





文摘
A20是脂多糖(LPS)诱导,细胞质锌指蛋白,抑制toll样receptor-activated核转录因子(NF) -κB信号deubiquitinating肿瘤坏死因子receptor-associated因子(TRAF) 6。A20的作用是通过复杂的形成与无名指蛋白质(RNF) -11,瘙痒和TAX-1绑定蛋白1 (TAX1BP1)。本研究调查是否表达A20是慢性炎症改变囊性纤维化(CF)气道上皮细胞。
CF患者鼻腔上皮细胞(F508del纯合),non-CF控制和家乡的上皮细胞(16 hbe14o和CFBE41o)与LPS刺激。胞质表达A20和NF-κB亚基的表达进行分析。A20泛素的形成编辑复杂也被调查。
CFBE41o -,峰值LPS-induced A20表达式被推迟与16 hbe14o相比,下降明显低于基础水平LPS刺激后12 - 24小时。这是确认在初级CF气道细胞。另外,一个重要的逆A20观察和p65表达之间的关系。抑制剂研究表明,A20不接受在CFBE41o——蛋白酶体降解。A20与TAX1BP1, RNF11 TRAF6 hbe14o 16 -细胞,但均未观测到这些交互CFBE41o -。
的表达A20 CF显著改变,和重要的交互与复杂的成员和目标蛋白质丢失,这可能导致慢性NF-κB-driven炎症的状态。
A20(由基因编码TNFAIP3)是一种可诱导的胞质锌指蛋白,负调节核转录因子(NF) -κB信号。组成型表达A20低在大多数细胞,但在应对迅速诱导一系列不同的刺激包括肿瘤坏死因子(TNF) -α和细菌的产品,如脂多糖(LPS)1]。的TNFAIP36 q23处轨迹与多种自身免疫和炎症有关。A20分子调节器是一种重要的炎症在类风湿性关节炎,系统性红斑狼疮,1型糖尿病、多发性硬化症、牛皮癣和炎症性肠病(IBD),尽管不同的发病机制(2]。在1型糖尿病intra-islet释放的炎症介质激活免疫细胞NF-κB-driven [3,4),和单核苷酸多态性分析已经确定A20作为1型糖尿病的易感性基因(5]。此外,缺乏表达A20基因敲除的小鼠实验发展严重的肠道炎症(6),这表明A20对肠道内稳态至关重要,抑制NF-κB-dependent炎症。符合这一点,最近7个常见的炎症性疾病的全基因组关联研究英国人发现A20作为克罗恩病的易感性基因(7]。更早的独立研究了来自139个家庭的260对IBD-affected还强调了作为候选基因表达A20 IBD的发展(8]。
A20相对独特的,因为它可以发挥ubiquitinating deubiquitinating对目标蛋白的影响(9]。这种双重功能分为n端结构域,充当deubiquitinating蛋白质和锌指(ZnF)包含c端域,它充当一个E3泛素连接酶(9]。A20的双重功能,促进有效抑制NF-κB首先deubiquitinating目标蛋白质使他们不活跃,随后ubiquitinating目标蛋白质的蛋白酶体降解[9,10]。A20和NF-κB之间的关系是复杂的,受到各种介质和刺激。A20终止NF-κB亚基的易位p65细胞核和终止健康唾腺上皮细胞炎症信号。相反,沉默表达A20 p65激活这些细胞增加和强化了炎症反应(11]。这是符合发现p65绑定到一个表达A20发起人具体NF-κB序列(12]。此外,A20抑制NF-κB激活的能力依赖于“A20泛素编辑情结”的形成与E3连接无名指蛋白质(RNF) -11和痒,和一个适配器蛋白质称为TAX-1绑定蛋白1 (TAX1BP1) [13,14]。没有任何复杂的成员,A20不能绑定,行动目标蛋白质,因此,不能防止NF-κB激活(13]。在上皮细胞和其他免疫细胞,LPS刺激触发toll样受体(TLR) 4 NF-κB通路的激活介导的。A20终止这个过程通过抑制TNF receptor-associated polyubiquitination和激活的因素(TRAF) 6早期通路(15]。
尽管广泛研究作为一个肿瘤抑制基因和突出的潜在生物标志物的发展慢性炎性疾病,包括克罗恩氏病和关节炎(16慢性肺疾病,A20仍然没有得到充分的研究。G在等。(17)表明,在终止TLR-2 A20至关重要和地介导白介素8 (IL)释放主气道上皮细胞,而Tiesset等。(18)发现A20迅速诱导的肺健康小鼠的挑战铜绿假单胞菌。然而,据我们所知,还没有调查A20慢性呼吸道疾病,如囊性纤维化(CF) [19]。CF是由突变引起的CF跨膜电导调节器(雌性生殖道,F508del突变是最常见的)基因位于顶端表面的上皮细胞。这些突变的影响最为明显的肺,这种不平衡在氯和钠运输导致脱水的上皮表面,降低黏膜纤毛的清除入侵的病原体,创建一个周期的慢性感染和炎症。modelling-derived假说表明,脱敏等慢性肺疾病的病原体识别CF可能继发于活跃TRAF6信号(20.]。事实上,持久NF-κB激活是一个CF气道炎症的标志,表明这个受到严格监管的信号通路可能是扭曲的。然而,A20的潜在作用慢性CF气道炎症还没有被调查。本研究的目的是确定的表达A20 CF上皮的改变是基于正常表达A20函数的假设损失或变异可能导致持久的和不受控制的CF气道炎症。
材料和方法
详情请参阅在线补充材料。
细胞培养和刺激
支气管上皮细胞系16 hbe14o和CFBE41o(纯合子F508del)获得d Gruenert(加州大学旧金山,旧金山,美国),生长在潜。所有的细胞系实验当细胞汇合的大约80%。原发性鼻上皮细胞(NEC)从CF患者获得纯合F508del和匹配控制志愿者如前所述[21)和完全分化的气液界面(ALI)。北爱尔兰的研究伦理委员会批准了研究和所有参与者提供知情同意(07年/ NIR02/23)。
流式细胞术
细胞质表达A20量化了流式细胞术在permeabilised 16 hbe14o和CFBE41o细胞。p50 NF-κB的异质二聚体和p65丰富在大多数细胞类型(22]。鉴于p65和表达A20之间的关系描述,上皮细胞呈阳性核p50 / p65异质二聚体单元的NF-κB使用一个适应流仪方法研究[23如前所述(24]。数据表示为平均荧光强度评估细胞质表达A20的荧光强度的变化,或核p50 / p65阳性细胞相比,同形像控制。数据采集和分析流式细胞分析仪(史诗XL;贝克曼库尔特,英国威科姆)。
确定引发
引发的浓度细胞系基底上层的顶端和泥沙从主要nec是衡量商用ELISA (PeproTech EC有限公司、伦敦、英国)。基底结合引发释放从顶端和nec的洗液。
抑制剂的研究
之前与LPS刺激,CFBE41o——细胞进行预处理1 h和选择性p65抑制剂(JSH-23 30μM),或为4 h和E1泛素激活酶的抑制剂(PYR-41, 25μM)和26 s蛋白酶体(mg - 132、10μM)。抑制剂都来自默克公司(达姆施塔特,德国)。
定量聚合酶链反应
从细胞总RNA提取(RNeasy微设备;试剂盒,克劳利,英国)和量化(NanoDrop;美国马热费希尔科学、沃尔瑟姆)。等量反向转录成RNA的cDNA (Sensiscript逆转录设备;试剂盒)。引物序列,基因加入数字和产品尺寸给出在线补充表S2。相对表达β-actin使用ΔΔCt方法计算。
免疫沉淀反应和免疫印迹
免疫沉淀反应实验、蛋白质溶解产物沉淀了抗体对全身的样子(sc - 52910,圣克鲁斯生物技术、达拉斯、TX,美国)直接使用IP包(热费希尔科学)。细胞溶解产物被稀释在核酸酶游离水和Laemmli加载缓冲区,装上Tris-HCl聚丙烯酰胺凝胶(热费希尔科学),由十二烷基硫酸钠聚丙烯酰胺凝胶电泳和转移到聚偏二氟乙烯膜。膜是孵化主要抗体(见在线补充材料),水洗,孵化与辣根peroxidase-conjugated二级抗体(英国)赫默尔亨普斯特德镇热费希尔科学、和视觉Bio-Rad化学Doc XRS系统(英国赫默尔亨普斯特德镇Bio-Rad实验室有限公司)。
统计分析
所有数据都意味着±扫描电镜。组之间的差异进行分析利用克鲁斯卡尔-沃利斯非参数方差分析邓恩的检测后,如果p < 0.05被认为是重要的。
结果
诱导表达A20峰值推迟CFBE41o -细胞
观察诱导表达A20峰值1 h LPS刺激后16 hbe41o -细胞。此后,A20表达式返回这些细胞基底的水平。然而,在CFBE41o -细胞表达A20表达显著诱导1 h LPS刺激后,与峰值表达观察4 h post-stimulation (p < 0.001)。虽然A20蛋白表达回到基础水平8 h post-stimulation表达显著下降(p < 0.05)低于基底与LPS水平治疗后12和24小时。这些研究结果是一致的表达A20 mRNA (图1一个)和蛋白质含量(1 b])。A20 mRNA表达也低于基础水平在初级nec CF患者F508del纯合子(图1 c)。
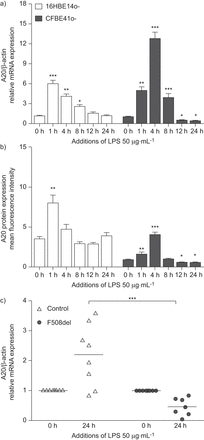
A20表达式在气道上皮细胞。A20) mRNA和b)蛋白表达以16 hbe14o和CFBE41o细胞定量PCR和流式细胞术。细胞被刺激μg·50毫升−1脂多糖(LPS)。数据意味着±扫描电镜。为定量PCR实验n = 6;流式细胞仪实验n = 5。*:p < 0.05;* *:p < 0.01;* * *:p < 0.001与相应的未经处理的控制(即。0 h)为每个单独的细胞系。c) mRNA表达A20也评估了定量PCR在初级鼻上皮细胞从患者纯合子F508del和年龄,sex-matched控制。表达式计算24 h后模拟的有限合伙人和数据均值±扫描电镜;n = 8。* * *:随着年龄的增长,相比p < 0.001 sex-matched控制。
减少表达A20表达式与CF细胞炎症信号相关联
我们检查了核的表达p50和NF-κB p65单元16 hbe14o和CFBE41o细胞。虽然基底p50 CFBE41o -表达高于16 hbe14o——细胞,p50感应的整体模式在LPS刺激两个细胞系没有差异(在线补充图S2A)。核p65表达迅速诱导16 hbe14o——和CFBE41o - 1 h LPS刺激后细胞。然而,p65表达式返回到基底的水平在16个hbe14o——细胞8 h post-stimulation但仍持续在CFBE41o调节(p < 0.001),细胞跨度为检查(图2一个)。这些发现被证实在初级nec CF患者为F508del纯合子的mRNA水平(图2 b)。另外,一个重要的(r = -0.709, n = 13, p = 0.007)逆A20和观察p65水平之间的关系(在线补充图S3)。
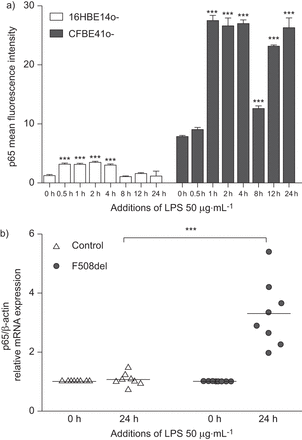
核因子(NF) -κB (p65)表达在气道上皮细胞。16 hbe14o和CFBE41o细胞暴露在脂多糖(LPS)为0,12和24小时)阳性细胞的百分比核p65亚基的表达NF-κB决定使用一个改编出版流仪法(25]。数据意味着±扫描电镜;n = 5。* * *:p < 0.001与相应的治疗(即。为每个细胞株0 h)控制。b)的mRNA表达p65评估原发性鼻腔上皮细胞从患者纯合子F508del和年龄——和sex-matched控制定量PCR。表达式计算24 h后仿真与有限合伙人和数据均值±扫描电镜;n = 8。* * *:随着年龄的增长,相比p < 0.001 sex-matched控制。
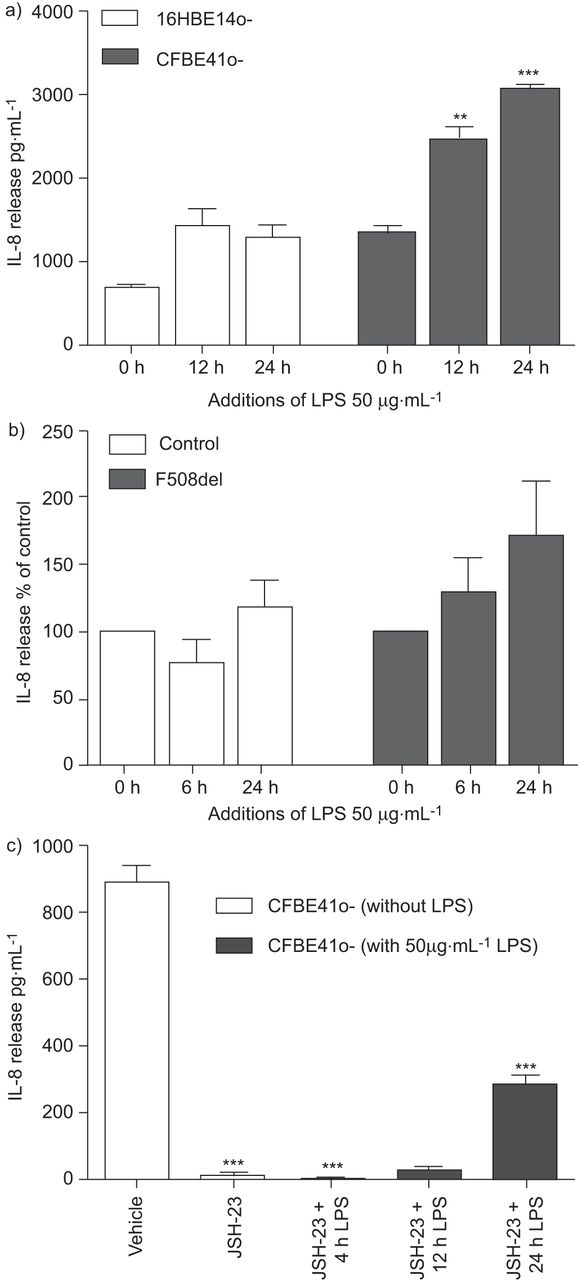
p65调节引发感应CFBE41o -细胞
LPS刺激后,观察时间增加引发释放CFBE41o -细胞(图3)。类似的趋势也发生在CF nec,但这些并没有达到意义(图3 b),与阿里文化普遍观察到的现象。先前的工作表明,p65牙龈上皮细胞中选择性地调节引发处理(26),但在CF的关系没有被调查。我们试图确定持久核表达p65 CFBE41o -细胞负责引发生产。CFBE41o——细胞使用选择性p65抑制剂(JSH-23)显著降低(p < 0.001)如果和LPS-induced引发释放(图3 c)。JSH-23被发现特定的抑制作用为p65 p50表达式没有影响(在线补充图开通)。
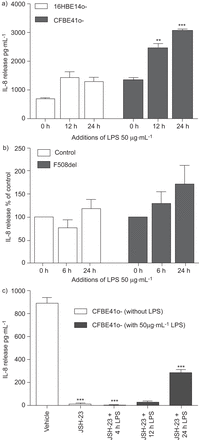
监管白介素8 (IL)处理。持久的表达p65也伴随着时间增加引发释放)CFBE14o -细胞(n = 5)和b)囊性纤维化鼻上皮细胞(n = 4),这是由ELISA。c)来确定p65选择性地调节引发的处理,CFBE41o——细胞使用商用和选择性p65抑制剂(JSH-23) 1 h与有限合伙人之前的挑战。引发释放到细胞上清液随后用ELISA (n = 3)。数据意味着±扫描电镜* *:p < 0.01;* * *:p < 0.001与相应的治疗(即。0 h)为每个细胞株或车辆控制。
减少表达A20 CFBE41o -不产生的蛋白酶体降解
我们提出这样子可能经历增加CF的蛋白酶体降解,这可以解释为什么A20表达低于CF LPS刺激后细胞基底的水平。为了验证这一点,CFBE41o——细胞处理商用E1 ubiquitin-activating酶抑制剂(PYR-41)和蛋白酶体降解(mg - 132)与LPS刺激之前,和表达A20调查定量聚合酶链反应和免疫印迹(图3)。然而,预处理与抑制剂没有影响A20 mRNA表达,它仍然显著(p < 0.05)低于基底如果水平有限合伙人治疗后24小时(图4)。也观察到类似的发现蛋白质水平(图4 b)。
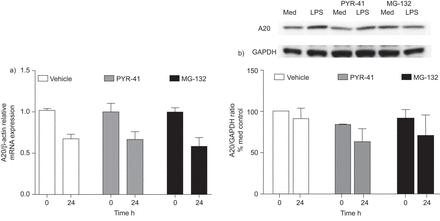
影响PYR-41(泛素抑制剂)和mg - 132 (26 s蛋白酶体抑制剂)表达A20表达式。16 hbe14o和CFBE41o细胞使用商用E1泛素激活酶的抑制剂(PYR-41)和26 s蛋白酶体(mg - 132) 4 h,之前挑战与脂多糖(LPS)进一步24 h)表达A20 mRNA表达当时检查定量PCR (n = 3)。数据意味着±扫描电镜。*:p < 0.05与相应的未经处理的控制(即。0 h)为每个抑制剂或车辆控制。b)表达A20蛋白表达也由免疫印迹(n = 3)。代表三个独立实验的形象。地中海:如果媒介控制。
改变的表达RNF11、瘙痒和TAX1BP1 CFBE41o -细胞
必须形成一个复杂的样子RNF11、瘙痒和TAX1BP1为了防止NF-κB激活(13]。因此,我们检查了这些复杂的成员使用定量PCR的表达。24小时后表达水平有限合伙人治疗所示图5变化,表达一个完整的时间进程的有限合伙人的治疗提出了在线补充图S4。数据提出了相对于未经处理的控制(1)基础水平正常化为每个细胞株或个别患者样本。在16个hbe14o——细胞,RNF11(的表达图5),瘙痒(图5 b)和TAX1BP1 (图5度)显著(p < 0.01−0.001)诱导后24 h (LPS刺激。峰值RNF11和TAX1BP1表达观察LPS刺激后24小时,而峰值痒表达式得到1 h治疗后仍明显高于基础水平,直到4 h post-stimulation,回到基础的水平之前(见在线补充图。S4)。然而,表达RNF11 CFBE41o和瘙痒保持不变——细胞,同时TAX1BP1表达显著下降(p < 0.05)低于刺激后基础水平。这些发现被证实(和发现更明显)在初级nec、RNF11 (图5 d),瘙痒(图5 e)和TAX1BP1 (图5 f)诱导控制nec和下降显著(p < 0.001)低于CF nec基底的水平。
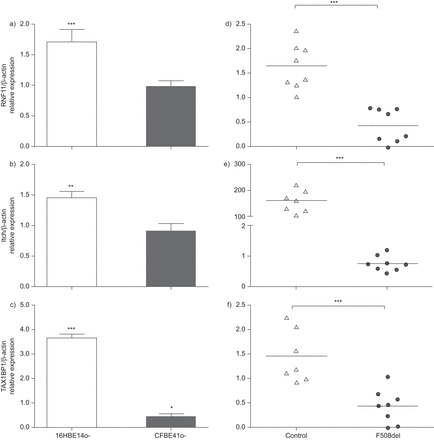
表达A20泛素编辑复杂的成员。的mRNA表达RNF11 (a和d),瘙痒(b和e)和TAX1BP1 (c、f)检查在细胞系(a、b和c)和原发性鼻腔上皮细胞(nec) (d, e, f)从患者纯合子F508del和年龄——和sex-matched控制定量PCR。表达式计算后24 h与脂多糖刺激。数据意味着±扫描电镜;n = 5细胞株和nec n = 8。a - c) *: p < 0.05;* *:p < 0.01;* * *:p < 0.001与相应的未经处理的控制为每个单独的细胞系。d-f) * * *:随着年龄的增长,相比p < 0.001 sex-matched控制。
样子不与RNF11或TRAF6 CFBE41o -细胞
评估A20泛素的形成编辑复杂,16 hbe14o——CFBE41o——细胞免疫沉淀反应的抗体完整长度的样子。A20与RNF11 1 h LPS刺激后16 hbe14o -细胞(图6),恰逢峰值A20表达在这个细胞株(图1)。这种交互似乎是短暂的,又观察了6和8 h后有限合伙人的待遇。此外,A20被发现与TRAF6 16 hbe14o——细胞跨度为检查,和1 h LPS刺激后最为明显。我们最初的实验并没有透露A20之间的交互和瘙痒或TAX1BP1 16 hbe14o——或者CFBE14o——细胞有限合伙人治疗后24 h。因为这些相互作用发生迅速和瞬变(14有限合伙人),我们执行一个额外的治疗后15分钟(图6 b)。A20互动TAX1BP1 15分钟后观察16 hbe14o——细胞,但与瘙痒仍不明显。没有描述CFBE41o -细胞相互作用被发现在任何时间点检查(图6)。
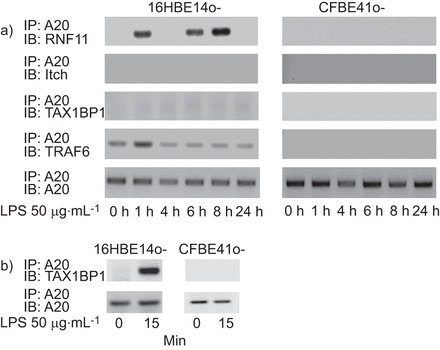
A20泛素编辑复杂的形成。核因子(NF) -κB抑制作用表达A20取决于A20“泛素编辑情结”的形成。)16 hbe14o CFBE41o,细胞被刺激与脂多糖(LPS) 0-24 h和检查形成的复杂的免疫沉淀反应(IP)。b)因为A20之间的相互作用和TAX1BP1已知发生迅速和瞬变(14)中所示的相同实验(a)有限合伙人0到15分钟后进行。所示图片代表两个独立的实验。IB:免疫印迹;RNF:无名指蛋白质;TAX1BP1: TAX1绑定蛋白1;TRAF:肿瘤坏死因子receptor-associated因素。
讨论
研究了锌指蛋白表达A20作为一个肿瘤抑制基因,参与自身免疫和炎症性疾病的进展,如克罗恩氏病和关节炎(16]。然而,这项研究是第一个调查A20表达在慢性炎症气道疾病。CF肺部疾病的特点是慢性细菌感染(特别是革兰氏阴性铜绿假单胞菌)和夸大NF-κB-driven炎症,导致组织损伤和肺功能的丧失。A20-deficient老鼠显示严重多器官炎症,激活炎症细胞增多和很少存活超过1周的年龄,暗示作用表达A20防止自发的先天免疫细胞介导炎症和组织破坏6]。此外,这些老鼠特别敏感外生TNF-α和有限合伙人,这表明A20在预防慢性炎症中起着至关重要的作用6]。使用上皮细胞来自患者CF (F508del纯合)和non-CF对照组,我们目前的研究结果表明,A20功能改变在CF,可能导致无法控制炎症。这些发现符合最近的报告在类风湿性关节炎(25和干燥综合征11]。
O的原创作品pipari等。(1使用鳞状细胞癌)细胞株(A431),发现峰A20 1 h与LPS刺激后或TNF-α表达式。与此一致的是,我们发现,峰值A20表达发生在non-CF LPS刺激后细胞1 h。然而,在CF细胞,峰值A20表达式是推迟到4 h post-stimulation, non-CF细胞相比,随后跌破基底(12 - 24 h post-stimulation)水平。此外,我们发现了一个显著负相关关系表达A20和p65 mRNA表达在初级CF nec。由于介质的数组,我们试图研究和有限的材料可以从我们的主要阿里文化,我们不能确认增加p65表达式或相关表达A20主要CF nec在蛋白质表达水平。然而,增加的核内蛋白表达p65发现占时间增加引发释放CFBE41o -细胞。虽然在CF细胞峰A20表达式被推迟,我们还观察到显著诱导蛋白1,4和8 h后LPS刺激。然而,没有明显的核NF-κB表达式或引发负面影响。这些发现提出问题的命运和行动表达A20 CF气道上皮细胞。
解决的问题为什么A20表达低于CF细胞基底的水平,我们首先检查是否表达A20 ubiquitinated或使用商用抑制剂针对蛋白酶体降解。泛素化是催化的顺序动作ubiquitin-activating酶(E1) ubiquitin-conjugating酶(E2)和泛素蛋白连接酶(E3)。PYR-41 cell-permeable化合物,特别是抑制普遍E1 ubiquitin-activating酶的活动防止进一步激活泛素化的序列,从而阻止ubiquitin-dependent细胞活动,包括蛋白质降解。mg - 132是一种强有力的、可逆的和cell-permeable 26 s蛋白酶体抑制剂。CF细胞用每个抑制剂预处理4 h与LPS刺激之前。然而,A20表达式不是治疗后恢复抑制剂和CF仍低于基础水平细胞,表明减少表达A20 CF扩展LPS刺激后细胞不产生的蛋白酶体降解。A20基因突变或甲基化被认为是一种失活(27),我们的研究结果,A20不接受增加退化CF基因可能指向一个固有的缺陷。另外,A20可能proteolytically裂解,这可能使它失效。例如,mucosa-associated淋巴组织淋巴瘤易位蛋白1(麦芽)最近被描述为具有蛋白酶活性和分裂A20 (28]。尽管这些替代A20失活机制目前投机,他们将是未来研究的主题。
我们下一个检查表达式的“泛素编辑情结”的成员。年代hembade等。(13)曾报道,NF-κB抑制作用表达A20依赖的形成是一个复杂的RNF11,瘙痒和TAX1BP1。沉默的任何成员复杂意味着A20不能结合,或采取行动,其目标蛋白质和无法阻止激活NF-κB [13]。除了促进行动表达A20 RNF11也证明独立抑制NF-κBκB激酶的抑制剂(IKK)级别(29日]。此外,TRAF6 RNF11防止泛素化,可能被证明是至关重要的,抑制TLR-induced NF-κB信号(29日]。TAX1BP1,或者叫TRAF6结合蛋白,是一个适配器蛋白质至关重要的表达A20 deubiquitination TRAF6应对细菌有限合伙人(30.),同时促进下游抑制NF-κB信号在病毒刺激反应(31日]。这里我们报告微分表达式CF和non-CF细胞之间的所有复杂的成员。这些差异更明显在初级nec细胞系。RNF11的表达,总的来说,瘙痒和TAX1BP1保持不变或者是调节控制nec LPS刺激后,但在CF nec显著下调。鉴于表达模式的差异,我们想知道是否表达A20泛素编辑复杂的被正确地形成于CF。尽管我们观察到A20之间的交互和TRAF6, RNF11和TAX1BP1 non-CF细胞,表达A20不存在与痒在任何细胞类型,或在任何时间点检测。此外,尽管显著诱导表达A20 CF细胞4 h LPS刺激后,与TRAF6互动,RNF11或TAX1BP1没有观察到在这个或任何其他时间点检查。
TRAF6的互动表达A20 RNF11和TAX1BP1 non-CF控制细胞和没有这些交互CF细胞可能有病理意义。TLR NF-κB通路的激活细胞膜始于receptor-ligand协会。这个新兵适配器蛋白质(骨髓分化主要响应(MyD) -88和receptor-associated激酶(伊拉克共和国))和TRAF6 ubiquitinated。积极表达A20暂停下游信号由deubiquitinating TRAF6 [14]。之前的研究表明,瞬态与RNF11 A20互动,瘙痒和TAX1BP1管理这个过程通过促进TRAF6 deubiquitination和扰乱TRAF6协会与泛素连接酶Ubc13,从而终止IKK和NF-κB激活(14]。A20-TRAF6轴被认为是必不可少的在预防LPS-stimulated NF-κB激活。沉默表达A20显示恢复TRAF6行动和NF-κB LPS刺激后激活(32]。这一事实表达A20泛素的不与任何成员编辑复杂,我们会假设,因此无法在CF与TRAF6细胞,可以部分解释为什么在这些细胞诱导表达A20似乎NF-κB活化的影响很小。之前的研究表明,抗炎活性表达A20可能归因于c端域包含七个ZnF蛋白质和充当E3泛素连接酶。ZnF4似乎特别重要,因为A20 ZnF4突变体无法抑制TRAF6泛素化(14]。未来的研究将调查c端域的酶活性在CF上皮细胞。
兴趣的行动,表明,潜在的治疗目标表达A20,近年来显著增加提供新颖的发现和出色的总结表达A20函数(参见Harhaj和Dixit(33])。而直接和永久抑制NF-κB可能使免疫系统无法应对病原体,瞬态感应的负面表达A20等监管机构将帮助目标炎症信号转导途径。有人建议,这种方法可能会提供一个更具体的和定制的方法治疗炎性疾病没有全球抑制免疫系统的34]。更好的理解这些过程,以及表达A20本身的规定,将大大提高A20-driven治疗炎症性肺部疾病的可能性。目前的研究是第一个报告管制A20表达式在CF和突出的潜在相关性ZnF蛋白质在慢性肺部炎症。
确认
作者要感谢d Gruenert(加州大学旧金山,旧金山,美国)的上皮细胞系,和诉布朗(英国贝尔法斯特女王贝尔法斯特大学)与流式细胞术寻求帮助。
脚注
可以从本文的补充材料www.ers.ersjournals.com
支持声明
这项工作是由的资助囊性纤维化信任英国公元前Schock (PJ541)。
感兴趣的语句
利益冲突的信息可以发现与本文的在线版本www.www.qdcxjkg.com
- 收到了2012年2月23日。
- 接受2012年8月24日。
- ©2013人队











{kind=link}
{kind=link}
![Nuclear factor (NF)-κB (p65) expression in airway epithelial cells. 16HBE14o- and CFBE41o- cells were exposed to lipopolysaccharide (LPS) for 0, 12 and 24 h and a) the percentage of cells positive for the nuclear expression of the p65 subunit of NF-κB was determined using an adapted published flow cytometric method [25]. Data are presented as mean±sem; n=5. ***: p<0.001 compared with corresponding untreated (i.e. 0 h) control for each cell line. b) mRNA expression of p65 was assessed in primary nasal epithelial cells from patients homozygous for F508del and from age- and sex-matched controls by quantitative PCR. Expression was calculated after 24 h simulation with LPS and data are presented as mean±sem; n=8. ***: p<0.001 compared with age- and sex-matched controls.](http://www.qdcxjkg.com/content/erj/41/6/1315/F2.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Formation of the A20 ubiquitin editing complex. The nuclear factor (NF)-κB inhibitory effect of A20 is dependent on the formation of the A20 “ubiquitin editing complex”. a) 16HBE14o- and CFBE41o- cells were stimulated with lipopolysaccharide (LPS) for 0–24 h and the complex formed examined by immunoprecipitation (IP). b) Since interactions between A20 and TAX1BP1 are known to occur rapidly and transiently [14] the same experiments shown in (a) were performed after 0 and 15 min LPS. Images representative of two independent experiments are shown. IB: immunoblot; RNF: ring finger protein; TAX1BP1: TAX1 binding protein-1; TRAF: tumour necrosis factor receptor-associated factor.](http://www.qdcxjkg.com/content/erj/41/6/1315/F6.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}