





摘要
特发性肺纤维化(IPF)是一种进行性且最终致命的疾病,其特征是肺实质纤维化和肺功能丧失。尽管IPF涉及的致病途径尚未完全阐明,但IPF被认为是由反复的肺泡上皮细胞损伤和修复失调引起的,其中肺成纤维细胞不受控制地增殖,成纤维细胞分化为肌成纤维细胞,细胞外基质(ECM)蛋白过度沉积在间质空间。包括血小板衍生生长因子(PDGF)、成纤维细胞生长因子(FGF)和转化生长因子-β在内的许多促纤维化介质被认为在IPF的发病机制中发挥重要作用。Nintedanib是一种有效的酪氨酸激酶PDGF受体、FGF受体和血管内皮生长因子受体的小分子抑制剂。数据从在体外研究表明,nintedanib干扰纤维化的活性过程,如成纤维细胞增殖、迁移和分化以及ECM的分泌。此外,在肺纤维化的动物模型中,nintedanib显示出一致的抗纤维化和抗炎活性。这些数据为nintedanib在IPF患者中的临床疗效提供了强有力的理论依据,最近已在III期临床试验中得到证实。
摘要
Nintedanib干扰纤维化过程,如。成纤维细胞增殖、迁移和分化http://ow.ly/Iae9z
简介
Nintedanib(国际非专利名称),其开发代码为BIBF 1120,是一种小分子,最初被设计为成纤维细胞生长因子受体(FGFR)-1和血管内皮生长因子受体(VEGFR)-2的atp竞争性抑制剂。这两种受体都是促血管生成受体酪氨酸激酶,而nintedanib被设计为一种用于癌症适应症的抗血管生成药物。nintedanib用于癌症适应症(包括非小细胞肺癌、结直肠癌和卵巢癌)的临床开发正在进行中。由于nintedanib也是一种血小板衍生生长因子受体(PDGFR)-α和β抑制剂,因此被选为特发性肺纤维化(IPF)的潜在治疗方法。IPF的病理生物学显示出若干惊人的相似性和与癌症生物学的联系(在[1])也支持了在IPF中探索nintedanib的基本原理。在两项INPULSIS®重复III期研究中[2], nintedanib被证明可以通过降低强迫肺活量(FVC)的年下降率来减缓IPF患者的疾病进展[3.].本文综述了目前对尼达尼布在纤维化肺疾病如IPF中的作用模式的认识。本文解释了nintedanib主要靶受体酪氨酸激酶在IPF中的作用。在体外来自IPF患者和对照供者的原代肺成纤维细胞的实验在活的有机体内综述了肺纤维化动物模型的研究。综上所述,这些研究证明了尼达尼布具有有效的抗纤维化和抗炎活性。这些特征可能解释了接受尼达尼布治疗的IPF患者疾病进展减缓的原因。
特发性肺纤维化
IPF是一种破坏性和致残的进行性肺部疾病,诊断后中位生存期仅为2-3年[4].IPF是特发性间质性肺炎最常见的形式[5],发病率在每10万人口0.22至17.4之间,取决于病例定义、区域和使用的方法[6].指规数通常出现在生命的第六个或第七个十年,男性比女性更常发生[4,7].在2011年发布的国际指规数管理指南中[4],强烈推荐的治疗方案仅限于补充氧气和肺移植。尽管有很高的医疗需求,但这些指南并未推荐任何特定的药物疗法用于IPF的慢性治疗[4].吡非尼酮于2008年在日本上市,用于治疗IPF,并基于两项III期临床试验(capability -1和2)的结果[8之后在欧洲、加拿大、韩国、中国、印度、墨西哥和阿根廷等多个国家和地区推出。另外一项III期研究(ASCEND)被要求支持其在美国的监管注册,该试验的积极结果最近已发表[9].随着nintedanib治疗IPF疗效和安全性的III期临床结果的发表(INPULSIS®-1和INPULSIS®-2)[3.]以及最近美国食品和药物管理局批准吡非尼酮和尼达尼布治疗IPF,经过十多年令人失望和失败的临床试验,医生们很快就会选择治疗IPF的方法。
IPF的病理特征是反复的微观肺泡上皮细胞损伤和修复失调,纤维化和细胞外基质(ECM)过度沉积,导致实质结构和肺功能的丧失[10,11].致病的伤害尚不清楚,但吸烟、环境和职业暴露、病毒感染、胃酸反流和遗传易感性已被报道为IPF的危险因素[4,7,12,13].在IPF中,成纤维细胞增殖不受控制,分化为肌成纤维细胞,过度产生ECM成分。聚集成簇的肌成纤维细胞(成纤维细胞灶)被认为是肺纤维化发展的标志细胞[14].肺纤维化中肌成纤维细胞的起源仍有争议[15].纤维化肺中的细胞池可能通过多种机制进行补充,包括上皮-间充质转化(EMT) [16],纤维细胞募集[17],周细胞转分化[18]、胸膜间皮细胞[19]或局部成纤维细胞数量的增加[20.].IPF患者肺部纤维化细胞池的凋亡受损,正常肺结构的再上皮化和恢复也随之受损[21].
IPF患者肺部微循环明显改变。对于一种以斑块状纤维化接近正常肺实质为特征的疾病,这些微血管异常是不均匀的,这并不奇怪。已发表的关于人类和实验性肺纤维化的报告表明,纤维化区域血管较少,而相邻的非纤维化组织高度血管化[22- - - - - -25].目前尚不清楚的是,在纤维化最少的区域毛细血管密度的增加是一种代偿反应,还是新生血管在IPF中失调的基质重塑中起关键作用[26].
白细胞介素(IL)-1β、肿瘤坏死因子-α、结缔组织生长因子(CTGF)、转化生长因子(TGF)-β、白细胞介素-13、血小板衍生生长因子(PDGF)和成纤维细胞生长因子(FGF)等多种原纤维化介质及其信号级联被认为在纤维化肺疾病的发病机制中发挥重要作用[27- - - - - -29].例如,IL-13是成纤维细胞增殖和ECM合成的有效刺激剂;它诱导纤维化前细胞因子,包括TGF-β、PDGF和CTGF,以及ECM成分,包括胶原蛋白1和纤维连接蛋白[30.].TGF-β1具有多种功能:促进成纤维细胞趋化和增殖,使成纤维细胞分化为肌成纤维细胞和EMT,保护肌成纤维细胞不发生凋亡[30.].TGF-β促进一系列前纤维化细胞因子和金属蛋白酶组织抑制剂(TIMPs)的产生,并抑制基质降解蛋白酶[30.].以下部分将重点讨论IPF纤维化病变中与成纤维细胞和肌成纤维细胞池的增殖、迁移和转分化有关的生长因子和生长因子受体,以及nintedanib的靶点。
PDGF/PDGFR信号在肺纤维化中的作用
PDGF包括一个二聚体蛋白家族,由4个多肽链组成:a, B, C和d。这些二聚配体与PDGFR-α和β的同二聚体或异二聚体相互作用。该受体的细胞外区域由五个免疫球蛋白样结构域组成,而细胞内部分是一个酪氨酸激酶结构域。配体结合导致受体的自磷酸化和随后的下游信号通过Ras, Raf,丝裂原活化蛋白激酶(MEK),细胞外信号调节激酶(ERK)和磷脂酰肌醇-4,5-二磷酸3-激酶(PI3K) (图1) [28].PDGF是成纤维细胞的有效有丝分裂原[31]并且似乎通过刺激增殖、迁移和存活在肌成纤维细胞的扩张中发挥着重要作用[28].当过于活跃或数量过多时,肌成纤维细胞会在间质空间沉积过多的结缔组织产物。其结果是肺泡结构扭曲,气体交换受损[4,32].在人肺中,PDGF主要由肺泡巨噬细胞和上皮细胞产生[28,33].对IPF患者肺部肺泡巨噬细胞的评估表明,这些细胞自发释放的PDGF是对照组肺泡巨噬细胞的四倍[34].
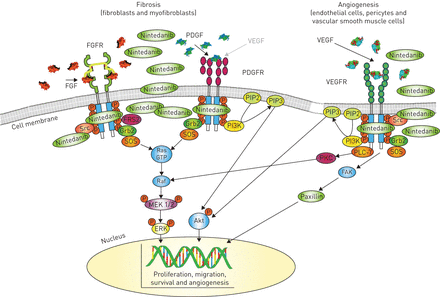
尼达尼布的多药理学及其下游信号通路。Nintedanib结合到成纤维细胞生长因子受体(FGFRs)、血小板源性生长因子受体(PDGFRs)和血管内皮生长因子受体(VEGFRs)的细胞内ATP结合袋,导致这些受体的自磷酸化和下游信号级联的阻塞。Nintedanib可能通过直接阻断Src和Lck等非受体激酶发挥额外的活性(未说明)。Nintedanib被证明可以抑制小鼠肺组织中PDGFR磷酸化和随后的蛋白激酶B (Akt)和细胞外信号调节激酶(ERK)1/2磷酸化。血管内皮生长因子(VEGF)可刺激血管生成通过也可以与成纤维细胞上的PDGFR结合,刺激增殖。nintedanib的抑制最终导致成纤维细胞的增殖、迁移和存活减少,并可能减弱肺中的血管生成。FAK:黏附激酶;FGF:成纤维细胞生长因子;FRS2: FGFR底物2;Grb2:生长因子受体结合蛋白2;MEK1/2:丝裂原活化蛋白激酶激酶1/2;PDGF:血小板衍生生长因子;PI3K:磷脂酰肌醇-4,5-二磷酸3-激酶;PIP2/3: phosphatidylinositol-2/3-phosphate; PKC: protein kinase C; PLC-γ: phospholipase C-γ; SOS: son of sevenless, a guanine nucleotide exchange factor that acts on the Ras GTPases.
继发纤维化的肺损伤动物模型对于确定PDGF在纤维化进展中的作用是非常宝贵的。pdgfr特异性酪氨酸激酶抑制剂减少小鼠辐射诱导的肺纤维化[35,36],以及五氧化二钒致大鼠肺纤维化[37].伊马替尼,一种PDGFR和c-Abl抑制剂,被证明可以减少博莱霉素诱导的小鼠肺纤维化[38,39],以及石棉致小鼠间质性肺炎[40].然而,在IPF和轻度至中度肺功能损害患者的II期临床试验中,伊马替尼或安慰剂治疗96周,伊马替尼不影响生存或肺功能[41].仅在48周时出现短暂的氧合改善[41].这些临床结果表明,尽管PDGFR抑制在动物模型中持续减少肺纤维化,但仅抑制PDGFR可能不足以治疗IPF。
FGF/FGFR信号在肺纤维化中的作用
在人类中,FGF家族有22个结构相关的家族成员。FGF家族中最重要的配体是FGF-1和FGF- 2。FGFs与硫酸肝素糖胺聚糖相互作用,22种已知的FGFs中有18种与FGFRs的胞外结构域结合,从而诱导受体激活[42].FGFR家族有四个成员:FGFR-1、2、3和4。每个受体由三个细胞外免疫球蛋白型结构域(D1-D3)、一个单跨膜结构域和一个细胞内分裂酪氨酸激酶结构域[43].FGFs和FGFRs调节重要的细胞功能,如细胞增殖、分化、迁移和存活[44].一旦FGFRs识别它们的FGF配体,它们就会经历二聚化,然后是自磷酸化和下游信号的激活通过成纤维细胞生长因子受体底物2,PI3K/蛋白激酶B (Akt), ERK1/2和Ras/Raf/丝裂原活化蛋白激酶(MAPK)通路对生长和细胞生存至关重要(图1) [45].
FGF-2是成纤维细胞的有效有丝分裂原[46],气道平滑肌细胞[47]和第II型肺泡上皮细胞[48].FGF-2诱导肺成纤维细胞胶原合成[46]和肌成纤维细胞[49].肺泡巨噬细胞、成纤维细胞、t淋巴细胞和内皮细胞能够产生FGF-2 [50- - - - - -52].然而,在IPF患者中,肥大细胞似乎是FGF-2的主要来源[49].在IPF患者的肺部检测到FGF-2水平增强,上皮细胞、内皮细胞和平滑肌细胞/肌成纤维细胞样细胞上FGFR-1表达增加,间质细胞上FGFR-2表达增加[49,53].FGF-9蛋白在活跃的细胞增生、化生和纤维化扩张区域升高[54].FGF-1和FGF-10也可能具有抗肺纤维化作用。人肺成纤维细胞的研究结果表明FGF-1可降低成纤维细胞的生长速度,诱导成纤维细胞凋亡并抑制向肌成纤维细胞分化[55].FGF-10已被证明促进肺泡上皮细胞修复[56]和FGF-10减毒博莱霉素诱导小鼠肺纤维化过表达[57].
TGF-β是FGF/FGFR信号级联的调节因子,证明了许多生长因子之间的复杂相互作用。TGF-β诱导人肺成纤维细胞FGFR-1和FGF-2上调[58]以及原发性肺成纤维细胞立即释放FGF-2 [59].TGF-β诱导的肺间质成纤维细胞增殖被抗fgf -2抗体阻断[59].
FGF/FGFR信号在肺纤维化动物模型中的作用也已被证明。FGFR-1在健康大鼠的肺中未检测到,但在百草枯加高氧诱导的弥漫性肺纤维化模型中强烈上调[60].体内,在博莱霉素处理的小鼠中,FGF信号的取消减少了肺纤维化并提高了生存率[61].综上所述,这些数据表明FGF/FGFR信号通路在肺纤维化疾病中起着重要作用。
VEGF/VEGFR信号在肺纤维化中的作用
血管内皮生长因子(VEGF)家族包括结构同源的分泌糖蛋白VEGF- a、B、C和D,以及胎盘生长因子[62].VEGF-A和B在血管生长调控中起关键作用,VEGF-C和D主要调控淋巴管生成[63].VEGF家族中最具代表性的成员是VEGF- a [62].VEGF家族的成员结合到三个VEGF受体:VEGFR-1, 2和3。vegfr在结构上相似,具有一个细胞外区域、一个跨膜螺旋和一个细胞质区域[62].VEGFR的激活导致受体的自磷酸化和下游信号通路的激活,包括Ras,磷脂酶C-γ,局部粘附激酶,p38和PI3K (图1) [63].肺内VEGF的主要来源为肺泡上皮细胞、支气管上皮细胞、气道平滑肌细胞、成纤维细胞、内皮细胞和肺泡巨噬细胞[64,65].VEGF-A也被证明可以刺激PDGFRs,从而调节间充质细胞的迁移和增殖(图1) [66].
据报道,在IPF患者中,基线血浆VEGF浓度与高分辨率计算机断层扫描上的纤维化评分呈正相关[67].此外,发生进展性疾病(从基线到第6个月FVC下降≥10%)的IPF患者基线时血浆VEGF浓度明显高于非进展者[67].与低浓度VEGF患者相比,高浓度VEGF患者的5年生存率更差[68].此外,血清VEGF水平似乎可以预测这些患者的肺活量恶化。然而,据报道IPF患者支气管肺泡灌洗液(BALF)中的VEGF水平降低[23,69,70].在肺纤维化大鼠模型中,VEGF在纤维化病变中几乎不表达,但在上皮细胞和内皮细胞中表达强烈[25].同样,在IPF患者中,纤维化病变的成纤维细胞和白细胞中VEGF表达较低,但在高血管化的肺泡间隔中毛细血管内皮细胞和肺泡II型上皮细胞中VEGF表达升高[22].大鼠实验证据表明,抑制VEGFR可减少纤维化[71]而VEGF的施用会加重纤维化过程[25].然而,VEGF被证明在肺中具有血管保护作用,并改善肺动脉高压作为IPF的合并症[24,25].因此,VEGF/VEGFR信号在IPF中的确切作用存在争议,有待进一步探讨。
Nintedanib是受体酪氨酸激酶PDGFR, FGFR和VEGFR的有效抑制剂
Nintedanib是一种吲哚啉酮衍生物,源自一种用于受体酪氨酸激酶抑制剂的化学先导优化程序(专利申请WO2001027081,例473)。Nintedanib最初是一种针对受体酪氨酸激酶VEGFR, FGFR和PDGFR的抗血管生成药物,用于治疗癌症[72].Nintedanib目前处于II期或III期临床开发阶段,用于多种癌症适应症(clinicaltrials.gov标识号:NCT00805194,NCT00806819而且NCT01015118).
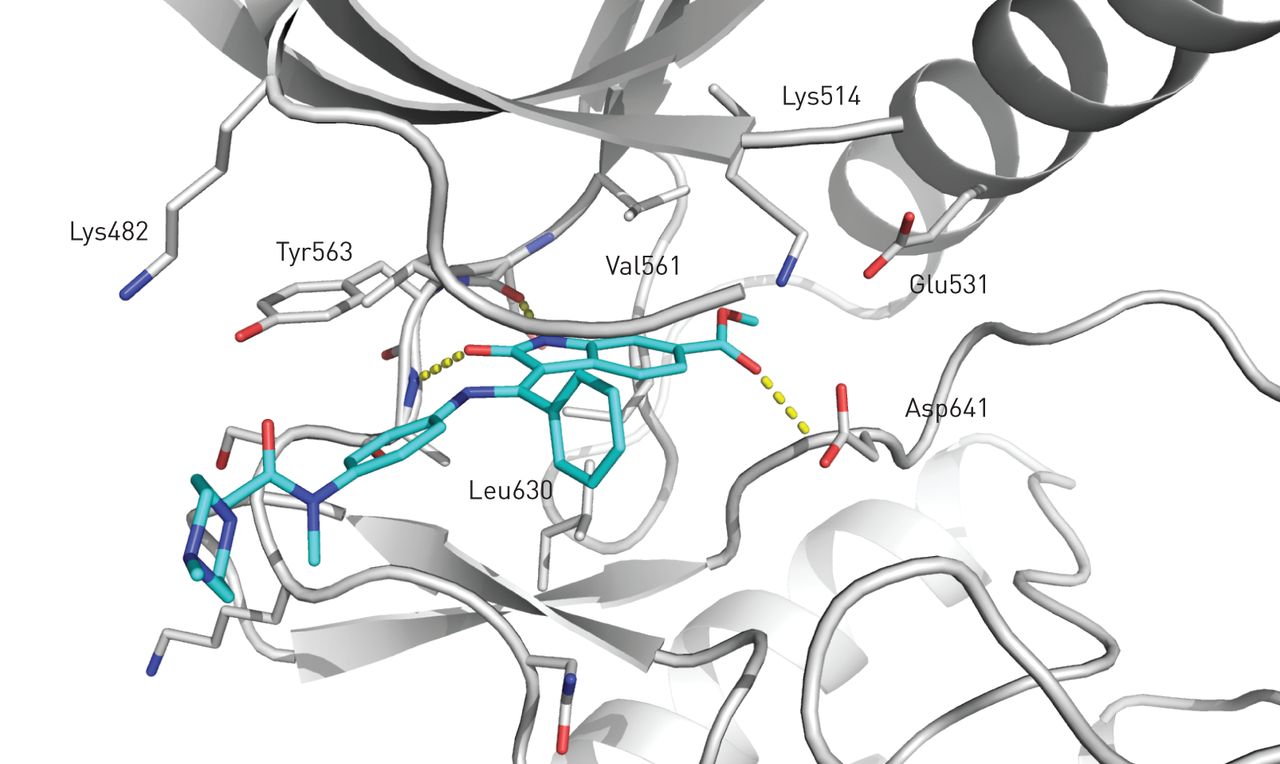
Nintedanib通过占据特定酪氨酸激酶的细胞内atp结合袋来阻断激酶活性。探索FGFR-1和VEGFR-2的结合模式(图2,在线补充材料A节和图S1)。只有细微的差异被发现,这在在线补充材料A部分进行了讨论。
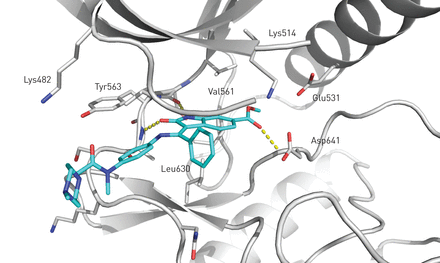
nintedanib的晶体结构与成纤维细胞生长因子受体-1激酶结构域的活性位点结合。沿活性部位排列的抑制剂和残基以棒状表示。氢键用黄色的圆点表示。关键残基被标记。
在使用人重组蛋白激酶结构域的酶促试验中确定了nintedanib的效力和选择性[72].Nintedanib是一种受体酪氨酸激酶PDGFR-α/β的抑制剂,具有一半最大抑制浓度(IC50)值分别为59和65 nmol·L−1,分别。Nintedanib通过IC抑制FGFR-1、2、3和450分别为69、37、108和610 nmol·L−1vegf -1、2、3与IC结合50值为34、21和13 nmol·L−1,分别[72].Nintedanib也通过IC抑制fms样酪氨酸激酶-3 (Flt-3)5026nmol·L−1[72].Flt-3在许多造血祖细胞表面表达,在造血中发挥重要作用[73].
Nintedanib还抑制IC的Src家族的非受体酪氨酸激酶50值为156 nmol·L−1Src为16 nmol·L−1Lck和195 nmol·L−1对于Lyn [72].Src在功能上参与控制多种细胞过程,如增殖、分化、运动和粘附。Src家族激酶抑制剂AZD0530已被报道在人肺成纤维细胞和博莱霉素诱导的小鼠肺纤维化中发挥抗纤维化作用[74].Lck是通过t细胞抗原受体激活t细胞所必需的功能[75]并且可能是t细胞存活所必需的[76].Lyn在b细胞抗原受体信号传导过程中既有正向作用,也有负向作用,在b细胞抗原受体信号启动和b细胞增殖过程中发挥作用[77].
Nintedanib干扰多种重要的纤维化过程在体外化验,在活的有机体内模型
对成纤维细胞增殖和迁移的影响
在来自IPF患者(IPF- hlf)和对照供体(N-HLF)的原代人肺成纤维细胞的细胞水平上证实了尼达尼布的抑制活性。Nintedanib抑制N-HLF中pdgf - bb刺激的PDGFRα和β自磷酸化50值为22和39 nmol·L−1和pdgf - bb刺激的增殖在同一组实验中使用IC5064nmol·L−1(图3) (78].在IPF-HLF中,PDGF-BB、FGF-2和VEGF引起显著的促增殖作用,而nintedanib显著逆转。在N-HLF中,即使较低浓度的尼达尼布也能显著拮抗生长因子诱导的促增殖作用。与N-HLF相比,IPF-HLF表现出对所有三种生长因子更强的迁移反应。生长因子诱导的细胞增殖在IPF-HLF和N-HLF中均被nintedanib显著拮抗[79].在时间推移显微镜研究中,nintedanib以浓度依赖的方式抑制PDGF和fgf刺激的成纤维细胞运动。Nintedanib减弱pdgf刺激的IPF-HLF和N-HLF与IC的运动能力50值为28和19 nmol·L−1和fgf刺激的IC运动50值为226和86 nmol·L−1,分别(图3b)。
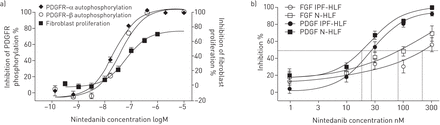
a) Nintedanib抑制血小板衍生生长因子(PDGF)- bb刺激的血小板衍生生长因子受体(PDGFR)-α和β自磷酸化和原代人肺成纤维细胞(HLFs)的增殖。用不同浓度的nintedanib孵育HLFs,并用PDGF-BB (50 ng·mL)刺激HLFs−1).采用酶联免疫吸附法检测磷酸化受体的PDGFR-α和β磷酸化情况。用溴脱氧尿苷掺入法测定增殖。浓度依赖性抑制数据以均数±表示扫描电镜(n = 3实验)。转载自[78,并得到出版商的许可。b) Nintedanib抑制特发性肺纤维化(IPF-HLF)和无IPF (N-HLF)患者的碱性成纤维细胞生长因子(FGF)和pdgf - bb刺激的原发性HLF的运动性。人肺成纤维细胞用不同浓度的nintedanib孵育30分钟,然后用基础FGF (20 ng·mL)刺激细胞−1)或PDGF-BB (50 ng·mL−1).使用cell - iq成像仪(CM Technologies Oy, Tampere, Finland)进行时间延迟显微镜检测细胞的运动性,并进行72小时的手动单细胞跟踪。平均细胞速度由三个实验计算。抑制表现为平均±扫描电镜.
EMT的影响
虽然上皮细胞能够分化为间充质细胞[80], EMT在IPF诊断时和进一步进展时的重要性仍存在争议[81].Nintedanib浓度≥300 nmol·L−1没有改变来自无IPF供体的原代肺泡II型上皮细胞的形态,对TGF-β诱导的(2 ng·mL−1) EMT(在线补充资料B部分)。
成纤维细胞向肌成纤维细胞转化的影响
Nintedanib抑制TGF-β诱导(10 ng·mL−1IPF患者的原代人肺成纤维细胞向肌成纤维细胞的转变,通过α-平滑肌肌动蛋白mRNA的表达作为肌成纤维细胞分化的标志物来确定,并估计IC50值为144 nmol·L−1[78].
对ECM组件的影响
Nintedanib可减少IPF患者原代人肺成纤维细胞培养48 h后TGF-β刺激的胶原分泌和沉积。Nintedanib还可降低TIMP-2分泌水平[78]并诱导原基质金属蛋白酶(MMP)-2的分泌[79].TIMP-2水平的降低,以及mmp -2的增加,可能会导致胶原蛋白的减少。nintedanib抑制成纤维细胞/肌成纤维细胞增殖也可能间接抑制ECM的分泌和沉积。
对凋亡的影响
据报道,在IPF患者中,肺成纤维细胞/肌成纤维细胞对阻止纤维化消退的凋亡刺激具有更高的抵抗力[82].nintedanib是否能诱导人肺成纤维细胞凋亡目前尚不清楚。Nintedanib诱导人脐血管内皮细胞(HUVEC)、人脐动脉平滑肌细胞(HUASMC)和牛视网膜周细胞凋亡。以裂解的caspase-3为凋亡指标。在VEGF或FGF-2刺激的HUVECs、PDGF-BB或FGF-2刺激的HUASMCs以及PDGF-BB和FGF-2刺激的周细胞中,nintedanib以浓度依赖的方式诱导凋亡[72].
nintedanib的抗血管生成活性
文献中已经讨论了抗血管生成活性在治疗IPF中的潜在影响,但尚未明确[26].实验证据证实,抑制VEGF、PDGF和FGF信号通路可减少肺部肿瘤血管生成[83].Nintedanib抑制三种有助于肺血管生成的细胞类型的增殖:内皮细胞、周细胞和平滑肌细胞[72].Nintedanib抑制vegf刺激的HUVECs、pdgf - bb刺激的HUASMCs和pdgf - bb刺激的IC牛视网膜周细胞的增殖50值为9、69和79 nmol·L−1,分别[72].此外,nintedanib可降低肿瘤微血管密度,并在小鼠异种移植物肿瘤动物模型中显示出临床前抗血管生成的功效[72].综上所述,实验证据支持nintedanib具有抗血管生成的功效;然而,这是否增加了其在IPF中的抗纤维化活性仍需进一步研究。
肺纤维化动物模型的抗纤维化和抗炎活性
的在活的有机体内在三种肺纤维化动物模型中探索了nintedanib的疗效:博莱霉素诱导的小鼠和大鼠肺纤维化和二氧化硅诱导的小鼠肺纤维化。研究背景和治疗方案总结在表1定性读数总结在表2.
简单地说,pdgf诱导的PDGFR磷酸化作为受体激活标记是通过小鼠肺组织匀浆的Western blotting确定的。在接受单次气管内PDGF剂量之前,用不同剂量的nintedanib治疗动物。用nintedanib对PDGFR磷酸化的抑制活性来建立nintedanib在小鼠体内的药代动力学/药效学相关性。实验证实,小鼠的有效剂量在30-100 mg·kg范围内−1[78].
博莱霉素对大鼠肺纤维化的影响
在气管内灌注博莱霉素后的第1天开始,Nintedanib口服,每日1次,预防性治疗方案抑制了大鼠的肺纤维化(在线补充材料C部分)。以剂量依赖的方式,在10、30和50 mg·kg剂量时,Nintedanib仅对肺纤维化变化产生最小抑制、部分抑制和几乎完全抑制−1,分别。10 mg·kg时,纤维化相关标记基因(TGF-β1和前胶原蛋白1)mRNA表达部分受到抑制−1.30和50 mg·kg−1检测到近乎完全的抑制(表2).从第10天开始,在治疗方案中给予nintedanib,治疗剂量为50 mg·kg−1根据组织学评估(图S2)和前纤维化标记物的基因表达(图S3),也导致了近乎完全的纤维化衰减。
博莱霉素对小鼠肺纤维化的影响
无论剂量如何,nintedanib的预防性给药均可减少博莱霉素诱导的肺部炎症。BALF中淋巴细胞数量减少,IL-1β水平下降,肺组织中髓样树突状细胞百分比下降,证明了这一点。此外,它降低了肺组织中的TIMP-1和总胶原蛋白水平,以及肺组织形态学分析中的纤维化评分。在治疗方案中,尼达尼布对炎症和纤维化也有显著的抑制作用[78].
对硅致小鼠肺纤维化的影响
预防性给药nintedanib每日一次,剂量分别为30和100 mg·kg−1通过灌胃减少中性粒细胞和淋巴细胞,但对BALF中的巨噬细胞计数无影响。Nintedanib显著降低肺匀浆中IL-1β、CXCL1/KC、TIMP-1和总胶原蛋白,减少肺部炎症、肉芽肿形成和纤维化。第10天开始的治疗减少了BALF中的中性粒细胞和淋巴细胞,总肺胶原蛋白和纤维化评分,与预防性治疗方式相当。在第20天开始治疗时,BALF中只有淋巴细胞显著减少。治疗组IL-1β、CXCL1/KC、TIMP-1、炎症评分、肉芽肿评分降低幅度均小于预防组[78].
临床疗效
第二阶段TOMORROW概念验证研究[84]提示IPF患者接受nintedanib 150 mg每日2次52周的治疗与FVC下降和恶化的减少以及与健康相关的生活质量的保持有关。两项设计相同的三期国际安慰剂对照双盲临床研究(INPULSIS®-1和INPULSIS®-2)研究了nintedanib对IPF患者的疗效和安全性[2,3.].总共有来自24个国家的1066名患者被随机分为3到2组接受nintedanib 150mg每日两次或安慰剂治疗52周,随后随访4周。主要终点是植被覆盖度的年递减率(mL·年)−1).主要次要终点为第一次急性加重的时间,以及52周内圣乔治呼吸问卷(SGRQ)总评分较基线的变化。与安慰剂相比,Nintedanib通过降低FVC的下降率来持续减缓疾病进展。在INPULSIS®-1中,植被覆盖度调整后的下降速率为114.7 mL·年−1nintedanib组为239.9 mL·年−1安慰剂组差异为125.3 mL·年−1(p < 0.0001)。在INPULSIS®-2中,植被覆盖度调整后的下降速率为113.6 mL·年−1nintedanib组为207.3 mL·年−1在安慰剂组,导致组间差异为93.7 mL·年−1(p = 0.0002)。这些结果的稳健性和一致性被各种敏感性分析以及额外的植被覆盖度结果所证实。此外,治疗效果的主要结果是一致的跨许多预先确定子群分析基于混合数据来自两个INPULSIS®试验,发现一致的治疗效果在子组定义按性别、年龄(< 65或≥65岁)、种族(白色或亚洲),基线FVC %预测(≤70%和> 70%),基线SGRQ总分(≤40或40 >)、吸烟状态(1)never-smoker或2)电流/烟民),全身使用糖皮质激素在基线(是或否),基线时支气管扩张剂使用情况(是或否)[85].在第52周时,INPULSIS®-2组SGRQ总分与基线的平均变化差为−2.69点(p<0.02), INPULSIS®-1组为−0.05点(p=0.97)。INPULSIS®-2中nintedanib组在52周内至少发生一次急性加重的患者比例(3.9%)低于安慰剂组(9.6%),导致风险比(HR)为0.38 (p=0.005),但INPULSIS®-1中没有观察到显著差异(HR 1.15;p = 0.67)。在一项预先指定的敏感性分析中,两项试验的汇总数据对首次确诊或疑似恶化的时间进行了分析,盲法评审委员会将其分类为nintedanib治疗患者的风险显著降低与安慰剂(HR 0.32;p = 0.001)。nintedanib组和nintedanib组中最常见的不良事件是腹泻,影响了62%的患者(安慰剂组为18%)。然而,在几乎所有病例中,腹泻的强度都是轻度或中度,导致使用尼达尼布治疗的患者中只有不到5%的人停止治疗。
总结与结论
Nintedanib是一种口服活性小分子酪氨酸激酶抑制剂,已在大型临床试验中评估用于治疗IPF, IPF是一种致命疾病,肺成纤维细胞不受控制的增殖和肌成纤维细胞的ECM沉积导致肺功能进行性丧失。本文综述了nintedanib在几种疾病中的抑制活性在体外研究肺纤维化病理机制的分析。nintedanib对FGF、PDGF和VEGF受体的多药理学作用,以及对Src等非受体激酶的多药理学作用,可对成纤维细胞和肌成纤维细胞的下游信号级联产生广泛的抑制活性,并可能对肺中参与血管生成的细胞也有抑制作用(图1).Nintedanib对原代人肺成纤维细胞的增殖和迁移具有强烈的抑制活性(图4).此外,在较高浓度下,nintedanib减弱成纤维细胞向肌成纤维细胞的转化和ECM沉积(图4).Nintedanib主要作用于PDGF, FGF和VEGF的下游,但不是唯一的,这些都是IPF发病机制中的主要生长因子。在肺纤维化的动物模型中,这些细胞活性转化为一致的抗纤维化和抗炎活性,独立于纤维化诱导刺激。综上所述,这些结果为nintedanib在IPF中的临床疗效提供了强有力的理论依据,最近已在两项重复的国际III期临床试验中得到证实。
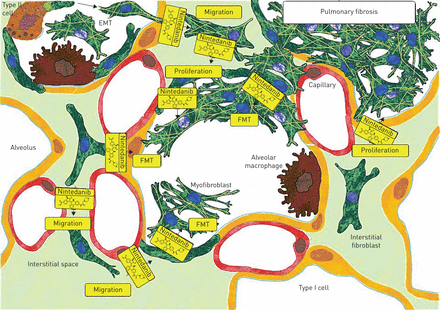
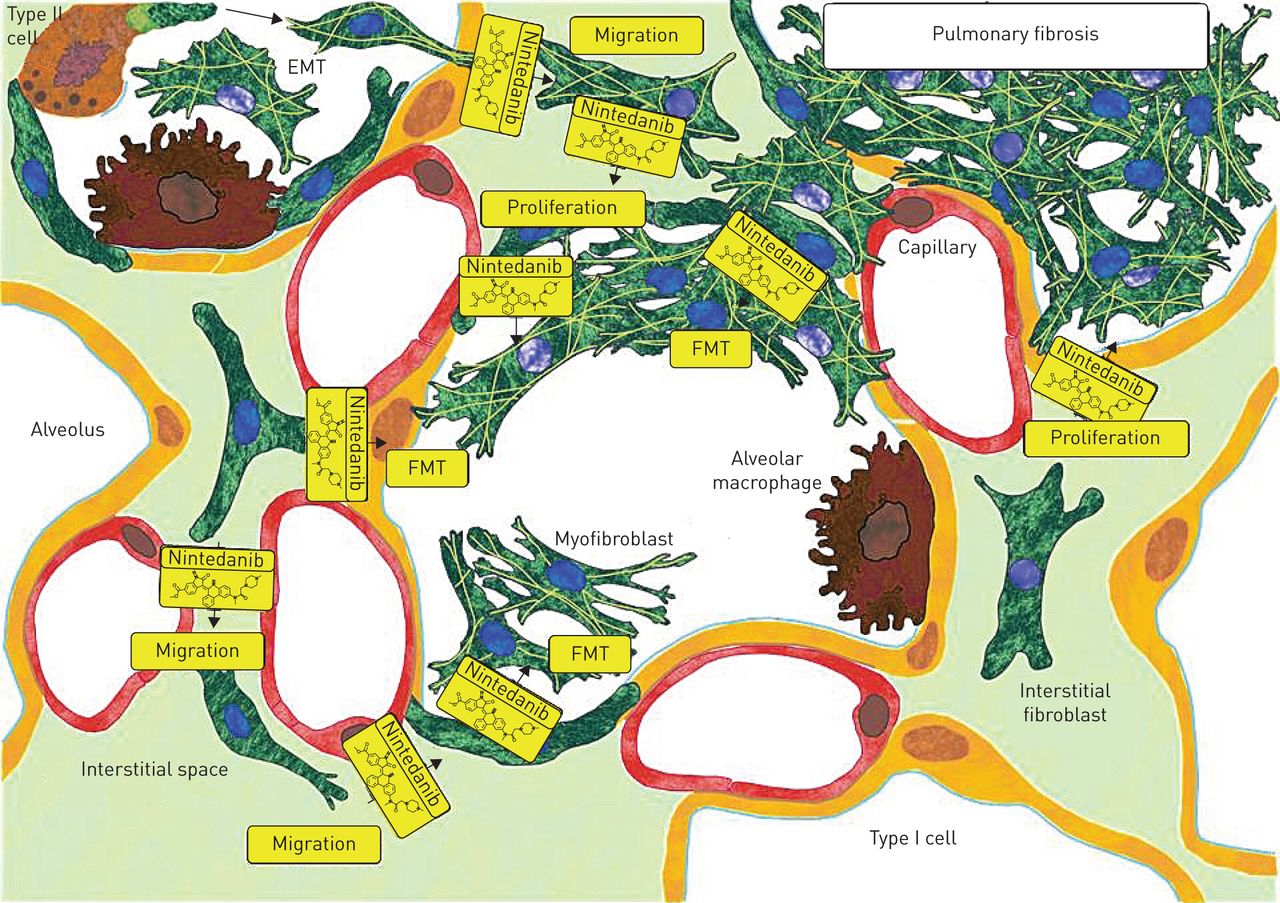
目前对尼达尼布在纤维化肺疾病中的作用模式的了解。该方案反映了特发性肺纤维化(IPF)病理学的持续过程。由于上皮损伤,肺泡上皮细胞发生凋亡,上皮型II细胞转化为肌成纤维细胞(上皮-间充质转化(epithelial - mesenchymal transition, EMT)),为初始修复过程提供间充质细胞。间质中残留的肺成纤维细胞开始增殖并迁移到损伤部位。过度的成纤维细胞增殖、迁移和转化为肌成纤维细胞(成纤维细胞到肌成纤维细胞转化(FMT)),以及细胞外基质(ECM)的合成和沉积是IPF纤维化病理的标志。Nintedanib(黄色分子)干扰成纤维细胞/肌成纤维细胞增殖、FMT和迁移。通过限制成纤维细胞/肌成纤维细胞的数量,减少了ECM的合成和沉积。Nintedanib对EMT无影响。尼达尼布对肺纤维细胞及其他结构细胞的影响有待进一步探讨。nintedanib的抑制活性以黄色显示。
确认
勃林格殷格翰资助的编辑协助,由英国伦敦弗莱什曼-希拉德集团有限公司的温迪·莫里斯在本手稿准备期间提供。作者完全负责所有内容和编辑决定,并参与了手稿发展的所有阶段,并批准了最终版本。
脚注
这篇文章有补充资料可从www.qdcxjkg.com
支持声明:本文和文中描述的nintedanib研究由勃林格殷格翰资助。本文的资助信息已存入FundRef.
利益冲突:可以在本文的在线版本中找到信息披露www.qdcxjkg.com
- 收到了2014年9月23日。
- 接受2015年1月5日。
- 版权所有©ERS 2015
ERJ开放文章是开放获取的,并根据创作共用署188滚球软件名非商业许可4.0的条款进行分发。











{kind=link}
{kind=link}
{kind=link}
{kind=link}
![a) Nintedanib inhibits platelet-derived growth factor (PDGF)-BB-stimulated platelet-derived growth factor receptor (PDGFR)-α and β autophosphorylation and proliferation of primary human lung fibroblasts (HLFs). HLFs were incubated with nintedanib at different concentrations and stimulated with PDGF-BB (50 ng·mL−1). PDGFR-α and β phosphorylation was determined by an ELISA specific for the phosphorylated receptors. Proliferation was determined by bromodeoxyuridine incorporation. Concentration-dependent inhibition data are presented as mean±sem (n=3 experiments). Reproduced from [78] with permission from the publisher. b) Nintedanib inhibits basic fibroblast growth factor (FGF) and PDGF-BB-stimulated motility of primary HLF from patients with idiopathic pulmonary fibrosis (IPF-HLF) and without IPF (N-HLF). Human lung fibroblasts were incubated with nintedanib at different concentrations for 30 min before the cells were stimulated with basic FGF (20 ng·mL−1) or PDGF-BB (50 ng·mL−1). Motility of the cells was determined by time lapse microscopy using a Cell-IQ imager (CM Technologies Oy, Tampere, Finland) and manual single cell tracking for 72 h. Mean cell velocity was calculated from three experiments. Inhibition is presented as mean±sem.](http://www.qdcxjkg.com/content/erj/45/5/1434/F3.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}