





文摘
自身免疫和肺动脉高血压之间的关系(PAH)一直在欣赏> 40岁,但自身免疫性损伤可能导致这种疾病的发病机制只有在一个特定的检查方式。
越来越清楚的是,各种不同的临床疾病,包括病毒感染、结缔组织疾病,可以在肺血管病理学是不可区分的高潮。有迄今为止未被欣赏的生物学联合这些看似无关的条件?
这个问题的答案可能来自于不断增加的证据涉及中央调控t细胞在防止不恰当的b细胞活动的重要性。两个条件之间惊人的相似之处与angioproliferative严重肺动脉高压有关缺陷的CD4 t细胞室和auto-antibody生产。致病性auto-antibodies针对内皮细胞能诱导血管内皮细胞凋亡,可能启动PAH的发展。
目前的审查将专注于什么是肺动脉高血压患者了解自身免疫现象,为了更好地考虑是否自我耐受性其次是自身免疫损伤的早期损失可能影响的早期发展严重angioproliferative肺动脉高压。
严重肺动脉高压(PAH)可以表现的胶原血管疾病和病毒感染。例如,∼12%的硬皮病患者将发展多环芳烃1。其他疾病,如系统性红斑狼疮、多发性肌炎、干燥综合征、Hashimo-to甲状腺炎都与发展相关的严重的多环芳烃2。它也知道,某些病毒感染,如。艾滋病毒和人类疱疹病毒8(疱疹),可以关联到的发展严重的多环芳烃3- - - - - -6。感兴趣的是,所有这些条件的特点是,或有一个倾向,自身免疫。自主免疫攻击可能发生由于监管CD4细胞的相对缺乏。它已经知道其中的一些疾病,有一个公认的思想负责监管子集的减少外周免疫耐受,CD4 + CD25 +细胞。目前的审查将讨论失去自我耐受性如何启动过程最终导致多环芳烃。
肺动脉高压:病理学和分类
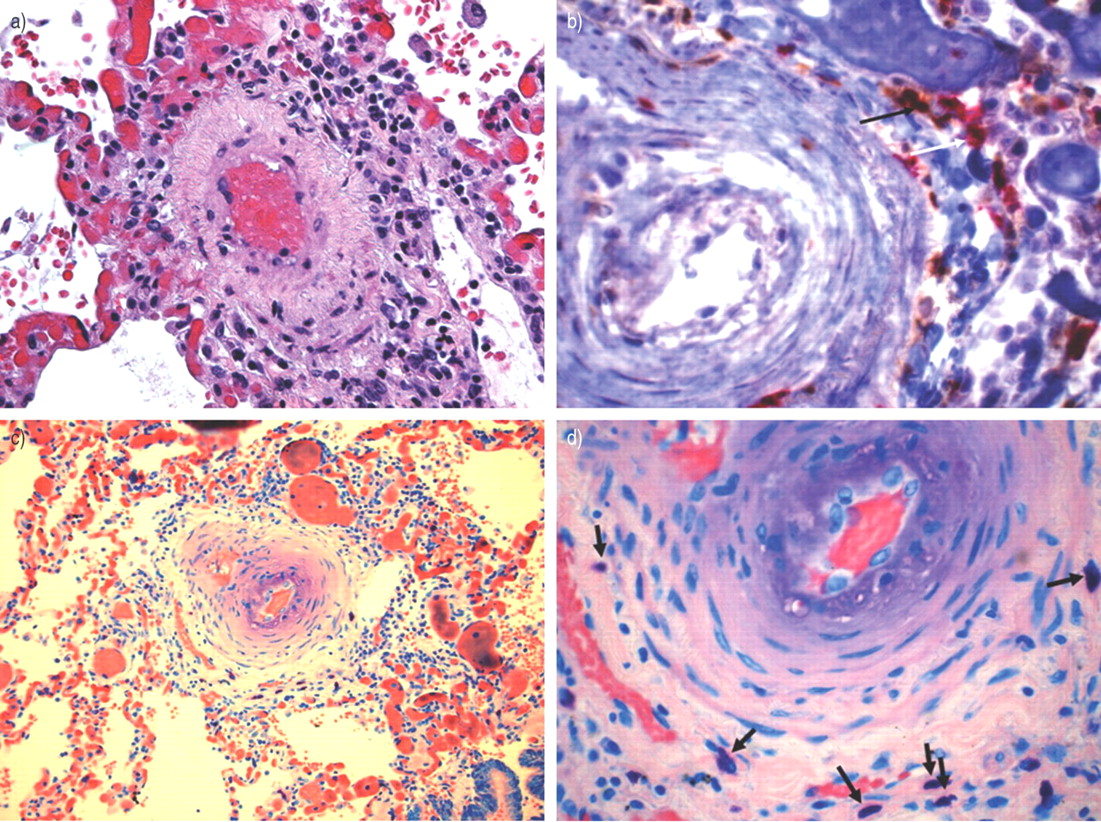
最近建立了肺高血压疾病的临床和病理分类在埃维昂会议7在威尼斯,意大利,2003年更新8。病理上,多环芳烃的特点是内皮细胞的增殖和扩张血管平滑肌和外膜细胞在肺动脉9。越来越多的升值,血管收缩的肺pre-capillary小动脉可能不是最重要的因素导致严重的肺血管重塑。虽然还没有被广泛接受,目前审查的作者使用术语“严重angioproliferative肺动脉高压”(SAPPH)10肺血管疾病之间的区分,明确,由于内皮细胞增殖和肺血管疾病发展主要是由于增加血管壁的muscularisation8。多环芳烃由内皮细胞病理学的例子包括特发性肺动脉高压(IPAH;原名主肺动脉高压)8,艾滋病病毒导致的多环芳烃和波峰(钙质沉着、雷诺氏现象、食管功能障碍,指硬皮病,毛细管扩张)相关SAPPH。的例子多环芳烃与严重的muscularisation pre-capillary小动脉,这证明内皮细胞病理学没有明确的证据,包括一些hypoxia-associated PAH疾病如慢性高山病11,12和新生儿多环芳烃13,14。术语SAPPH首选有几个原因。1)SAPPH提供pathobiological概念,(即。血管生成或angioproliferation)。2)SAPPH统一这两个所谓的主要和次要形式的多环芳烃,“严重”的旗帜下和同事的条件复杂的肺血管病变,包括丛状的病灶。3)SAPPH可能提供预后和治疗信息,目前,这些严重的治疗形式的angioproliferative PAH非常相似10。SAPPH特征也影响周围的炎症细胞和肺血管。希斯15描述中肥大细胞的存在网状病变患者的主要多环芳烃> 30年前。进一步的工作态度et al。16,亨伯特et al。17描述的存在炎性浸润血管病变的多环芳烃。图1⇓和2⇓说明免疫病理学IPAH患者淋巴细胞和肥大细胞浸润,以及免疫球蛋白G沉积在缩小和闭塞的血管腔。
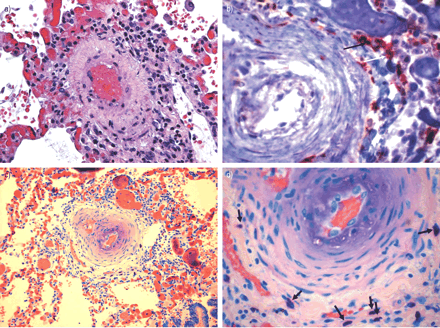
免疫病理的肺小动脉特发性肺动脉高血压病人。38-yr-old(抗核抗体阳性,人类疱疹病毒8阳性)与特发性肺动脉高血压女性过期。苏木精和伊红染色显示影响周围小动脉栅栏样的单核细胞。快速b) CD4 +(红底物(红染色);白色箭头)和CD8 + (3,3′-diaminobenzidine(棕色染色);黑色箭头)细胞病变。c)吉姆沙染色剂的肥大细胞指示peri-arteriole渗透。d)病变的放大视图,肥大细胞中黑色箭头所示。

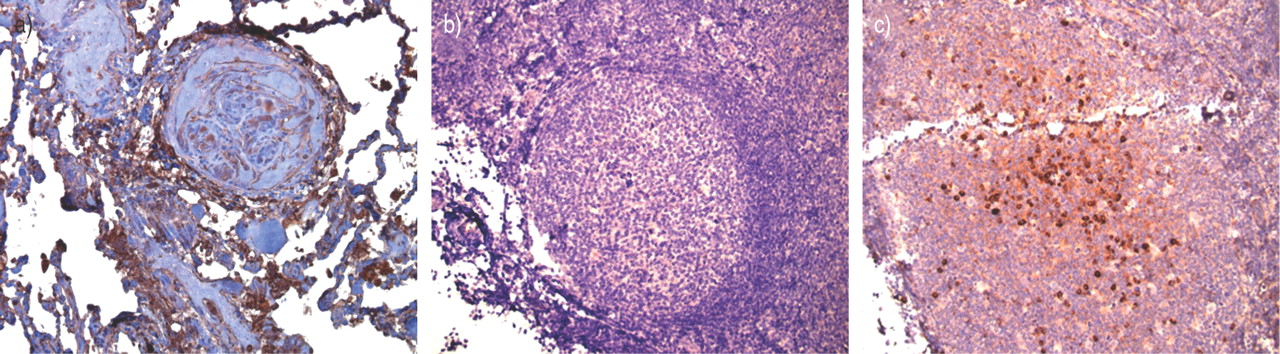
免疫球蛋白G染色的丛状的特发性动脉肺动脉高压病人的病变。38-yr-old(抗核抗体阳性)与特发性肺动脉高血压女性过期。一)免疫球蛋白G积极性在丛状的病灶(3、3′-diaminobenzidine;棕色染色)。b) -抗体控制外周淋巴结。c)阳性抗体控制外周淋巴结。
多环芳烃:自身免疫和免疫调节
是否存在炎症和免疫细胞,如T - B -淋巴细胞病变16SAPPH原因或结果依旧不明,可能讨论一段时间。然而,它已被认可> 40岁,有自身免疫性疾病之间的联系和严重的多环芳烃。进展甚微,在当前的了解免疫损伤可能参与SAPPH的发病机理。除了承认自身免疫和SAPPH之间的联系,还有一个免疫机能不全和SAPPH之间的联系,因为HIV阳性病人和艾滋病患者开发多环芳烃和血管性病变组织学IPAH区分开来。最近的潜在HHV-8的描述18患者IPAH进一步引出了一个问题的免疫系统调节PAH的发展。
这个问题的答案可能是大多数情况下与SAPPH与CD4 t细胞室一个缺陷,这意味着这些条件的特点是绝对不足CD4细胞,CD4 / CD8比值下降和/或CD4 + CD25 +细胞的比例相对减少,假定监管t细胞(Treg)的子集。具体地说,艾滋病毒3,HHV-84,5和丙型肝炎病毒6,19- - - - - -21都是与CD4缺陷相关的感染,自身免疫现象(包括auto-antibodies)和SAPPH的发展。同样,其他PAH-associated条件,包括结缔组织疾病,也与CD4细胞缺陷有关。例如,硬皮病22,23系统性红斑狼疮,24,25、多肌炎26,桥本甲状腺炎27和干燥综合征28都能表现出选择性CD4细胞缺陷和自身免疫(同样包括auto-antibodies)。更具体地说,硬皮病和红斑狼疮与减少外围CD4 + CD25 +细胞,假定的Treg人口23- - - - - -25。此外,多环芳烃脾切除术后被描述29日。最后,它最近报道,患者自身免疫性polyendocrinopathy-candidiasis-ectodermal萎缩症,这是由于基因突变导致的损失函数的自身免疫调节器(问卷调查)蛋白质,IPAH死于致命的30.。中部的涂画或基因thymus-dependent自我耐受性发展的重要性。t细胞、b细胞、巨噬细胞和肥大细胞浸润是一个特色patholological特性SAPPH丛状病变的患者16,31日。此外,据估计,30 - 40%的患者IPAH抗核抗体阳性,而另一个10 - 15%的患者可以表达antiphospholipid抗体32,33。统一这些累积的发现是,假设地址设置的相对或绝对免疫缺陷(包括减少Treg活动),免疫失调发生并导致致病性autoreactive b细胞和t细胞的激活。
如果自身免疫触发SAPPH的发展,那么为什么PAH的患病率相对较低的某些自身免疫性疾病如系统性红斑狼疮(6.2%)34吗?如果自身免疫现象导致终末器官损害在起作用,为什么肺血管不普遍参与这些自身免疫性结缔组织疾病呢?虽然这个问题无法回答,有几种可能性。众所周知,在某些免疫性疾病,基因型可以带来显著的风险而不完整的疾病外显率。例如,在1型糖尿病风险增加的基础上赋予HLA-DR-DQ基因型,只有不到10%的易感个体或30 - 40%的同卵双胞胎的1型糖尿病患者将开发这种疾病35。环境因素如饮食或病毒感染已经调用必要的“第二次打击”这种疾病的发展。类似地,很可能一个两面夹攻的现象可能需要这样自身免疫导致血管损伤和多环芳烃。当前作者假定,PAH-associated条件的共同因素是免疫调节的损失,第二个“宽容”因素可能是患者的基因型和/或伴随的血管损伤由于感染或临时的高剪切应力。总之,不完全外显率的多环芳烃在自体免疫条件下与多环芳烃可能由于两个或两个以上的要求额外的因素存在于少数的病人。
硬皮病:自身免疫性多环芳烃的范式
硬皮病是一种结缔组织疾病的特点是过度的皮肤中胶原蛋白的积累和内脏器官。正如上面提到的,据估计,硬皮病患者将开发SAPPH∼12%1。硬皮病研究者相信,内皮细胞凋亡可能是第一个事件在硬皮病的发病机制36。Anti-endothelial抗体在硬皮病患者的循环37,38,他们的存在与该疾病的临床进展38。硬皮病的煽动损伤内皮细胞,可能会引发此类auto-antibody形成可能是病毒感染39。一些调查人员发现病毒感染的证据,比如巴尔病毒,细小病毒B19和丙型肝炎,E, G硬皮病患者40- - - - - -45。虽然巨细胞病毒(CMV)的角色在硬皮病的发病机理讨论46、间接证据CMV-specific抗体的作用还讨论了这种疾病的发展47,48。不仅是绝对的淋巴细胞计数降低硬皮病49,50,但硬皮病患者也有相对较少的CD4 + CD25 +细胞与健康对照组相比末梢循环23。在减少亚这个设置,b细胞也观察到的失调51。丛状的病灶在动脉壁硬皮病PAH患者包括炎症浸润52包括巨噬细胞、t细胞、b细胞和肥大细胞53- - - - - -55。总之,硬皮病是一种自身免疫性疾病,与病毒感染有关,内皮损伤,减少亚群,b细胞特异表达,丰富的肥大细胞和auto-antibodies。本文随后将讨论Treg活动是如何与b细胞活动,如何与肥大细胞浸润auto-antibodies的生产,最后,这些过程如何在多环芳烃可能达到高潮。
AUTOREACTIVE b细胞活化的t细胞的缺乏监管
两个发现,近30年前,强烈与t细胞是负责控制反应细胞和b细胞在维护自我耐受性。1969年Nishizuka Sakakura56表明,新生儿的正常小鼠胸腺切除术,尤其是在出生后一天2和4之间,导致自身免疫性破坏卵巢。1973年,Penhaleet al。57显示胸腺切除术的成年老鼠跟着几个曝光亚致死的X-irradiation导致自身免疫性甲状腺炎的发展。作为正常的CD4 +细胞的接种预防这些疾病,两组怀疑抑制t细胞的损耗是一个假定的自身免疫的发展机制58,59。作为这个群体最终被缩小至CD4 + CD25 +细胞,很明显,这个相对较小的族群的损耗(5 - 10%的CD4 +细胞)足以打破自然自我耐受性和煽动慢性和破坏性的自身免疫性疾病。这种失去自我耐受性与各种疾病特性auto-antibodies相关的外观60。完全消除CD4 + CD25 +细胞,发生系统性自身免疫所体现的多器官炎症和auto-antibody生产61年。
除了控制t细胞活动,亚群影响b细胞反应。例如,CD4 + CD25 + treg已被证明直接抑制lipopolysaccharide-induced b细胞的增殖在体外。在一个过继转移系统,CD4 + CD25 + t细胞表达下调T-cell-mediated反应抗体的生产。激活淋巴细胞产生CCL5和吸引CD4 + CD25 + t细胞在体外,假定b细胞的CD4 + CD25 + t细胞可能会限制b细胞自身免疫反应62年。CD4 + CD25 +细胞防止anti-DNA抗体转基因系统的激活63年。因此,亚群调节对自我和异物抗原抗体反应。亚群可能对b细胞产生直接的抑制作用62年或者可以抑制t细胞分化64年。因此,在t细胞缺乏适当监管,auto-antibodies可以出现,可以开发和自身免疫性疾病。因此,失去Treg-mediated自我耐受性,在t细胞耐受不仅损失,但b细胞耐受的崩溃。在缺乏这些亚群,其他细胞大概提供刺激信号相关的反应b细胞,救他们脱离细胞凋亡,促进他们形成致病性抗体65年。
传统的理解自身免疫性疾病引起的不当有力的t细胞室自身免疫性疾病如糖尿病和多发性硬化症66年。相反,自身免疫性疾病如肖格伦疾病和系统性红斑狼疮,杰出的致病性auto-antibody生产显然Treg妥协的设置功能。多环芳烃的自身免疫可能更像是后者的疾病。正如上面所讨论的,Treg活动通常是负责防止auto-antibody生产。抗体针对血管内皮当然可以促进内皮细胞凋亡。内皮细胞凋亡,可能继发于自身免疫损伤,可以启动不正常内皮细胞增殖的高潮在多环芳烃以同样的方式,内皮细胞凋亡,血管内皮生长因子引起的对立,导致内皮细胞增殖和多环芳烃67年。Anti-endothelial存在抗体在系统性红斑狼疮等自身免疫性疾病相关的多环芳烃68年、混合结缔组织病69年和硬皮病38。红斑狼疮、干燥综合征、抗体和补充存款本地化的墙壁PAH患者的肺动脉70年,71年。所以,简而言之,这种形式的效应细胞自身免疫可能是b细胞(后分化为浆细胞)产生anti-endothelial抗体。尽管autoreactive b细胞的作用是强调,t细胞在炎性病变的存在表明t细胞特异表达也在多环芳烃导致自身免疫损伤。
它是有用的考虑antiphospholipid综合症可能是临床场景有许多上述PAH模型的元素。antiphospholipid综合症患者改变了早期肠在外围,尤其是CD4 + CD25 +显著降低的人口72年。antiphospholipid综合症通常与病毒有关与多环芳烃相关症状,如艾滋病和丙型肝炎,有免疫调节作用73年。假说提出了缺陷Treg人口(可能发生病毒感染后)会导致失去Treg活动后续生产auto-antibodies和血管内皮损伤有关。antiphospholipid综合症与antiphospholipid抗体绑定并激活内皮细胞。这种抗体参与最终导致血管内皮细胞的凋亡74年,75年。最后,unmanipulated无胸腺的裸小鼠,还缺一个孤立的T细胞,已经被证明是自发发展antiphospholipid抗体,而严重联合免疫缺陷小鼠,既缺乏T - b细胞,没有antiphospholipid抗体76年。因此,在antiphospholipid综合症和其他自身免疫性疾病,最重要的效应细胞可能产生auto-antibodies的b细胞特异表达血管内皮因为正常监管t细胞活性降低或缺席。
Treg活动减弱时,肥大细胞,而不是t细胞,会增强b细胞活化通过白介素(IL) 4 (即。肥大细胞可能替代作为il - 4的来源,增加当地的b细胞活化77年)。值得注意的是,肥大细胞是non-T-cell il - 4的一个重要来源78年,il - 4已经牵连的扩张器autoreactive b细胞79年细胞因子的重要性在硬皮病的实验模型80年- - - - - -82年。肥大细胞在丛状的病灶及周边的存在一直观察到15,31日。肥大细胞在PAH最初认为是那样重要83年,折扣,因为它后来证明缺乏肥大细胞并没有阻止实验多环芳烃84年,而过量的肥大细胞85年,86年没有与临床相关的多环芳烃。然而,肥大细胞确实是出现在PAH患者的炎性病变31日和monocrotaline-induced PAH在无胸腺的裸体老鼠缺乏t细胞87年。后者是有趣的因为monocrotoline-induced PAH的缺失加剧了亚(即。引人瞩目的是,当不存在)t细胞和肥大细胞浸润在丛状的病灶周围的炎症。现在提出,肥大细胞的存在可能不需要所有亚型的多环芳烃,但是,和其他自身免疫性疾病一样,肥大细胞可能是一个重要的先天和适应性免疫反应之间的联系88年,89年。而不是绝对必需的发展多环芳烃,肥大细胞可能是重要的主持人增效autoreactive b细胞的免疫反应。鉴于假定的细胞因子和趋化因子的重要性一代的适应性免疫反应,重要的是还要注意,血清il - 1和il - 6水平明显升高严重的多环芳烃和显著可能导致这种疾病的炎症环境90年。
骨形态形成蛋白受体2:一个新的解释在多环芳烃的发展的作用
基因的基础最近决定家族PAH的某些情况下,即。遗传突变的参与骨形成蛋白受体2 (BMPR2)91年。BMPR2属于肿瘤生长因子(TGF) -β受体家族,是骨形成蛋白(bmp)的配体2,4,6和7,但不是TGF-β92年。针对性的当前模型是BMP2和4角色发展,增长潜力和T和b细胞凋亡。BMP2和BMP4(及其受体可能,BMPR2)也对胸腺细胞分化至关重要93年- - - - - -95年,适当的胸腺Treg发展是至关重要的,以避免自身免疫性疾病60。BMP2介导生长逮捕和B谱系细胞的凋亡96年,97年。最后,BMP4对b细胞祖细胞的生成至关重要(除了erythro-myeloid集落形成细胞和自然杀伤细胞祖细胞)98年。可想而知,作为每个位置,这些受体BMPR2密切参与这些免疫效果。这已经显示BMP对胸腺发育的影响93年。因此,是BMPR2可能突变可影响正常发展,成熟,增长逮捕和淋巴细胞的死亡。的方式类似于发展中PAH患者因为涂画或基因突变30.部分,生殖系BMPR2突变可能导致PAH因为他们导致外周免疫耐受(一个根本性的缺陷即。不恰当的Treg发展删除b细胞)和/或异常的能力。这个想法打开有趣的途径的研究关注的角色BMPR2在免疫耐受而不是简单地对平滑肌细胞生长的影响。
SAPPH开发的一个集成模型
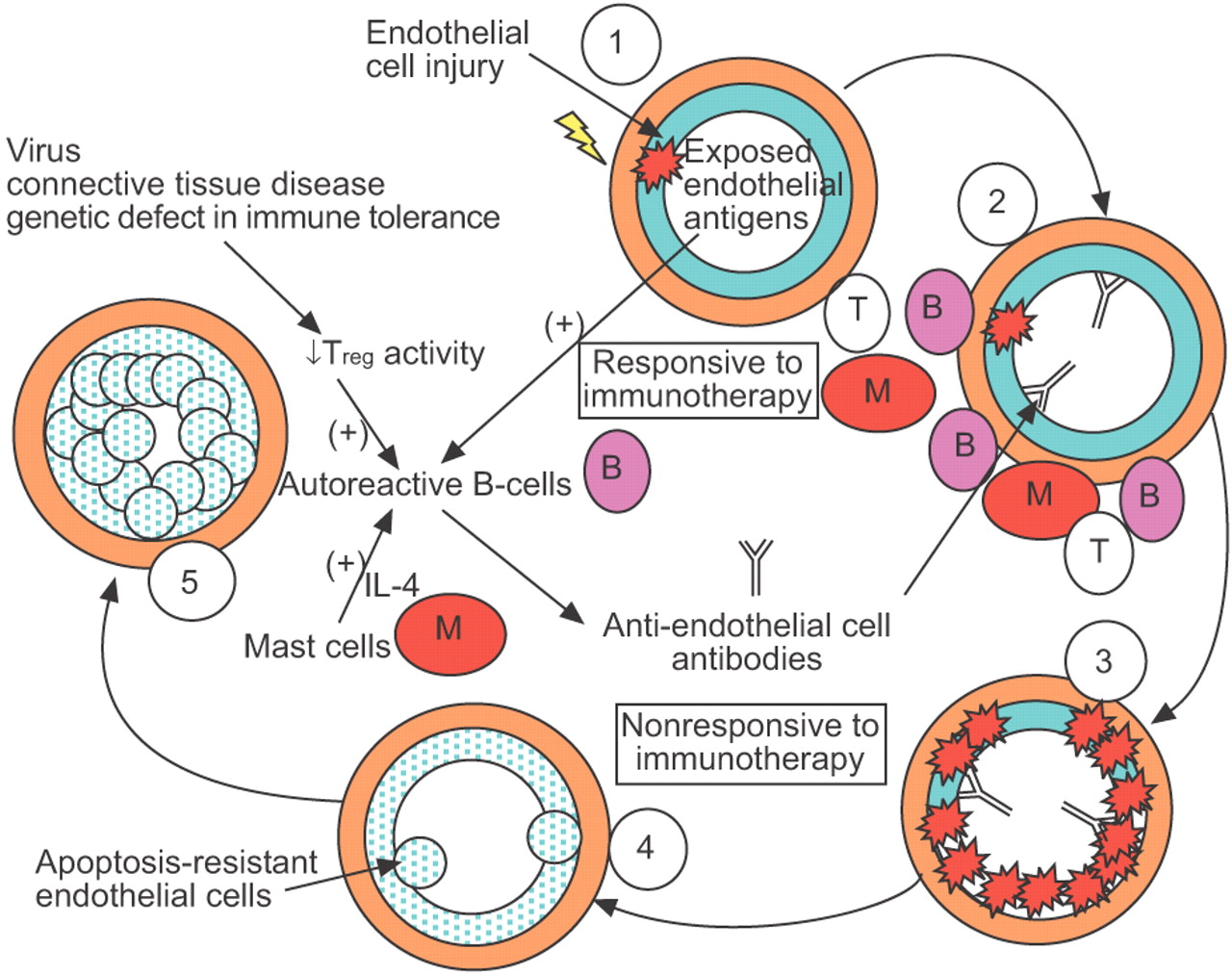
PAH的事实一般不应对免疫抑制可能可以解释为什么调查人员没有全面探讨自身免疫损伤和多环芳烃之间的因果关系。然而,很可能不适应的反应初期炎症损伤导致组织损伤反应延迟的生物完全不同于触发免疫侮辱和不再适应免疫疗法,它可能是在“起始阶段”响应。的多环芳烃,内皮细胞破坏的免疫介导的损伤可能导致apoptosis-resistant内皮细胞的生成和恶性细胞分享功能99年- - - - - -101年。在这个场景中,单层的“法律”坏了,这些内皮细胞增殖,最终成为“堆”,掩盖了容器内腔16。这些想法在图3中表示⇓。
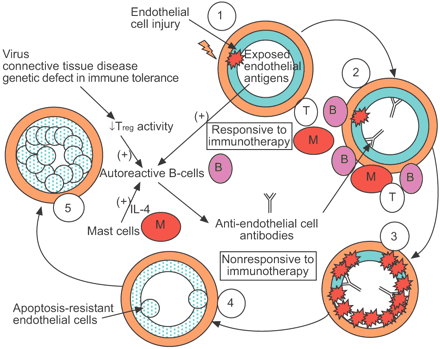
示意图显示自身免疫进化的肺动脉高血压(PAH)。1)损伤血管内皮(如。从血管营养病毒或剪切应力)内皮抗原暴露。在病毒感染、结缔组织疾病或遗传缺陷在免疫耐受,减少监管t细胞(Treg)活动可能被观察到。当血管损伤的“两支安打”和减少外周免疫耐受同时发生,这可能会导致失去正常的控制对autoreactive b细胞。作为直接天生的宿主防御的一部分血管损伤,内皮细胞肥大细胞浸润血管周的空间接近受伤。肥大细胞富含白介素4 (IL),以及其他因素,能够刺激autoreactive b细胞分泌auto-antibodies包括anti-endothelial细胞抗体。2)临床PAH的特点是B细胞(B), T细胞(T)和肥大细胞(M)浸润丛状的病变,和antibody-complement存款位于PAH患者的肺动脉。有可能在这炎症初期,病变与免疫治疗可以逆转。3)抗体沉积可能导致持续的内皮细胞凋亡。4)作为组织修复反应,持续的内皮细胞凋亡导致的生成apoptosis-resistant内皮细胞恶性表型。这可能是一段时间的伤害,不再适合免疫疗法。 5) Apoptosis-resistant endothelial cells become “heaped-up”. The resulting vascular remodelling leads to vascular occlusion, an increased vascular resistance and worsening of PAH.
结论
国立卫生研究院注册中心数据表明,零星的PAH患者的生存中值是2.8年,存活率在1、3和5年是68年,48和34%,分别102年。而前列腺素类治疗是改善生存对某些形式的多环芳烃103年,多环芳烃仍然是一个经常致命疾病的相对神秘的起源。> 40岁已经意识到,自身免疫现象与PAH相关联,但它从来没有先前表明,自身免疫,本身,可能是某种形式的多环芳烃的根源。多环芳烃的人力和财力代价是巨大的。例如,硬皮病的流行在美国在1996∼9000名患者104年,如上所述,12%的硬皮病患者将开发多环芳烃105年- - - - - -108年。1997年美国照顾硬皮病患者的成本是15亿美元/年的大部分钱走向PAH的治疗109年。目前的治疗策略亚型与自身免疫性疾病相关的多环芳烃,目前没有不同于多环芳烃没有建立自身免疫性协会管理110年。如果自身免疫是真正重要的发病机制中的多环芳烃,然后高危患者(如硬皮病或多环芳烃BMPR2突变的家庭成员)可以有针对性的免疫疗法,旨在防止自身免疫损伤的建立和传播。了解自身免疫的发展可以触发PAH将宝贵的PAH发病机理的形成模型的设计,可以促进新治疗免疫失调的具体地址。目前的审查中概述的概念试图解释一些常见的临床观察。最值得注意的是,这里的假设先进联合免疫缺陷和自身免疫的概念经常被观察到与多环芳烃相关条件。此外,提出的模型可以解释细胞群(如肥大细胞)与疾病密切相关,但以前没有pathogenetically集成。
总之,通过识别关键免疫的基础上对许多形式的肺动脉高血压治疗的合理的设计目标,这群经常致命的疾病将大力推动。
- 收到了2005年4月15日。
- 接受2005年7月12日。
- ©人期刊有限公司










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}