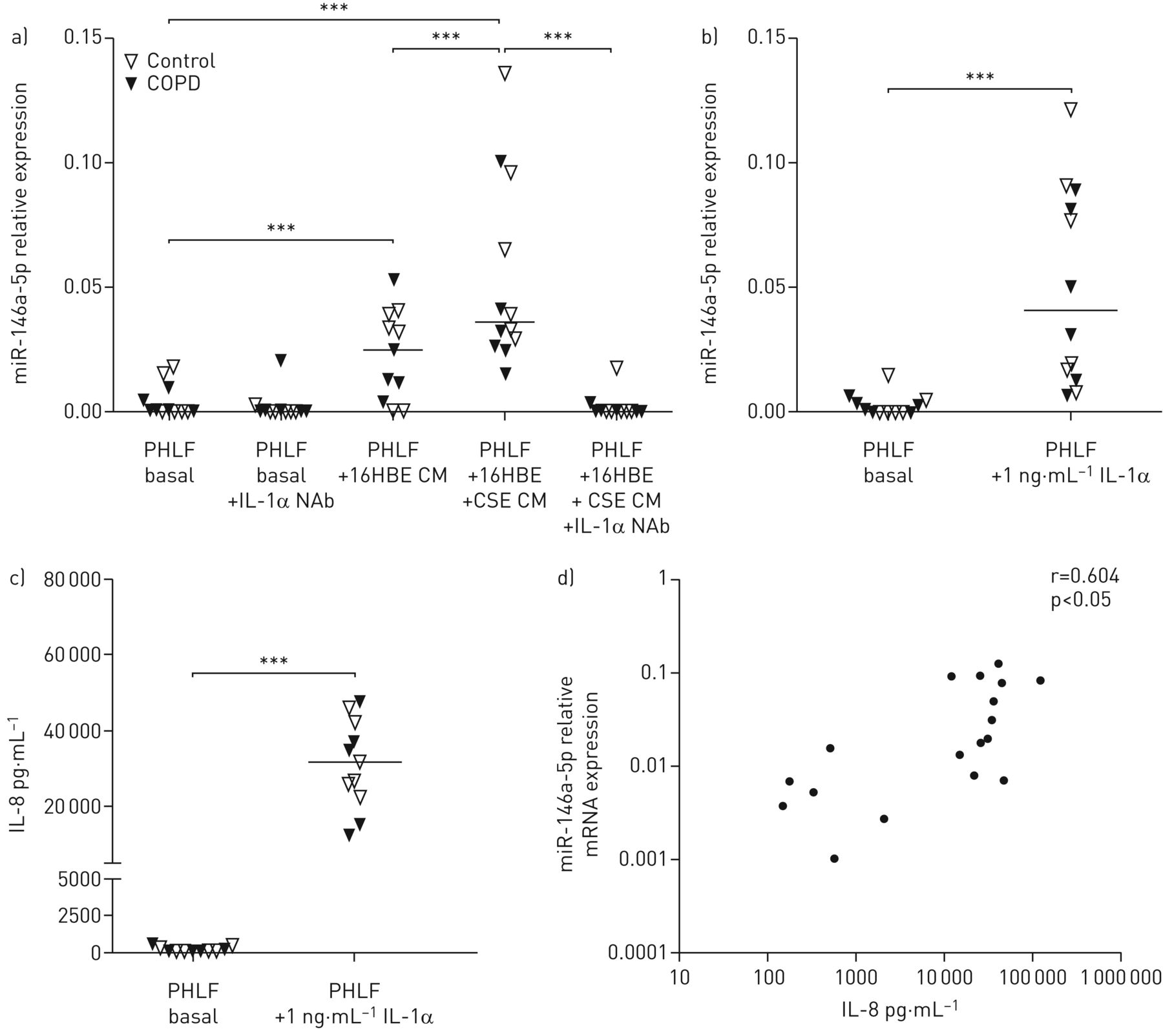
我们之前的研究表明,来源于气道上皮细胞(原代细胞和16HBE14o-细胞)的IL-1α是肺成纤维细胞来源的炎症的重要调节因子[14],我们首先确定上皮来源的IL-1α是否会诱导肺成纤维细胞中的miR-146a-5p。我们用先前已测定IL-1α水平的16HBE14o条件培养基刺激非COPD对照组和COPD患者的PHLFs [14]在IL-1α中和抗体存在或不存在的情况下。我们发现miR-146a-5p的表达在来自16HBE14o-细胞的条件介质处理过的PHLFs中增加,而在对16HBE14o-细胞进行CSE预刺激后进一步增强(图1一个).此外,添加IL-1α中和抗体(图1一个).我们还评估了rhel -1α刺激对PHLFs中miR-146a-5p表达的直接影响,并发现,与16HBE14o细胞条件介质一样,PHLFs中miR-146a-5p表达显著增加(图1 b).最后用1 ng·mL刺激−1(图1 c)或0.01 ng·mL−1(补充图S1) rhel -1α导致PHLFs中IL-8浓度的显著释放(图1 c),与miR-146a-5p在PHLFs中的表达升高显著相关(图1 d).经16HBE14o条件介质或rhIL-1α刺激后,COPD与对照组衍生的PHLFs之间miR-146a-5p的表达水平和IL-8的释放均无显著差异。
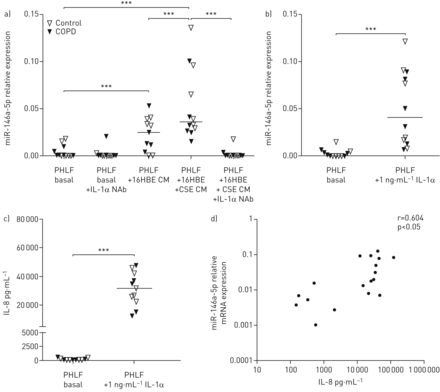
上皮来源的白细胞介素(IL)-1α是肺成纤维细胞miR-146a-5p表达增加的原因。来自对照供体(n=6)和慢性阻塞性肺疾病(COPD)患者(n=6)的原代人肺成纤维细胞(PHLFs)被培养到融合并在一夜之间被剥夺血清。a)在4µg·mL存在或不存在香烟烟雾提取物(CSE)预处理的16HBE14o- (16HBE)细胞中,用条件介质(CM)刺激后PHLFs中miR-146a-5p的表达−1IL-1α中和抗体(NAb)。b)加/不加1 ng·mL刺激后,miR-146a-5p在无血清PHLFs中的表达−1重组人IL-1α 24 h。miR-146a-5p表达水平与管家性非编码RNU48相关,表达为2——ΔCt.c) PHLF释放的IL-8浓度。表示中位数。***: p<0.001,在指示值之间。d) IL-1α刺激后PHLFs释放的IL-8浓度与PHLFs中miR-146a-5p表达的相关性。数据以对数刻度显示。
COPD患者与16HBE14o-细胞共培养的PHLFs中miR-146a-5p表达降低
我们之前在共培养模型中表明,copd衍生的aec通过更高的IL-1α诱导肺成纤维细胞更促炎[14].因此,我们对对照组和copd衍生的PHLFs中miR-146a-5p表达的调控感兴趣。在我们的共培养模型中,来自对照组和COPD供体的phlf中,16HBE14o-细胞显著上调了miR-146a-5p的表达。有趣的是,在与16HBE14o-细胞共培养后,与对照组相比,在copd来源的phlf中miR-146a-5p的诱导显著降低(图2).
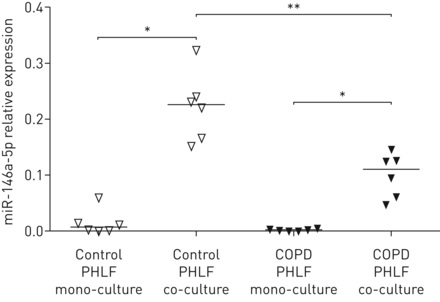
miR-146a-5p在原代人肺成纤维细胞(PHLFs)与16HBE14o-细胞共培养时的表达。对照供体或慢性阻塞性肺疾病(COPD)患者的PHLFs单独培养或与16HBE14o-细胞共培养72小时,之后去血清。miR-146a-5p在对照组供体和COPD患者的PHLFs中表达,在单培养和与16HBE14o-细胞共培养时,与管家非编码RNU48相关,表达为2——ΔCt.表示中位数。*: p < 0.05;**: p<0.01,在指示值之间。
COPD成纤维细胞中miR-146a-5p的低诱导与单核苷酸多态性rs2910164有关
为了解释COPD与对照成纤维细胞之间miR-146a-5p诱导的差异,我们考虑了两种可能性。1) NF-κB转录因子家族成员RelB在小鼠肺成纤维细胞中调控miR-146a-5p表达的差异[20.].2)主miR-146a-5p序列中SNP rs2910164的存在差异,因为该SNP已被证明会导致成熟miR-146a-5p表达的降低[21].
1)我们检测了来自共培养模型的PHLFs中RelB的表达,我们发现在对照组和copd衍生的PHLFs中RelB的mRNA表达没有显著差异(图3一).2)我们比较了共培养实验中使用的phlf的rs2910164 SNP基因型,发现GG基因型的供体在共培养后比CG基因型的供体具有更低的miR-146a-5p诱导能力(图3 b).重要的是,GG基因型的供体除一人外均为COPD患者。
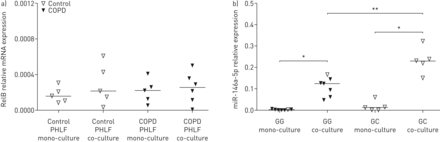
共培养中RelB表达和rs2910164多态性对miR-146a-5p表达的影响来自对照供体或慢性阻塞性肺疾病(COPD)患者的原代人肺成纤维细胞(PHLFs)单独培养或与16HBE14o-细胞共培养72小时,之后去血清。a) RelB在对照组供体和COPD患者的PHLFs中表达,分别在单独培养和与16HBE14o-细胞共培养时表达。b)与16HBE14o-细胞共培养前后,单核苷酸多态性rs2910164对PHLFs中miR-146a-5p表达的影响。mRNA水平归一化至管家基因β2-微球蛋白和蛋白磷酸酶1α,而miR-146a水平与管家性非编码RNU48相关,均表达为2——ΔCt.表示中位数。*: p < 0.05;**: p<0.01,在指示值之间。
miR-146a-5p过表达对肺成纤维细胞具有抗炎作用
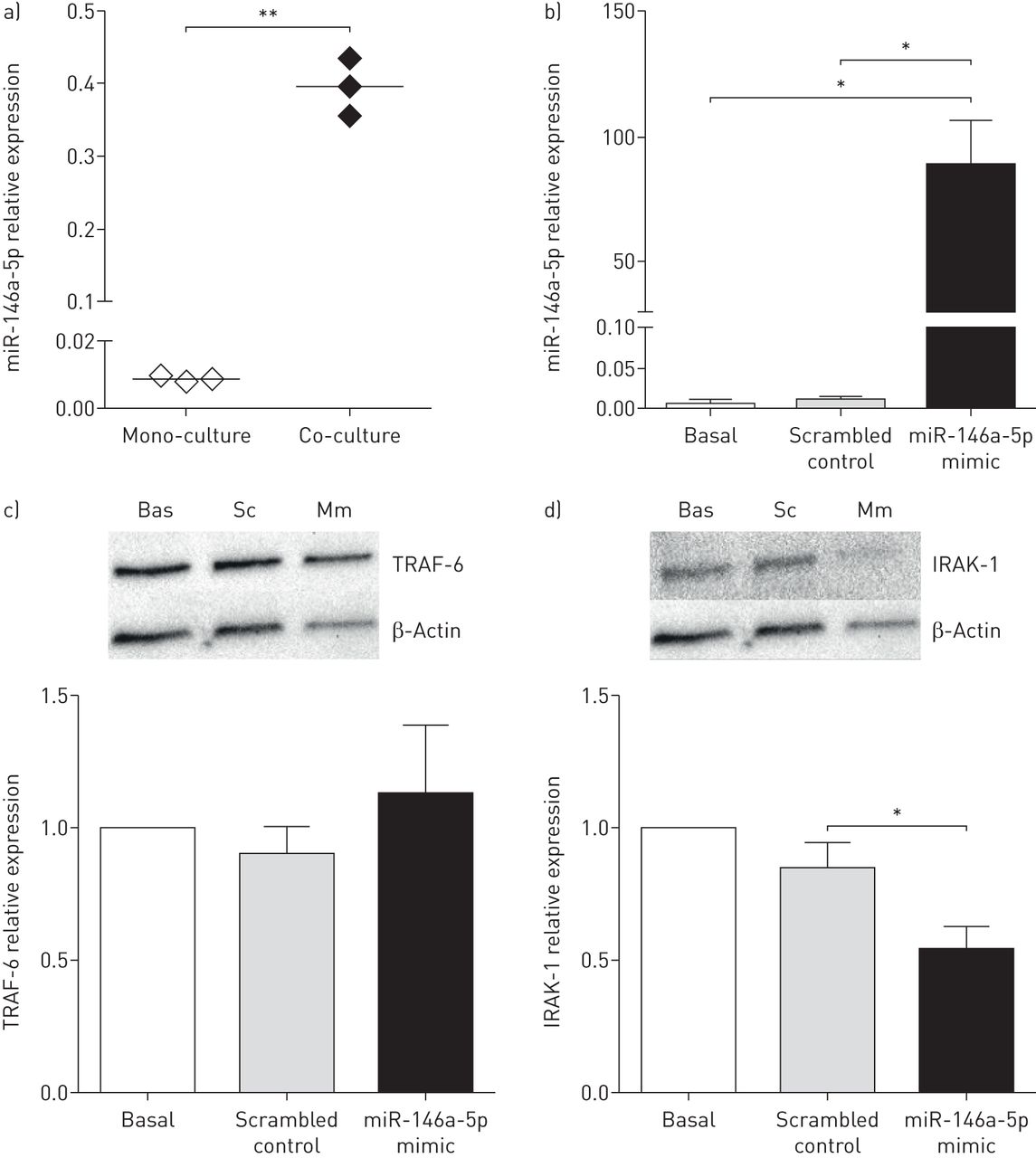
据报道,miR-146a-5p通过靶向IL-1受体相关激酶(IRAK)-1和TNF受体相关因子(TRAF)-6的蛋白质发挥抗炎作用,这两个蛋白是细胞对IL-1反应的关键下游介质[22].因此,我们有兴趣研究miR-146a-5p在人肺成纤维细胞中的抗炎机制。对于miR-146a-5p的过表达实验,我们使用了人肺胎儿成纤维细胞(MRC-5)。首先,我们检测了miR-146a-5p在与16HBE14o-细胞共培养的MRC-5成纤维细胞中的表达,并发现在共培养时miR-146a-5p在MRC-5成纤维细胞中的相似诱导(图4一)。接下来,我们通过miR-146a-5p模拟物处理MRC-5成纤维细胞,与打乱的非靶向对照模拟物相比,成功地过表达miR-146a-5p水平(图4 b).然后,我们评估了过表达miR-146a-5p后MRC-5成纤维细胞中IRAK-1和TRAF-6的蛋白表达水平。我们发现,与打乱对照相比,miR-146a-5p过表达后IRAK-1的蛋白表达显著降低,但TRAF-6未受影响(图4 cd).这表明miR-146a-5p确实在人肺成纤维细胞中调控IL-1受体下游信号。
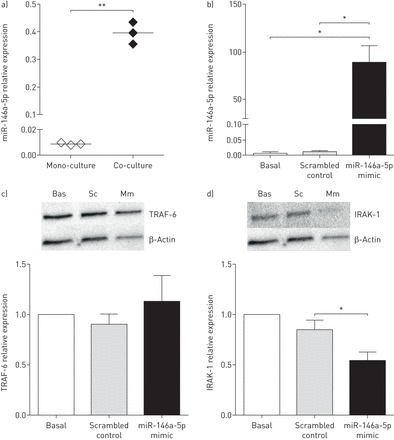
miR-146a-5p在肺成纤维细胞中具有抗炎作用。a) MRC-5成纤维细胞单独培养或与16HBE14o-细胞共培养。miR-146a-5p在成纤维细胞中的表达与管家性非编码RNU48相关,表达为2——ΔCt.表示中位数。b)将MRC-5成纤维细胞播种,立即用25 nM miR-146a-5p mimic或打乱对照转染48小时。miR-146a-5p在成纤维细胞中的表达与管家性非编码RNU48相关,表达为2——ΔCt.数据以均数±表示扫描电镜(n=3或4个独立实验)。c)肿瘤坏死因子受体相关因子(TRAF)-6和d)转染miR-146a-5p mimic (Mm)或打乱对照(Sc)后,MRC-5成纤维细胞中白细胞介素-1受体相关激酶(IRAK)-1蛋白表达的代表性印迹和密度测定。Bas:基底。以β-肌动蛋白作为蛋白表达的负载对照。数据以均数±表示扫描电镜(n=3或4个独立实验)。*: p < 0.05;**: p<0.01,在指示值之间。
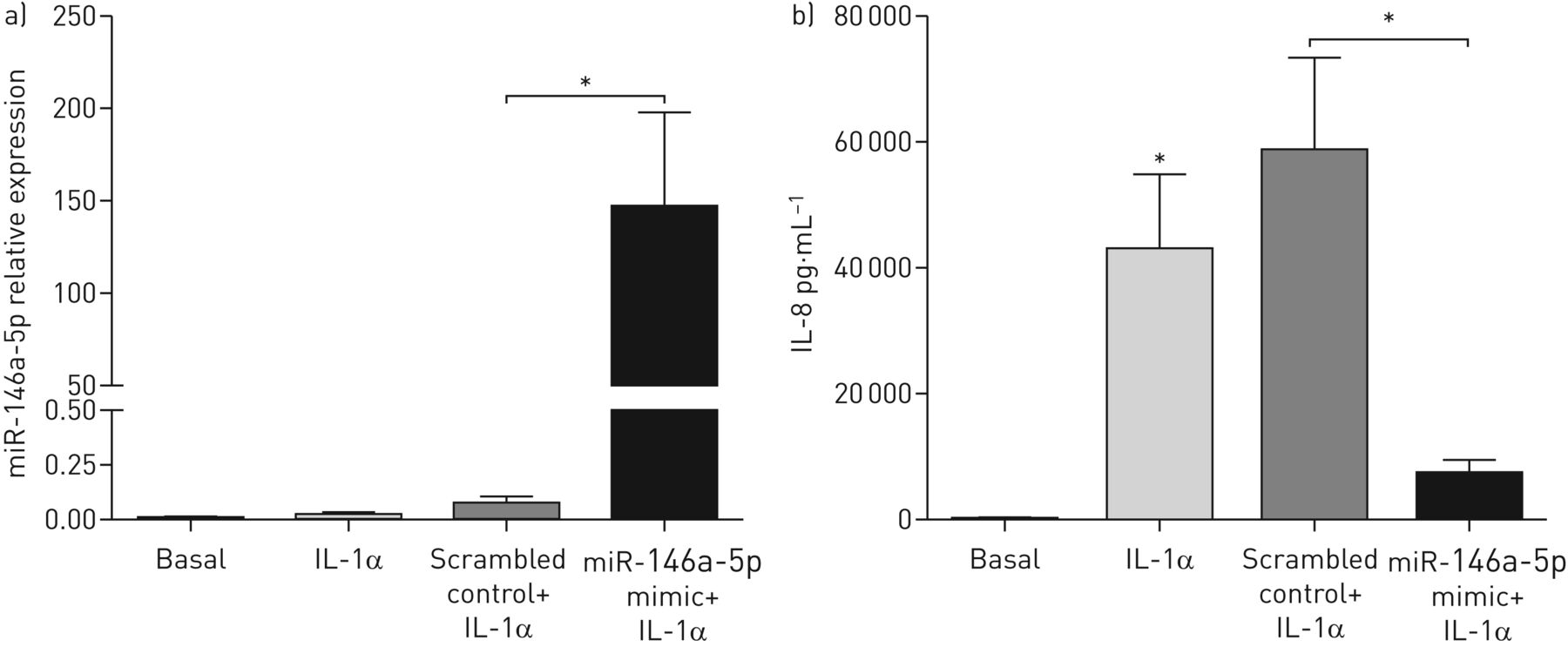
为了确定miR-146a-5p在抑制il -1α诱导的IL-8释放中的作用,我们在miR-146a-5p过表达后用rhIL-1α处理MRC-5成纤维细胞(图5一个).在这里,我们发现当miR-146a-5p过表达时,与杂乱对照处理的细胞相比,MRC-5成纤维细胞中IL-8的释放显著减少(图5 b).综上所述,我们的数据显示miR-146a-5p在肺成纤维细胞中调节il -1α诱导的促炎反应(图6).
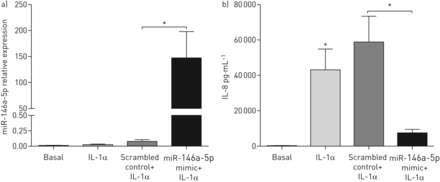
miR-146a-5p减少白细胞介素(IL)-1α-诱导的肺成纤维细胞IL-8的释放。将MRC-5成纤维细胞播种,立即用25 nM miR-146a-5p mimic或打乱对照转染48小时。然后将MRC-5成纤维细胞剥夺血清过夜,用1 ng·mL或不加1 ng·mL刺激−1重组人IL-1α 24 h。a) miR-146a-5p mRNA在MRC-5成纤维细胞中的表达与管家性非编码RNU48相关,表达为2——ΔCt.数据以均数±表示扫描电镜.b) MRC-5成纤维细胞释放IL-8。数据以均数±表示扫描电镜(n=3或4个独立实验)。*: p<0.05,在指示值之间。
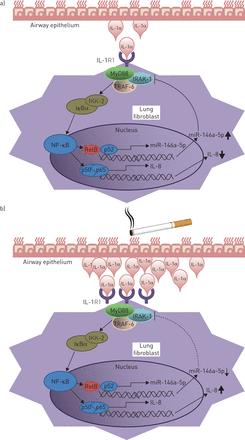
miR-146a-5p在气道上皮细胞(AECs)和肺成纤维细胞之间交叉对话中的作用[12,14,20.,22,24].a)在对照组中,上皮来源的白细胞介素(IL)-1α诱导miR-146a-5p表达以及肺成纤维细胞释放IL-8。然后,miR-146a-5p结合并下调IL-1通路下游的IL-1受体(IL-1R)相关激酶(IRAK)-1的表达,以抑制NF-κB激活和上皮来源的IL-1α对肺成纤维细胞的炎症作用。b)在慢性阻塞性肺疾病(COPD)中,吸烟暴露导致aec中IL-1α的释放增加,从而进一步增加成纤维细胞中IL-8的释放。然而,在copd来源的成纤维细胞中,与对照来源的成纤维细胞相比,il -1α诱导的miR-146a-5p表达的增加较低,这有助于降低NF-κB激活的反馈抑制和夸大的促炎反应。MyD88:髓系分化第一反应基因88;肿瘤坏死因子受体相关因子;IKK: IκB激酶。
讨论
我们研究了miR-146a-5p在COPD患者气道上皮细胞和肺成纤维细胞之间异常IL-1α信号通路中的作用。我们发现PHLFs与AECs共培养显著增加了miR-146a-5p的表达,而miR-146a-5p完全依赖于上皮来源的IL-1α。我们证明miR-146a-5p的表达具有抗炎作用,通过下调IL-1通路下游的IRAK-1的表达,随后减少肺成纤维细胞IL-8的释放。此外,我们发现miR-146a-5p在COPD成纤维细胞中的诱导显著降低,这与miR-146a-5p基因中的SNP rs2910164 (GG等位基因)有关。
miR-146a-5p在先天免疫和适应性免疫中作为细胞功能的调节因子已被充分研究[23].具体而言,miR-146a-5p被认为靶向各种炎症途径,包括toll样受体和IL-1受体信号转导[23,24].在肝脏中过表达miR-146a-5p可以阻止促炎细胞因子的释放,并通过靶向和降低IL-1通路下游的IRAK-1和TRAF-6的蛋白表达来保护小鼠免受缺血-再灌注损伤[22].P越橘et al。[13]表明miR-146a-5p的过表达降低了单个培养的肺泡上皮细胞中il -1β诱导的IL-8的产生。此外,Bhaumiket al。[25]发现miR-146a-5p在衰老的人类新生儿包皮成纤维细胞中比静止细胞高表达。这种高表达反映了一种负反馈机制,由于成纤维细胞中强大的衰老相关分泌表型活性,调节il -1α诱导的IL-6和IL-8的分泌[25].与我们目前的研究一致,这种效应与IRAK-1的抑制有关,但与TRAF-6无关。25].这一发现尤其重要,因为肺部上皮细胞和成纤维细胞等各种细胞类型的衰老已被证明有助于COPD的发病机制[26].
IL-1α是先天性免疫反应的重要驱动因素[27].该细胞因子组成性地存在于肺上皮中,作为免疫防御吸入颗粒的一部分,并负责释放趋化因子,如IL-8,负责中性粒细胞募集[27,28].COPD患者与对照组相比,痰液和支气管肺泡灌洗液中IL-1α水平升高,表明IL-1α释放增加[29].我们之前的研究表明,与对照组相比,暴露于CSE诱导COPD患者气道上皮中IL-1α的表达更强,导致肺成纤维细胞共培养时IL-8和IL-6等促炎介质的释放增强[14].此外,我们发现IL-1α在基线时分泌,并在肺成纤维细胞中负责促炎开关[14].
在本研究中,miR-146a-5p的过表达减少了肺成纤维细胞il -1α诱导的IL-8分泌。目前存在几种调节IL-1α作用的调节机制在活的有机体内,例如天然存在的IL-1受体拮抗剂(IL-1Ra)和诱饵IL-1R2受体的分泌[27,30.].除了这些机制外,miR-146a-5p已成为IL-1通路的关键调节因子[25].与之前的研究一致[22,25],我们发现miR-146a-5p的诱导依赖于IL-1α的刺激,也会导致IRAK-1蛋白表达下调。IRAK-1是一种重要的丝氨酸/苏氨酸激酶,在刺激时与IL-1R1受体复合物结合[31].这最终导致激活蛋白AP-1和NF-κB等转录因子的激活,随后导致包括IL-8在内的几种炎症介质的诱导和释放[31].因此,我们假设il -1诱导miR-146a-5p的升高在与aec共培养时以负反馈回路调节观察到的肺成纤维细胞促炎活性。CSE预刺激上皮细胞后,miR-146a-5p的诱导进一步增强。这表明对miR-146a-5p诱导的需求增加,作为一种负反馈机制,以抵消吸烟者气道上皮细胞IL-1α释放的增强。
有趣的是,与对照肺成纤维细胞相比,与上皮细胞共培养时观察到的miR-146a-5p表达的增加在copd来源的肺成纤维细胞中更小。与我们之前的研究一致[14], miR-146a-5p的诱导减少可能导致对成纤维细胞来源的炎症的反馈抑制受损,这是由暴露于CSE的copd来源的上皮中IL-1α的较高产量引起的(图6).年代atoet al。[12]还发现,与健康受试者相比,IL-1β/TNF-α刺激后,COPD患者成纤维细胞中miR-146a-5p的诱导更少。这种下调与环氧化酶-2酶的表达增加和炎症介质前列腺素E的产生增加有关2在慢性阻塞性肺病患者的痰中[12].我们的模型中PHLFs中miR-146a-5p的诱导差异仅在共培养72 h后可见,但在上皮条件培养基刺激PHLFs 24 h后未见。这表明,需要长时间暴露于IL-1α来诱导COPD和对照来源的成纤维细胞之间的差异miR-146a-5p诱导,这可能是COPD肺上皮-间充质营养单位的代表,慢性暴露于香烟烟雾和由此产生的上皮来源的IL-1α [14,19,32].这也符合P越橘et al。[13],他提出了IL-1对miR-146a-5p表达的时间和浓度依赖效应。
为了阐明copd衍生的PHLFs与上皮细胞共培养后降低miR-146a-5p诱导的潜在机制,我们研究了RelB的可能参与,RelB是NF-κB家族的成员,已被证明可调节miR-146a-5p的表达[20.].虽然年代heridanet al。[33]报道,与非吸烟者相比,患有或不患有COPD的吸烟者的PHLFs中RelB的表达较低,在我们的共培养模型中,我们没有发现非COPD和COPD衍生的PHLFs中RelB mRNA的表达差异。主要miR-146a-5p序列中常见的G>C SNP rs2910164与成熟miR-146a-5p表达的降低有关[21].有趣的是,我们发现来自SNP rs2910164 GG等位基因纯合子的供者的PHLFs(除了一个来自COPD患者外)在共培养后比来自该等位基因杂合的供者的成纤维细胞诱导miR-146a-5p的能力更低。这表明,在我们的共培养模型中,该SNP的表达与copd来源的肺成纤维细胞中miR-146a-5p诱导降低有关。有趣的是,W盎et al。[34]的研究表明,这种特殊的SNP也与COPD相关的低miR-146a-5p表达相关。
总之,这项研究表明,在我们的共培养模型中,由上皮细胞-成纤维细胞相互作用失调导致的COPD肺成纤维细胞的促炎表型可能(至少部分)是由于COPD来源的成纤维细胞上调miR-146a-5p来反调节促炎活性的能力降低。miRNAs可能具有治疗潜力[5] miRNA疗法最近进入了临床试验阶段[35].因此,我们的发现可以为进一步针对COPD慢性炎症的研究提供基础。
补充材料
披露的信息
确认
作者贡献:E.T. Osei, L. Florez-Sampedro, D.S. Postma, W. Timens, T.L. Hackett, I.H. Heijink和C-A。Brandsma参与了研究设计和概念化。E.T. Osei, L. Florez-Sampedro, H. Tasena, J.A. Noordhoek和A. Faiz进行了实验。A. Faiz进行SNP分析。E.T.欧塞分析了实验数据。c - a。Brandsma和I.H. Heijink参与了实验的监督。E.T.欧塞写了手稿。c - a。Brandsma和I.H. Heijink修改了手稿。 D.S. Postma, W. Timens and T.L. Hackett edited the manuscript. The final version was critically reviewed and approved by all authors.
脚注
这篇文章有补充资料可从www.qdcxjkg.com
支持声明:本研究由Jan Kornelis de Cock Stichting和Stichting Astma Bestrijding资助。本文的资助信息已存入交叉参考基金注册.
利益冲突:可以在本文旁边的网站上找到信息披露www.qdcxjkg.com
- 收到了2016年5月10日。
- 接受2017年1月29日。
- 版权所有©ERS 2017











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Proposed role of miR-146a-5p in the cross-talk between airway epithelial cells (AECs) and lung fibroblasts [12, 14, 20, 22, 24]. a) In controls, epithelial-derived interleukin (IL)-1α causes an induction of miR-146a-5p expression as well as release of IL-8 from lung fibroblasts. miR-146a-5p then binds to and downregulates the expression of IL-1 receptor (IL-1R)-associated kinase (IRAK)-1 downstream of the IL-1 pathway in a feedback loop to dampen the NF-κB activation and the inflammatory effects of the epithelial-derived IL-1α on pulmonary fibroblasts. b) In chronic obstructive pulmonary disease (COPD), cigarette smoke exposure causes an increased IL-1α release from AECs which further increases IL-8 release from fibroblasts. However, in COPD-derived fibroblasts, the IL-1α-induced increase in miR-146a-5p expression is lower compared with control-derived fibroblasts, which then contributes to lower feedback inhibition of the NF-κB activation and an exaggerated pro-inflammatory response. MyD88: myeloid differentiation primary response gene 88; TRAF: tumour necrosis factor receptor-associated factor; IKK: IκB kinase.](http://www.qdcxjkg.com/content/erj/49/5/1602538/F6.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}