





抽象的
吸烟是慢性阻塞性肺疾病(COPD)的主要病因,可导致气道上皮结构和功能异常。到目前为止,潜在的机制尚未解决。
我们研究了香烟烟雾提取物(CSE)对COPD患者、非吸烟者和健康吸烟者的人支气管上皮16HBE细胞和原代支气管上皮细胞(PBEC)的上皮屏障功能和伤口再生的影响。
我们证明,CSE可快速且短暂地损害16HBE屏障功能,主要是由于细胞间接触的中断。CSE可诱导PBEC出现类似但更强且更持久的缺陷。特异性表皮生长因子受体(EGFR)的应用抑制剂AG1478表明,EGFR激活有助于CSE诱导的16HBE细胞和PBEC缺陷。此外,我们的数据表明,内源性蛋白酶钙蛋白酶通过紧密连接蛋白降解介导这些缺陷。CSE还延迟了伤口愈合过程中16HBE细胞间接触的重建,并在伤口愈合过程中引起细胞凋亡评估伤口再生时PBEC屏障功能。这些结果在吸烟者、健康吸烟者和COPD患者的PBEC之间具有可比性。
总之,我们首次证明CSE降低了上皮完整性,可能是由于EGFR和calpain依赖性的细胞间接触中断。这可能会增加对环境伤害的敏感性,例如吸入病原体。因此,EGFR可能是改善吸烟相关疾病黏膜屏障功能的治疗策略的一个有前途的靶点。
慢性阻塞性肺疾病(COPD)是一种与气道重塑和/或肺气肿相关的炎症性气道疾病,导致肺功能加速下降。尽管吸烟是COPD的主要危险因素,但仍不清楚同一香烟烟雾暴露导致的不同COPD表型是如何发展的。吸入烟草烟雾首先遇到气道上皮形成高度调节的屏障。在病理条件下,这种屏障的破坏可能导致污染物和病原体进入粘膜下层的机会增加。上皮屏障功能由紧密连接(TJs)维持,TJs包括相互作用的蛋白质闭塞素、闭塞带(ZO)-TJs限制细胞旁通透性并实现栅栏功能,使基底外侧区与顶端区分离。受伤时有效的TJ修复可能对上皮内环境稳定至关重要。
吸烟可通过诱导结构改变,如慢性阻塞性肺病患者气道中的黏液增生,从而极大地干扰上皮连接[1].吸烟不仅与分泌黏液的细胞增多有关[2,3.],但也增加了粘膜通透性[4以及增加对过敏原的渗透性体外[5,6].同样,吸烟可能会促进病毒和细菌的跨上皮细胞交叉,并产生关键的后果,因为近50%的COPD加重与细菌感染有关[7].因此,了解烟雾诱导屏障功能障碍的机制非常重要。
烟雾诱导的渗透性增加最初归因于烟雾的细胞毒性作用。最近,吸烟以及6天暴露于香烟提取物(CSE)体外,已被证明可导致气道上皮中顶端连接复合物基因的下调[8].此外,主流香烟烟雾降低人气道腺癌Calu-3细胞的跨上皮抵抗并诱导occludin的酪氨酸磷酸化[9,10].此外,香烟烟雾冷凝物通过激活表皮生长因子受体(EGFR)/细胞外信号相关激酶(ERK)通路和随后TJ蛋白的离定位机制对人支气管上皮BEAS-2B细胞造成损害[11].这种效应可以归因于CSE中存在的亲脂化合物(包括酚类化合物、醛类和多环芳香烃),它们能够通过上皮质膜[12].CSE作为屏障功能的读取器对上皮抵抗的影响尚未被研究。我们的目的是评估无毒浓度的CSE对TJ形成和上皮屏障功能的影响,并阐明吸烟诱导上皮屏障功能障碍的潜在机制。
我们研究了CSE暴露在人类支气管上皮细胞系16HBE以及COPD患者和健康非吸烟和吸烟供体的原发性支气管上皮细胞(PBECs)中的影响。我们证明,CSE诱导了上皮屏障功能的短暂缺陷,并降低了损伤后16HBE细胞与细胞接触的重建,这可能是由egfr依赖性的TJs破坏造成的。据我们所知,这项研究首次表明,CSE在PBEC中引起了类似的缺陷,但发病延迟和恢复时间延长。非吸烟者、健康吸烟者和慢性阻塞性肺疾病患者的PBECs对创伤和CSE的反应没有明显差异。
方法
上皮细胞培养
16HBE细胞由D.C. Gruenert(加利福尼亚大学,旧金山,CA,美国)和蔼地提供,并按照先前描述的方法培养。13].从全球慢性阻塞性肺疾病倡议(GOLD) III期和IV期的3名重度COPD患者中获得了PBECs [14通过支气管刷拭(包括≥10包/年的吸烟、1秒内用力呼气量(FEV)1)/强迫肺活量<70%和FEV1小于50%的预测年龄为50-62岁;表1)根据标准指引[15].荷兰格罗宁根大学医学中心的医学伦理委员会批准了这项研究,参与者签署了知情同意书。此外,来自4名健康吸烟者(40-48岁)和3名健康非吸烟者(52-56岁)的PBECs来自Lonza (Walkersville, MD, USA)。PBECs在2.5 mL激素补充的支气管上皮生长培养基(BEGM;龙沙)上的胶原蛋白/纤维连接蛋白涂层烧瓶[16]实验前,通过台盼蓝染色评估细胞活力(刷毛细胞的平均活力为95±0.6%,Lonza细胞的平均活力为93±1.6%)。
细胞被重复接种到电池衬底阻抗传感(ECIS)阵列,室载玻片(LabTek;Thermo Fisher Scientific Inc., Waltham, MA, USA)或24孔板。在80-90%的融合度下,将Eagle最低必需培养基(EMEM)/10%胎牛血清(FCS)或BEGM (Lonza)用EMEM/0.5% FCS(用于16HBE细胞)或基础培养基(BEBM;龙沙)/0.5% FCS (PBECs)。在线补充材料中描述了上皮细胞抗性测定、免疫印迹和免疫荧光的方法。
CSE的准备
如前所述制备香烟烟雾提取物(CSE)[17简而言之,肯塔基2R4F研究参考卷烟(肯塔基大学烟草研究所,莱克星顿,KY,美国)使用无滤嘴,两支香烟的烟雾通过25毫升培养基(100% CSE)鼓泡,新鲜提取,用0.22μm过滤器消毒,30分钟内使用。
上皮细胞刺激
分别用或不用EGFR酪氨酸激酶抑制剂AG1478 (2 μM;Calbiochem;calpain inhibitor II (ALLM, 10 μM;随后,用5% CSE、表皮生长因子(EGF) (10 ng·mL)处理细胞−1;Sigma-Aldrich)或载体(培养基/DMSO) 0-24小时,并由ECIS监测,固定用于免疫荧光染色或收获用于细胞裂解液制备。台盼蓝染色检测细胞活力。5% CSE不影响细胞活力(数据未显示)。
欧洲互通性系统委员会
如前所述,使用ECIS(美国纽约州特洛伊市应用生物物理公司)测量融合或损伤上皮的电特性[18,19]接种后,分别在400 Hz和40 kHz下测量电阻和电容。通过电穿孔(5 V,40 kHz,30 s)添加CSE/载体后立即损伤细胞。
统计分析
数据分析使用非参数秩和Mann-Whitney u检验进行受试者组之间的分析,配对观察的t检验比较组内或16HBE细胞系的条件。使用双向方差分析来检验在上皮耐药性测量中控制和治疗时间曲线之间的显著差异。
后果
CSE诱导16HBE细胞的电阻瞬时降低
我们首先检查了CSE对16小时细胞单层屏障功能的影响。CSE的添加诱导大量瞬态落下的低频电阻,最敏感的参数来监测屏障紧密度[18,19].抗性水平立即开始下降,在4 - 6小时内效果最大(最大平均±扫描电镜减少26±3%;N =5),然后在约15小时后缓慢恢复到控制值(图1一个溶媒处理井的阻力也在4-6小时内下降(最大平均值±)扫描电镜抑制14±3%),但CSE组显著降低(n=5;p = 0.011)。我们还测量了高频电容,高频电容对细胞-基质接触变化敏感,但对细胞-细胞接触变化相对不敏感[18,19].在这里,CSE产生的影响不太明显(平均值±扫描电镜最大增加9±1%),表明细胞-基质接触受影响的程度小于细胞-细胞接触(图1 c).
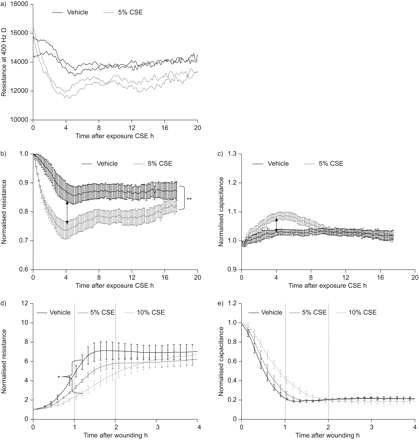
香烟烟雾提取物(CSE)对16HBE电池电阻的影响。16HBE细胞在细胞底物阻抗传感(ECIS)阵列中重复接种,培养3-4天,置于低血清中,5%或10% CSE或载体中,监测48小时或立即电穿孔。使用ECIS在400 Hz下测量电阻,在40 kHz下测量电容。a)给出了典型实验的绝对电阻值。b)归一化电阻值(平均值±扫描电镜)如图所示(n=5)。将数值标准化为添加CSE/车辆之前的水平。**:对照组与CSE曲线、双向方差分析之间的p<0.01。c) 归一化电容值(平均值±扫描电镜)显示(n=5)。d)规正电阻值(平均值±扫描电镜)如图所示(n=3)。受伤后立即将数值标准化为水平。*:对照组与10%CSE曲线、双向方差分析之间的p<0.05。e) 归一化电容值(平均值±扫描电镜)显示(n=3)。
CSE延迟电气穿孔损伤时16HBE细胞的上皮再生和重构细胞 - 细胞触点
除了对16HBE屏障功能的影响外,通过电穿孔损伤后CSE受损的上皮屏障重构。使用这种类型的伤口,上皮单层能够在几小时内恢复,由于细胞迁移/蔓延,而不是来自增殖[19].16HBE细胞的屏障功能在2小时内完全恢复(图1 d).细胞在1小时内重新填充电极,这反映在高频电容的稳定(图1 e).CSE显着推迟了伤害时上皮抗性的恢复。虽然当CSE以5%加入CSE时,观察到显上皮抗性的趋势,但加入10%CSE显着受到上皮屏障的恢复(图1 d).同样,CSE(5和10%)对高频电容的影响小于对低频电阻的影响。在伤后1-2小时,与对照处理的井相比,低频电阻仍然显著降低,而5和10% CSE (图1 e)这表明CSE影响上皮细胞-细胞接触的重建,而不是影响创伤后上皮细胞的迁移、扩散和/或附着。
CSE诱导的屏障功能缺陷可通过抑制EGFR活性而逆转
因为之前的研究提出了egfr依赖的信号在cse诱导的TJ蛋白再分配中的作用[11],并且EGFR活性的增加与慢性阻塞性肺病有关[20.,我们接下来研究EGFR活性是否参与了cse诱导的上皮屏障功能缺陷。用EGFR酪氨酸激酶抑制剂AG1478处理16HBE细胞显著且几乎完全阻止了cse诱导的低频电阻变化(图2a和c)。同样,AG1478减弱了cse引起的高频电容增加,在30-120 min内显著(图2bd,但不是在CSE发挥最强作用的时间点,即。添加CSE后4小时(图1 c).
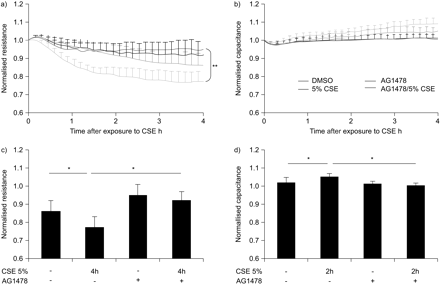
香烟烟雾提取物(CSE)对16HBE细胞屏障功能的影响依赖于表皮生长因子受体的活性。16HBE细胞在细胞底物阻抗传感阵列中重复接种,培养3-4天,低血清放置,加或不加AG1478 (1 μM)预处理30分钟,5% CSE或载体暴露。在400hz测量电阻,在40khz测量电容。数值被归一化到增加CSE/车辆之前的水平。a和c)归一化电阻值(平均值±扫描电镜)显示(n=5)。**:指示曲线(CSE和CSE+AG1478)间p<0.01,双向方差分析;*:表示值之间p<0.05。b和d)归一化电容值(平均值±扫描电镜)如图所示(n=5)。*:指示值之间的p<0.05。
根据EGFR活性在CSE诱导的屏障功能障碍中,我们发现CSE增加了磷磷酸普夫特(TYR 1173)以及EGFR下游信号分子磷酸ERK-1/2,但在6时,不是全EGFR水平。H (图3b).此外,添加EGF可暂时降低上皮抵抗,在4-6 h左右达到最大效果,之后细胞恢复缓慢(图3 c和d)。
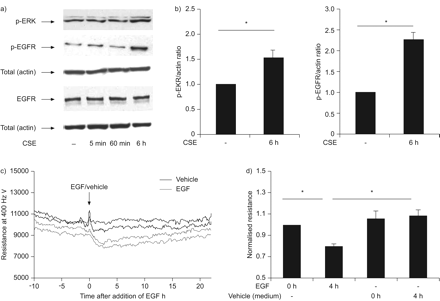
香烟烟雾提取物(CSE)诱导表皮生长因子受体(EGFR)依赖的信号传导,表皮生长因子(EGF)影响16HBE细胞的电阻。16HBE细胞重复接种于24孔板或细胞底物阻抗传感阵列中,培养至融合3-4天,置于低血清中,用或不用5% CSE或EGF (10 ng·mL)刺激−1)静置5分钟至24小时。a)制备细胞总裂解液,用western blotting检测磷酸化细胞外信号相关激酶(phospho-ERK)、磷酸化EGFR和EGFR(箭头)。肌动蛋白作为负荷对照。给出了4个独立实验的代表。b)密度测定,磷酸化erk和磷酸化egfr水平与肌动蛋白相关。比率(平均±SEM;n=4)。c)显示了代表性实验的绝对电阻值。d) 显示了在添加EGF/载体之前标准化的电阻值(平均值±)SEM;n = 3)。*:P <0.05在指示值之间。
CSE诱导TJs依赖egfr的分解
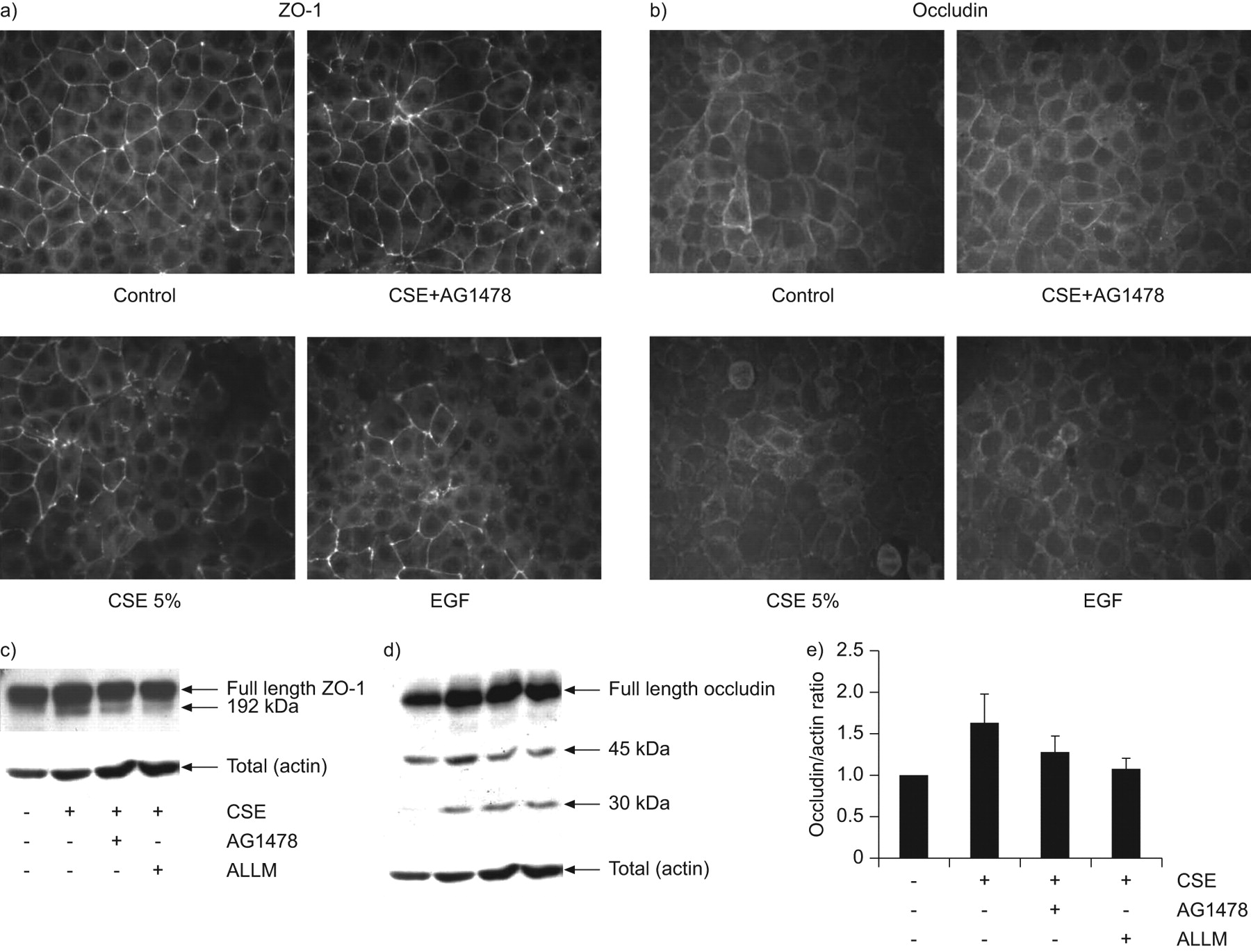
为了明确cse诱导屏障功能障碍的机制,我们研究了屏障功能的改变是否与细胞间连接的改变平行。我们观察到细胞外和细胞内的TJ成分,即。在静息状态下,ZO-1和闭塞素分别定位于细胞间连接,形成一个连续的环(图4).CSE暴露后,ZO-1染色(图4),以及较小程度上的occludin (图4 b)AG1478的存在部分逆转了这些效应,导致ZO-1和闭塞素的环向染色增多(图4b)。在同意的假设中,EGFR活性在CSE诱导的TJ拆卸中发挥作用,EGF诱导ZO-1和occludin的临时化(图4b)。
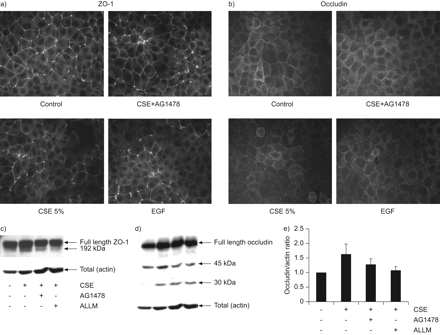
香烟烟雾提取物(CSE)和表皮生长因子(EGF)诱导紧密连接的解体。16HBE细胞在室载玻片或24孔板中培养3天,置于低血清中,用或不用AG1478(1μM)或ALLM(10μM)预处理1小时,并用或不用CSE(5%)或EGF(10 ng·mL)孵育−1a)免疫荧光染色检测封闭带-1 (ZO-1);b)免疫荧光染色检测Occludin。c)制备细胞总裂解液,用western blotting检测ZO-1(箭头)。给出了四个独立实验的代表。d)制备细胞总裂解液,western blotting检测occludin(箭头)。肌动蛋白作为负荷对照。e)对30 kDa occludin片段进行密度测定,其水平与肌动蛋白相关;比率(平均±SEM;n = 4)进行描述。
接下来,我们研究了CSE和EGF是否也影响occludin和ZO-1的总水平。CSE诱导了部分ZO-1切割,表现为一个较小的片段(~ 192 kDa)的出现,以及occludin从其全长形式(~ 65 kDa)部分切割成较小的切割片段(~ 45和~ 30 kDa;图4c-e)我们没有观察到粘附连接(AJ)蛋白E-钙粘蛋白的裂解(数据未显示)。AG1478的存在减少了较小ZO-1和闭塞蛋白片段的出现(图4c-e).此前,内源性半胱氨酸蛋白酶calpain的激活与EGFR信号通路有关[21]钙蛋白酶被描述为能切割连接蛋白[22].钙蛋白酶抑制剂II也减少了cse诱导的occludin和ZO-1切割成更小的片段。因此,暴露于CSE似乎以egfr依赖的方式破坏TJs,我们的数据表明,这涉及calpain介导的TJ蛋白切割。
CSE诱导非吸烟者和健康吸烟者PBEC屏障功能中egfr依赖的缺陷
接下来,我们对CSE对原代上皮的影响以及不吸烟个体和当前吸烟者之间可能存在的差异感兴趣,以评估长期吸烟是否存在上皮异常。PBEC在建立单层细胞后表现出比16HBE细胞低得多的抗性水平,不吸烟者和吸烟者之间没有显著差异(平均值±扫描电镜4257±1422 Ω、5238±1350 Ω)。暴露于CSE可导致非吸烟者和吸烟者上皮屏障功能下降约50% (图5和5b),分别为1,937±563Ω和2,942±9555Ω的水平。与16HBE细胞相比,CSE诱导的屏障功能障碍的发作延迟,在CSE暴露后,12-18小时的效果在12-18小时之间,在CSE暴露后直至25-35小时。两组之间没有观察到显着差异。
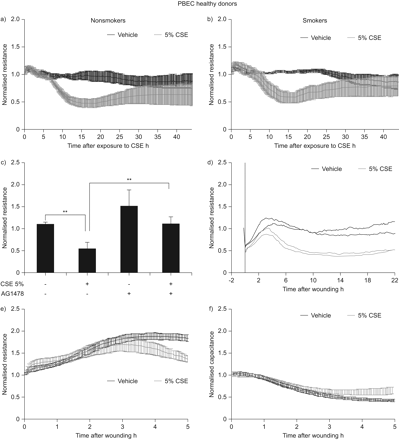
来自非摩托车和吸烟者的原发性支气管上皮细胞(PBECs)在电池基板阻抗传感阵列中替加替换,生长至汇合3-4天,生长因子/激素剥夺过夜,暴露于5%香烟烟雾提取物(CSE)或载体并监测48小时或立即通过电穿孔伤害。在400hz测量电阻,在40khz测量电容。数值被归一化到增加CSE/车辆之前的水平。当可获得足够的细胞时,在CSE暴露之前,用或没有Ag1478预处理细胞。a)归一化电阻值(平均值±扫描电镜),非吸烟者的多氯联苯(n=3)。数值被归一化到增加CSE/车辆之前的水平。b)归一化电阻值(平均值±扫描电镜)吸烟者的PBEC(n=4)。c)标准化电阻值(平均值±扫描电镜(吸烟者和非吸烟者的PBECs;n = 4)。**:指示值之间p<0.01。d)吸烟者的PBECs规正电阻值(代表)。数值恢复到受伤前的水平。e)规格化电阻值(平均值±扫描电镜) (n=3)。受伤后立即恢复正常。f)归一化电容值(平均值±扫描电镜)吸烟者的PBEC数量(n=3).
接下来,我们测试了EGFR活性的参与情况。与16HBE细胞类似,AG1478在9-28 h显著降低了CSE诱导的PBEC屏障功能缺陷(数据显示在12 h, CSE的影响最明显的时间点;图5度),在AG1478存在的情况下,CSE不再能够显著降低上皮耐药性。在没有CSE的情况下,AG1478也增加了上皮耐药性;然而,这种效果直到添加30小时后才达到统计学意义。此外,AG1478消除了CSE对高频电容的影响,尽管CSE对低频电阻的影响不如对高频电容的影响显著,且仅在3 ~ 5 h时显著(图1在线补充资料)。
此外,我们研究了PBECs对电穿孔损伤的反应,观察到这些细胞比16HBE细胞再生更慢,因为细胞需要约3小时才能恢复其屏障功能(如图所示,健康吸烟者的上皮;图5 d和e)。同样,我们没有观察到吸烟者和非吸烟者上皮细胞之间的差异(图2在线补充资料)。CSE暴露降低了新建立的单分子层的电阻,一旦未处理细胞的电阻值稳定下来,电阻进一步下降(图5 e).对高频电容的影响也不太明显(图5 f).
CSE在慢性阻塞性肺疾病患者中诱发了与PBECs类似的屏障功能缺陷
为了进一步评估我们研究结果的相关性,我们使用了三名重度COPD患者(均为戒烟者)的PBECs。这些细胞的单层膜建立了与健康PBECs相似的低频电阻,即。3281±926Ω。此外,CSE引起的∼屏障功能降低50%(图6),结果为1516±163 Ω。与正常PBECs进一步一致的是,CSE对低频电阻的影响比高频电容更显著(数据未显示)。
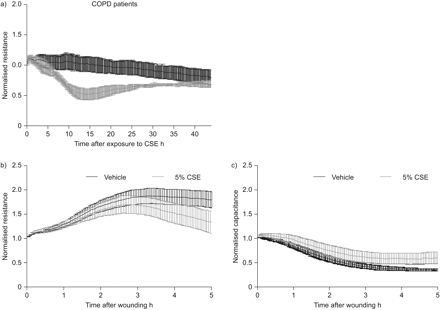
支气管上皮细胞来源于慢性阻塞性肺疾病IV期慢性阻塞性肺疾病患者全球倡议中的支气管刷。将细胞重复接种在电细胞-底物阻抗传感阵列中,生长至融合3-4天,隔夜去除生长因子/激素,暴露于5%CSE或载体中,并监测48小时或立即通过电穿孔损伤。电阻在400Hz下测量,电容在40kHz下测量。a) 标准化电阻值(平均值±扫描电镜)显示(n=3)。数值被归一化到添加CSE或车辆之前的水平。b)归一化电阻值(平均值±扫描电镜)显示(n=3)。受伤后立即恢复正常。c)归一化电容值(平均值±扫描电镜)如图所示(n=3).受伤后立即恢复正常。
此外,COPD患者的PBEC和健康PBEC对电穿孔损伤的反应相似,其抗性水平在10分钟内恢复到其原始值∼3小时后,CSE再次降低了新形成的单层损伤时的低频电阻(图6 b),对高频电容的影响较小(图6 c).
我们的数据在一起表明CSE暴露损害了气道上皮的完整性,主要是通过破坏细胞间接触,在COPD和健康上皮中具有类似的模式。
讨论
慢性阻塞性肺病患者的上皮重塑和异常上皮修复反应可能会干扰粘膜屏障功能。香烟烟雾可以增加上皮细胞的通透性,尽管其潜在机制尚不完全清楚。我们的研究表明,CSE导致16HBE细胞和PBECs的气道上皮屏障功能短暂但实质性的下降。这种作用依赖于EGFR的激活,我们为calpain介导的TJs破坏屏障功能障碍的新机制提供了证据。此外,CSE降低了伤口再生过程中16HBE细胞-细胞接触的恢复,减弱了创伤时PBECs的屏障功能,COPD患者与健康对照组之间无显著差异。我们的数据可能有重要的意义,例如对于具有COPD的吸烟者的加剧,因为降低的上皮完整性可以促进病毒和细菌的运输,这对COPD恶化作出了重要贡献。
CSE在COPD患者、健康吸烟者和非吸烟者的PBECs中同样影响上皮耐药。考虑到EGFR在cse诱导的缺陷中的作用,这与我们的观察相符,即EGFR水平在受试者之间没有差异(图3在线补充资料)。与16HBE细胞相比,PBECs细胞的作用更强、更持久。由于细胞间接触较弱,PBECs可能更容易受到cse诱导的损伤[19]在气液界面(ALI)培养时,原代细胞能够形成紧密的屏障,允许分化为极化的粘液纤毛上皮[23].不幸的是,ECIS系统不允许在ALI条件下进行测量。我们之前已经证明,在ALI条件下培养显著增加了PBECs中连接蛋白的膜表达,从而产生了相当于16HBE细胞水平的上皮耐药[24].因此,在研究上皮屏障函数时,我们认为16小时细胞作为可靠的模型[19]事实上,据报道16HBE细胞在水下条件下极化并形成功能性AJ和TJ[25]在16HBE细胞和PBEC中,EGFR活性似乎有助于CSE诱导的缺陷。此前,EGFR活性已被证明可诱导连接蛋白的去定位和钙蛋白酶的激活[21,26,27],可能导致TJ蛋白降解[28]我们的数据支持EGFR在CSE诱导的TX分解中的作用,因为EGFR酪氨酸激酶抑制剂AG1478消除了CSE诱导的屏障功能缺陷。此外,CSE增加了16HBE细胞中EGFR的磷酸化。因此,EGFR通过其配体EGF的激活暂时降低了上皮屏障功能,CSE和EGF均诱导16HBE细胞中TJ蛋白occludin和ZO-1的离域和裂解。
CSE诱导的EGFR激活可能有几种机制。首先,紧密连接打开后,顶端分泌的EGF配体与基底外侧定位的EGFR的通路增加[29].然而,这似乎不是事实,因为EGFR本身的活性是TJs的破坏所必需的。另外,吸烟被描述为激活A崩解素和金属蛋白酶(ADAM)17, EGFR配体转化生长因子(TGF)-α的主要脱落酶[30.]我们观察到CSE诱导16HBE细胞产生ADAM17和EGFR依赖的白细胞介素-8(图4,表明CSE确实可以激活ADAM17。然而,在CSE暴露时,我们无法检测到TGF-α。此外,ADAM17/10抑制剂GW280264的存在并没有阻止cse诱导的屏障功能缺损,也没有阻止cse诱导的EGFR活化(数据未显示),从而降低了ADAM17发挥作用的可能性。因此,更合理的解释可能是Src激酶活性的诱导和随后的配体独立EGFR磷酸化,这在A549细胞中已被描述[31]我们将在未来的研究中探讨这种可能性。
除了对细胞-细胞接触的影响外,EGFR活性还可以通过诱导钙蛋白酶活性和纤维连接蛋白的分裂和/或局部粘附成分talin来影响细胞-基质接触[32- - - - - -34].在我们的环境中,适度的CSE诱导的高频电容增加增加对细胞 - 矩阵触点的影响,并且AG1478的存在废除了这些效果。在16HBE细胞和PBEC中,我们观察到比高频电容低频率电阻更明显的效果,表明香烟冒烟诱导的EGFR活性影响细胞间接触的比细胞矩阵触点更多。
我们的数据表明,CSE也影响响应伤口的细胞间触点的重构,从而导致16HBE细胞中屏障函数的延迟。在PBECS中,CSE在重新建立单层而不是在恢复过程中受损障碍功能。我们没有对CSE引起的PBEC中的CSE引起的缺陷发出的解释。这可能是这涉及CSE对两种细胞类型中的CSE对细胞内信令和/或细胞架构的差异影响。再次,我们没有观察在CSE的存在或不存在的非莫克者和健康吸烟者的上皮反应的显着差异。这表明长期吸烟不会引起文化过程后仍然存在的变化,例如与上皮细胞再生有关的蛋白质。此外,与健康的COPD患者相比,COPD患者的PBECs在受伤和/或CSE暴露反应方面没有显著差异。然而,我们不能排除在COPD患者和健康人之间的ali极化PBECs存在差异的可能性。在一项单独的、未来的研究中,进一步探索COPD上皮细胞可能的内在差异将是有兴趣的,该研究包括更大数量的COPD患者和来自于年龄和吸烟史匹配的健康供体支气管刷拭的上皮细胞。
综上所述,我们证明香烟烟雾可能通过内源性蛋白酶calpain的机制,诱导egfr依赖性的TJs破坏,并在损伤时降低气道上皮完整性。我们认为,吸烟可能对慢性阻塞性肺病的细菌和病毒感染具有重要的意义,这两种情况都是通过促进病原体的进入以及在病原体诱导损伤后损害黏膜免疫屏障的恢复。因此,靶向EGFR可能为COPD加重的治疗提供新的途径。
致谢
我们感谢H.G. de Bruin(荷兰格罗宁根大学医学中心)对免疫检测实验的帮助。
脚注
这篇文章有补充资料可从www.www.qdcxjkg.com
利益陈述书
有关本研究的兴趣声明,请访问www.www.qdcxjkg.com/site/misc/statements.xhtml
- 收到了2010年12月15日。
- 接受2011年6月27日。
- ©2012人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}