





摘要
大量的实验证据现在支持这一观点,即过敏性疾病的特征是免疫系统向t辅助细胞2型(Th2)表型倾斜。
使用人类和小鼠模型系统的研究为Th2细胞因子在推动过敏性炎症的许多特征中所起的作用提供了关键证据。此外,Th2细胞因子对气道靶细胞发挥作用的信号通路正在被迅速阐明,Th2通路的拮抗剂正在积极开发中。
在这篇综述中,总结了目前关于t -辅助性细胞2型在哮喘中的作用的知识,重点是t -辅助性细胞2型如何以及在哪里从naïve前体分化。参与t辅助细胞2型分化的信号分子和转录因子将被详细回顾,试图将使用转基因小鼠的研究转化为对哮喘和其他过敏性疾病的有意义的见解。
“炎症和免疫细胞的信号和转录调节:肺生物学和疾病的重要性”系列
编辑:K.F. Chung和I.M. Adcock
本系列的第二题
大量证据支持过敏性哮喘是一种免疫介导的疾病,部分是由针对航空过敏原和病毒的t细胞驱动的1。特别是CD4+ t辅助细胞(Th) 2型细胞,分泌白细胞介素(IL)-4, IL-5, IL-9和IL-13,被认为有助于哮喘的许多病理生理特征,包括气道炎症,粘液分泌和气道高反应性(AHR)。2。目前的大部分研究旨在了解Th2细胞因子如何作用于居住的肺细胞,包括气道上皮细胞、肌成纤维细胞和平滑肌,以诱导哮喘表型,参与这一过程的信号通路和转录因子正在迅速阐明3.。th2偏向免疫反应的精确分子基础是复杂的,在任何给定的个体中都可能是多因素的。遗传易感性是由Th2集群中的单核苷酸多态性(SNPs)和单倍型以及Th2信号分子和不同的过敏性疾病之间的关联所暗示的(表1)⇓)。不明确的环境暴露也会导致Th2偏倚,部分原因是环境暴露作用于树突状细胞和其他影响t细胞激活和扩张的细胞。由于Th2偏倚通常是特异反应的特征,似乎有可能存在疾病特异性易感基因和环境暴露,最终影响特定特异反应表型的发展(如。特应性皮炎与哮喘);这方面的候选人已开始被确定4–6。根据这个模型,th2偏倚的免疫系统可以被认为是过敏性哮喘发展的必要条件,但还不够,这需要一个“敏感气道”(图1)⇓)。这篇文章回顾了导致th2偏向免疫反应的遗传和潜在的环境因素。它主要来自使用小鼠模型的研究,这些研究确定了与Th2分化有关的关键分子,但将强调未来研究应该增强对人类特应性疾病的理解的领域。
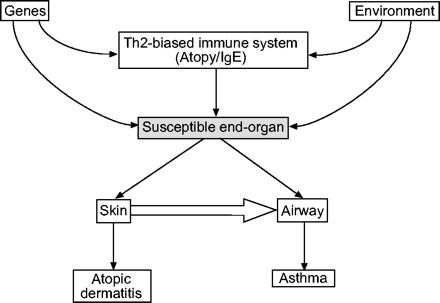
t辅助细胞2型(Th2)偏倚免疫应答与器官特异性特应性疾病的关系该模型强调,th2偏倚可以被认为是免疫失调的系统性标志,但不同疾病的临床表现受器官特异性因素的影响。特应性疾病从皮肤到肺部的进展(如。在童年时期观察到的“特应性进行曲”)由大箭头表示。搞笑:免疫球蛋白。
Th2细胞,哮喘和特异反应
一些研究记录了哮喘患者肺部活化CD4+ t细胞数量的增加,并且,在迄今为止的大多数研究中,这些细胞被发现表达Th2细胞特有的细胞因子或转录因子22–32。在一些研究中,哮喘中这种明显的Th2偏倚延伸到气道外的循环淋巴细胞33–35。此外,在小鼠模型上的研究已经确定了Th2细胞因子在哮喘病理生理学中发挥的关键作用,包括粘液高分泌和AHR36–50最近的一项研究强调了IL-4和IL-13在慢性过敏原暴露时气道功能障碍和重塑中的重要性50。在人体研究中,给药重组IL-4导致哮喘受试者出现AHR51而IL-12抑制血液和痰中的嗜酸性粒细胞(不影响AHR)52)。此外,临床上有用的药物,包括吸入糖皮质激素,可有效抑制Th2细胞因子基因的表达53,54。综上所述,这些观察结果支持了一种假设,即气道中Th2细胞的扩张和激活是哮喘病理生理学的关键组成部分55。这一假设的一个推论是,详细了解导致哮喘Th2偏倚的因素应确定未来的治疗靶点。
鉴于Th2细胞和细胞因子在哮喘病理生理学中发挥的关键作用,最初认为在肺中诱导Th1反应将具有保护作用。然而,现在已知Th1细胞及其标志性细胞因子干扰素(IFN)-γ在肺中具有复杂和潜在的促炎作用。使用小鼠模型的研究提供了关于Th1细胞和IFN-γ在气道炎症和AHR中的潜在作用的相互矛盾的结果。在一些研究中,Th1细胞和IFN-γ被发现可以抑制过敏性气道炎症56而在其他研究中发现Th1细胞和IFN-γ具有促炎作用57–59。在病毒诱导的AHR和气道重塑小鼠模型中,IFN-γ缺陷小鼠的这些参数没有衰减60。有趣的是,有证据表明,哮喘患者外周血CD8+ t细胞中IFN-γ的表达与AHR相关61最近的一项研究发现,脐带血CD8 t细胞中IFN-γ的产生可以预测随后的过敏致敏62。一些哮喘患者的肺或痰中存在IFN-γ表达淋巴细胞63,64,在病毒引发的病情加重期间,分泌IFN-γ的细胞溶解性CD8+细胞可能会被招募到肺中。尽管有一些证据表明CD8+在哮喘加重期间招募到肺部65令人惊讶的是,目前对这些细胞的细胞因子特征知之甚少。综上所述,这些研究表明IFN-γ在哮喘中的潜在作用是复杂的,取决于表达这种细胞因子的细胞位置和疾病的阶段。需要使用特定的细胞因子拮抗剂进行研究,以帮助阐明IFN-γ和相关细胞因子在人类哮喘受试者中的作用。
t细胞个体发育和th2分化
目前还不清楚Th2细胞在特应性个体中的确切位置。Naïve t细胞在经历了阳性选择和逃脱了阴性选择后,带着高效重新排列的t细胞受体(TCR)离开胸腺。由于它们的归巢受体的表达,包括外周淋巴结地址,naïve t细胞通过中央免疫器官(包括淋巴结)循环,直到它们遇到抗原呈递细胞(APC),其表面TCR识别肽片段。如果APC被适当激活,naïve t细胞会接收到额外的信号,启动部分由IL-2自分泌驱动的克隆扩增。有趣的是,最近的研究表明,除了对t细胞增殖的影响外,长时间暴露于IL-2也促进了Th2分化66。随着克隆扩增的进行,许多活化的t细胞经历活化诱导的细胞死亡,这限制了随后的免疫反应。一些t细胞存活下来并分化为效应细胞或记忆细胞,然后被招募到外周粘膜部位以协调细胞免疫。基于该模型,似乎可以合理推测,过敏原特异性Th2细胞的分化是从区域淋巴结开始的。因为特应症患者会变得敏感通过皮肤和呼吸道对环境过敏原的影响,Th2分化可能发生在引流皮肤和肺部的淋巴结中。然而,最近在小鼠模型中的发现表明,在没有淋巴结的情况下,肺具有启动初级免疫反应的内在能力67–69;这些模型中幼稚t细胞教育的确切位置,以及它们如何应用于人类过敏性疾病,仍有待确定。尽管在技术上具有挑战性,但在未来的研究中,研究不同过敏性疾病的人类过敏原特异性t细胞的致敏途径和命运将是有趣的。沿着这些思路,对职业性哮喘患者的研究可能具有深刻的见解,因为其致敏途径各不相同,而且这种疾病在成年期发病。
t细胞亚群的极化程度取决于极化信号的强度和持续时间,既涉及细胞内在因素,也涉及外源信号。细胞内在因子在Th分化中的相对贡献(即。在预先提交的前体细胞的“选择”中)很难确定(有关综述见70)。表达IL-4或IL-13的单个等位基因的决定似乎是一个随机事件71。然而,现在很清楚的是,外源性信号(包括细胞因子)的“指令”在驱动naïve t细胞分化中起着关键作用。IL-4是Th2分化最重要的细胞因子,激活转录因子信号换能器和转录激活因子(Stat)6,以及其他下游效应物(见下文)。启动Th2分化的IL-4的原始来源一直存在争议。观察到naïve t细胞可以快速产生IL-4,这表明IL-4可能来自naïve t细胞本身72。在这个模型中,如果naïve t细胞激活期间提供的精确信号优先诱导IL-4而不是IFN-γ的表达,那么这可以前馈放大Th2分化。然后预测有利于早期IL-4产生的遗传多态性可以增强Th2分化,并有助于易感特应性个体的Th2偏倚(图2)⇓)。
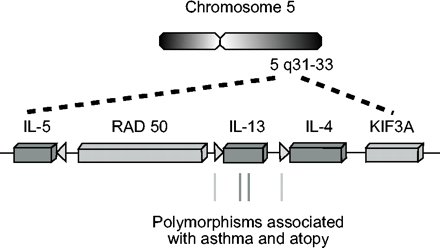
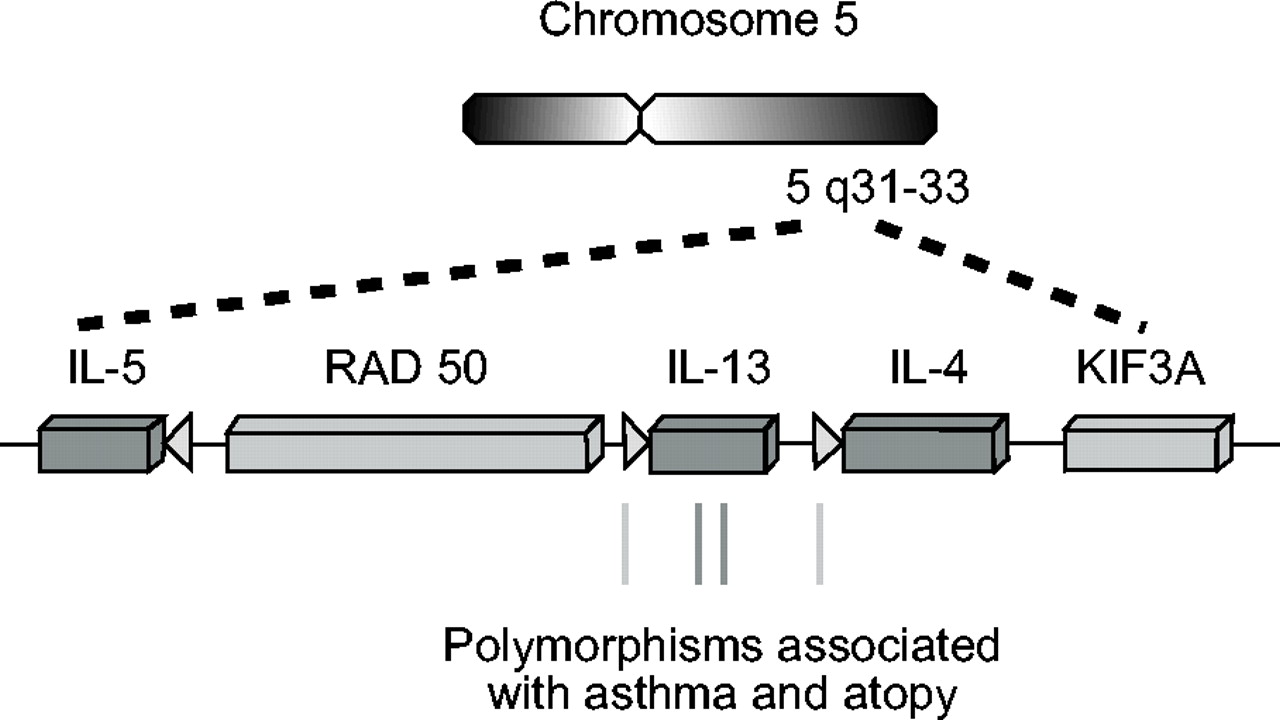
5号染色体上t辅助细胞2型(Th2)细胞因子基因簇的组织。图中显示了Th2细胞因子基因(矩形)及其启动子(箭头)的相对位置。位于启动子和编码区与不同哮喘和特异反应表型相关的单核苷酸多态性显示在底部。这个图是示意图,不是按比例画的。IL:白介素;RAD:辐射灵敏度50;KIF:驱动蛋白家族成员。
虽然IL-4的非t细胞来源不需要被调用来启动Th2分化,但值得注意的是,与人类特应性疾病相关的另外两种细胞类型也可以产生IL-4,即嗜碱性粒细胞和嗜酸性粒细胞。人嗜碱性粒细胞在IgE交联后分泌大量IL-4,并在过敏肺中产生IL-473,74。此外,使用转基因小鼠进行的实验发现,在Th2反应中,大多数GFP阳性细胞被招募到肺中,是嗜酸性粒细胞75。嗜酸性粒细胞在过敏原驱动的气道炎症和重塑中的重要作用最近通过使用两个嗜酸性粒细胞缺乏小鼠的新模型显示出来76,77。尽管嗜酸性粒细胞组成性表达IL-4表达所需的转录因子78但是,这些细胞分泌IL-4蛋白的能力仍然存在争议。这两种细胞类型是否影响naïve Th细胞的分化将取决于它们获得naïve t细胞的能力。沿着这些思路,最近的研究表明,嗜酸性粒细胞存在于气道引流淋巴结中,这表明它们实际上可能是抗原呈递细胞,但这是一个有争议的领域79。
Th1微分和t-bet
与IL-4相反,驱动Th1分化的关键细胞因子是IL-12。IL-12的主要来源是树突状细胞,在主要组织相容性复合体的背景下,树突状细胞处理并向t细胞呈递可溶性抗原。IL-12激活转录因子Stat4,部分通过结合到IFN-γ基因附近的调控序列来增强IFN-γ的表达80。另一个驱动Th1极化和IFN-γ基因表达的关键转录因子是T-bet。最近发现T-bet是IFN-γ和IL-12Rβ2基因表达所必需的谱系特异性转录因子,这是对Th1分化理解的重大进展81,82。CD4+细胞中调节T-bet的信号是复杂的,包括IFN受体(通过Stat1)和t细胞受体通过对途径了解不足83。T-bet似乎部分在染色质重塑水平上诱导IFN-γ基因表达84但目前还不清楚IFN-γ基因内和周围T-bet的精确结合位点。T-bet参与效应CD8+细胞和自然杀伤细胞的生成85,86,也调节b细胞同型转换87。有趣的是,IFN-γ基因表达存在T-bet独立通路88。
Finottoet al。89据报道,t- bet缺陷小鼠自发发展为哮喘样综合征,伴有气道炎症、重塑和高反应性。此外,过敏性哮喘患者肺部t细胞中T-bet的表达减少89。本研究提示过敏性哮喘可能是由T-bet和/或Th1细胞产生缺陷引起的。鉴于IFN-γ在哮喘中的复杂作用(如上所述),这一结果很有趣,这是否反映了T-bet对IFN-γ以外基因的影响值得进一步研究。哮喘患者T-bet表达减少的精确分子基础目前尚不清楚。有趣的是,多态性TBX21最近发现,T-bet基因与哮喘儿童吸入皮质类固醇的反应更强有关90。TBX21是否是哮喘或其他特应性疾病的易感基因目前尚不清楚。
树突状细胞与th2分化
树突状细胞(dc)是激活naïve t细胞的关键抗原呈递细胞,人们对破译dc衍生的影响Th分化的信号有很大的兴趣(最近的优秀评论已经深入讨论了这一主题,见91,92)。一般来说,DC对Th1分化的控制比Th2分化更好理解,这是由于DC暴露于细菌或其他启动DC成熟的强烈危险信号所致。DC成熟的特点是内吞作用减少,抗原提呈增强,趋化因子受体表达改变,从外周粘膜位点归巢到区域淋巴结,以及免疫调节细胞因子(包括IL-12)的产生增加。这些危险信号中的许多是由模式识别受体的toll样受体(TLR)家族转导的,TLR家族在dc和先天免疫系统的其他细胞上表达。强烈的危险信号倾向于通过诱导分化产生大量IL-12和相关的促进Th1的细胞因子(包括IL-23、IL-27和1型ifn)的dc来促进Th1免疫应答(图3)⇓)。
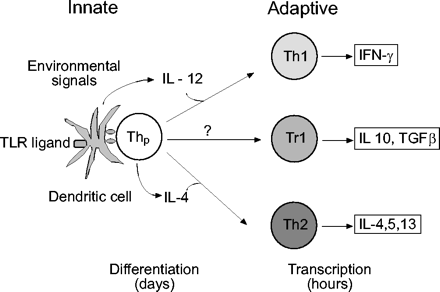
树突状细胞(DCs)提供的信号对t细胞分化的调节。强toll样受体(TLR)信号通路增强IL-12的产生,导致t辅助细胞(Th) 1型分化。相反,当naïve t细胞中IL-4基因转录被促进时,Th2分化导致IL-12缺失。是否存在dc衍生信号可以积极促进Th2分化,目前还不完全清楚(详情见正文)。Naïve t细胞也可以在外围分化为可下调免疫的调节表型:参与调节t细胞分化的信号尚不清楚,但可能包括IL-10(未显示)。干扰素:干扰素;TGF-β:转化生长因子-β。
树突状细胞不产生促进Th2的细胞因子IL-4,可能间接启动Th2分化。一种可能是激活dc产生少量的IL-12 (IL-12)罗DCs会促进Th2分化(图3)⇑)。因此,导致IL-12成熟的环境信号罗dc可被认为是特异反应中Th2偏倚的易感因素。一些信号已经被发现,允许dc成熟为IL-12罗/ Th2-promoting表型。这些包括低剂量内毒素、TLR2配体、糖皮质激素和许多g蛋白偶联受体激动剂(如。组胺,霍乱毒素,前列腺素E2, PGD2其他的见表2⇓)。这些信号和其他信号究竟如何影响哮喘患者过敏原驱动的DC成熟尚不完全清楚。需要注意的是,糖皮质激素(GCs)可能通过直接作用于dc而诱导th2偏向。一些研究表明,在发育中的dc中,gc抑制IL-12的产生,并且gc暴露的IL-12罗dc与t细胞共培养时诱导Th2表型93,94。然而,在这些研究中,DCs在与t细胞共培养(在没有GCs的情况下进行)之前被清洗。当GCs出现在DC: t细胞共培养过程中,一种更生理的t细胞激活模型,IL-4的表达被强烈抑制95。这一观察结果与已知的gc抑制IL-4和Th2基因表达的能力一致在活的有机体内,这与这些药物治疗哮喘的有效疗效相一致54。因此,吸入GCs导致哮喘Th2偏倚的结论是不成熟的,并且反映了一种人为因素在体外使用培养系统。⇓
IL-12的产生或活性降低是过敏性疾病患者Th2致敏的危险因素,这一观点得到了一定的支持99,尽管这方面还需要进一步的研究。然而,应该注意的是,在缺乏IL-12的情况下,t细胞不会自动默认使用Th2通路(如。il -12缺陷小鼠)。因此,尽管DC IL-12的产生减少可以被认为是Th2致敏的风险因素,但激活的DC启动Th2分化还有其他机制。这些可能包括分泌因子和接触依赖信号。IL-12的区别罗和未成熟的dc并不总是清晰的,dc也可以通过诱导调节性t细胞的分化来下调免疫(如。分泌IL-10、转化生长因子β或其他细胞因子的细胞108,109)。调节性t细胞在哮喘和特异反应中的潜在作用已开始得到重视,但超出了本综述的范围。
Th2招募和生存
Th2细胞在哮喘中的扩张是由于循环t细胞不断招募到肺中,还是由于驻留气道效应细胞的扩张和持续存在,仍有待确定。最近对小鼠的研究表明,在解释效应细胞在肺和其他非淋巴器官中的积聚时,持续的t细胞募集比驻留淋巴细胞的扩张重要得多110,111。例如,在过敏致敏和挑战模型中,哈里斯et al。110显示被激活的t细胞从局部淋巴结转移到肺和气道,在那里它们分泌IL-4,但不能进一步分裂。分泌IL-4的Th2细胞的特异性招募可能是由Th2吸引趋化因子介导的,如胸腺和活化调节趋化因子(TARC或CCL17)和巨噬细胞来源的趋化因子(MDC或CCL22)。112,113。TARC和MDC的配体是C-C趋化因子受体4 (CCR4),它优先表达在一些Th2细胞上。最近发现PGD增强了MDC的表达2在哮喘小鼠模型中114。有趣的是,PGD2也可以通过结合受体CRTh2直接招募Th2细胞,CRTh2受体选择性地表达在Th2淋巴细胞上。综上所述,这些研究表明拮抗Th2募集为哮喘提供了一个新的治疗靶点。
尽管与naïve t细胞分化的调控相比研究较少,但记忆t细胞在外周的持久性也在特异反应中的Th2偏倚中发挥作用。控制记忆细胞长期存活的因素包括稳态细胞因子,如IL-7和IL-15115。Th2细胞似乎比Th1细胞存活时间更长,这可能容易发生凋亡。例如,在Wu最近的一项研究中et al。116,在积极分泌IFN-γ的细胞中,记忆CD4+ Th1细胞的生成受到抑制,这与IFN-γ促进t细胞凋亡的已知能力一致117。这些研究表明,不管初始分化信号如何,Th2细胞可能在特异反应中优先扩增。尽管有一些实验证据支持这一观点118,119因此,需要进一步研究Th2细胞在特异反应中持续存在的因素。这些因素的遗传多态性也可能导致人类过敏性疾病中的Th2偏倚(见表3)。⇑。
Th2转录调控
无论是由于增强募集还是持续性,在过敏气道中积累的t细胞倾向于表达Th2细胞因子。在过去的十年中,细胞因子基因Th2亚群选择性表达的分子基础受到了严格的审查,并且已经确定了参与该过程的几个关键信号通路和转录因子(图4)⇓)。本综述的其余部分将涵盖参与Th2基因调控的转录因子及其在过敏性疾病患者基因表达失调中的潜在作用。
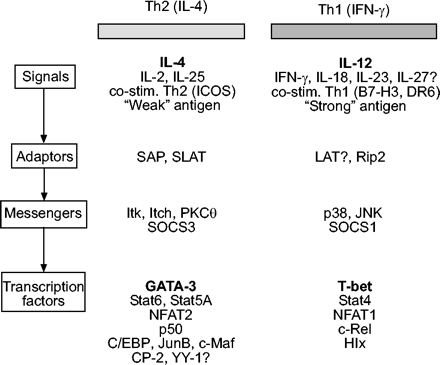
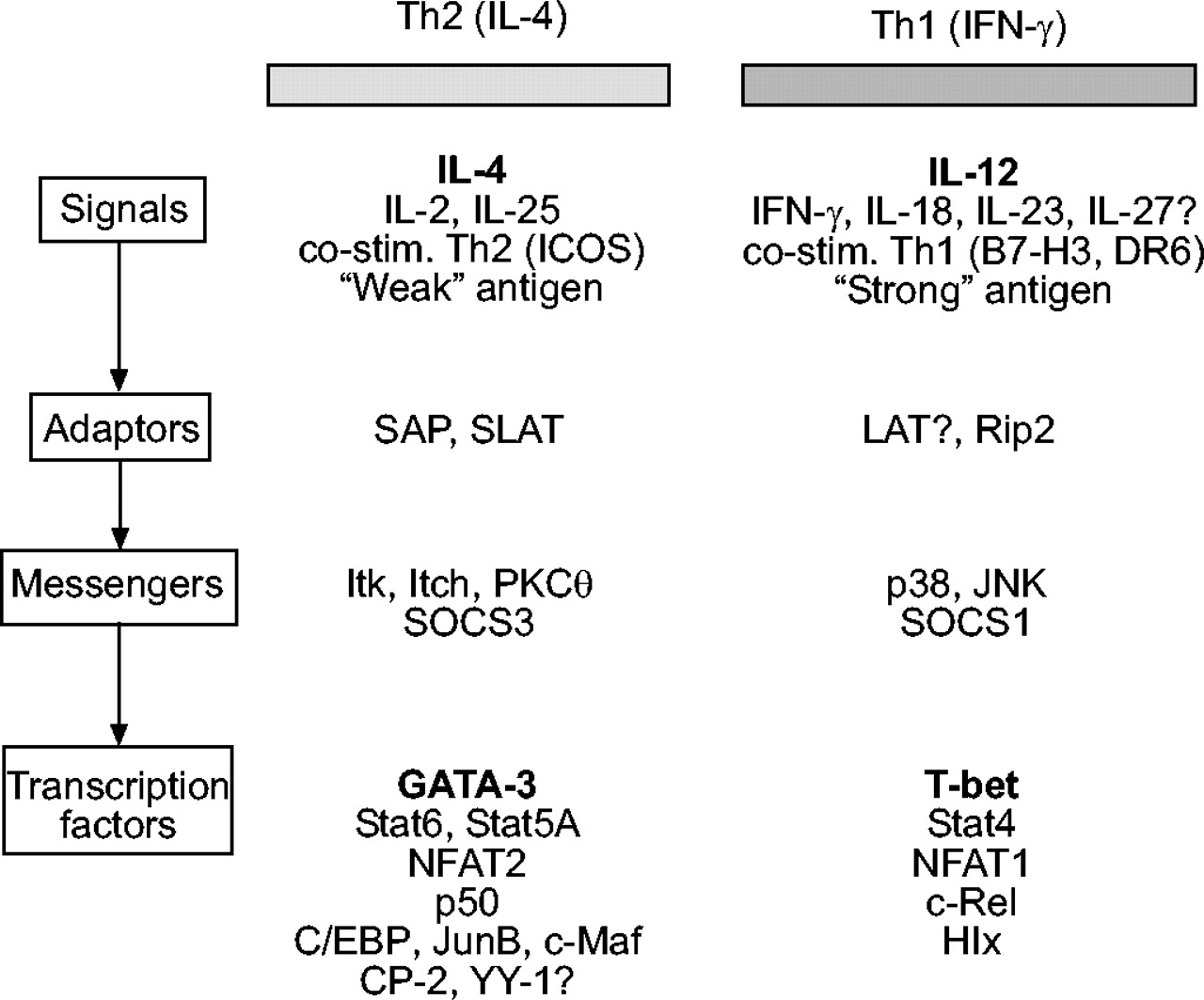
参与t辅助细胞(Th) 1型的分子与Th2两极分化。这一数字是使用基因靶向小鼠和转基因小鼠的研究汇编,并不意味着是全面的。这种格式是由Ho和格里姆彻121,里面包含了一些主要的参考资料。其他参考文献包括:白细胞介素(IL)-23122, IL-25123, IL-27124, B7家族125,抗原强度(126,127和其中的参考文献),信号淋巴细胞活化分子相关蛋白(SAP)128, swat -70样T接合分子(SLAT)129t细胞激活连接剂(LAT)130, Rip2131, Itk132,痒133,蛋白激酶C (PKC)-θ134,135,丝裂原活化蛋白激酶(MAPK)综述136和细胞因子信号抑制因子(SOCS)137。文中未讨论的其他转录因子参考文献包括:核因子-κB p50138,139,:140,和Hlx141。对于某些分子,目前尚不清楚它们是作为一种途径的主要激活剂还是另一种途径的抑制剂(如。IL-27142和纬度130)。IFN-γ:干扰素γ;ICOS:诱导共刺激器;Stat:信号转换器和转录激活器;NFAT:活化t细胞的核因子;T-bet: t细胞表达的T-box;c/EBP: CCAAT增强子结合蛋白。
Th2基因位于染色体5q上的一个簇中(图2⇑)。IL-4和IL-13具有相似的内含子/外显子结构和序列同源性,被认为是由原始基因复制事件引起的。尽管IL-4、IL-5和IL-13基因的5 '侧区都含有TATA序列(包含RNA聚合酶ii的基础起始复合物的组装所必需),但整体启动子序列的同源性是有限的。这表明,每个启动子将允许独特的转录因子复合物的组装,并且给定的Th2细胞因子的表达可以在启动子驱动的转录起始水平上进行微调。为了支持这一观点,个体Th2细胞因子及其等位基因可以选择性地表达,当以单个细胞为基础检查细胞群时71,120。
由于最初描述了IL-4启动子中活化t细胞的转录因子核因子(NFAT)的结合位点143在其中,许多其他因素的离散结合位点已被鉴定出来(最近的综述见144)。基于物种间的序列守恒,IL-4启动子的5 '边界通常被认为在转录起始位点上游包含数百个碱基对,但这不可避免地是一种武断的区分。在使用报告结构的瞬时转染分析中,IL-4启动子可诱导响应细胞刺激,这意味着一些负责增强IL-4基因表达的因素在启动子水平上起作用138。使用组合在体外dna结合测定(如。在使用野生型和突变启动子报告结构的功能研究中,一些因素已被证明可以结合并增强IL-4启动子活性,包括NFAT、激活蛋白1 (AP-1)和C/增强子结合蛋白(EBP)家族的成员145–147。与IL-4启动子结合的其他核因子包括信号传感器和转录激活因子(Stat6)、CCAAT-box结合蛋白2 (CP-2)和y -1148–150目前该领域的一个挑战是确定不同的因素如何以组合的方式相互作用,从而导致特定的基因表达模式。值得注意的是,这个列表中没有Th2细胞的标志性转录因子GATA3151。目前几乎没有证据表明GATA3直接与IL-4启动子中的高亲和力位点结合,过表达的GATA3不会转激活IL-4启动子152)。GATA3在Th2细胞中增强IL-4基因表达的确切机制是复杂的,将在下面进一步考虑。
活化t细胞的核因子
NFAT细胞由一个钙敏感转录因子家族组成,该家族对许多t细胞因子的表达至关重要,包括IL-2和IL-4,以及诱导耐受153,154。NFAT蛋白构成细胞质,但在钙活化磷酸酶钙调神经磷酸酶去磷酸化后转移到细胞核(图5)⇓)。IL-4启动子包含至少6个富含嘌呤的NFAT位点(称为P元件P0-P5),目前认为dna结合的NFAT蛋白与其他转录因子相互作用,以提高活化t细胞中IL-4的转录率145,155。在某些P元素上,NFAT与AP-1家族成员相互作用形成异三聚体转录因子复合物。AP-1被一个独特的信号通路激活(涉及蛋白激酶C (PKC)/ras激活),这些所谓的复合NFAT位点代表了细胞核中的信号整合点156。NFAT AP-1相互作用的结构基础已经被解决,涉及到NFAT和AP-1之间相互作用的扩展表面,也覆盖了DNA模板的~ 15个碱基对157。NFAT和AP-1之间的这种密切相互作用有助于解释AP-1一致位点在支持组合相互作用的IL-4启动子P元件上不易检测到这一事实155。没有AP-1的NFAT激活(如。仅用钙离子载体刺激细胞后)诱导t细胞无反应性状态,称为无能(或耐受)。158。这是由于不同分子的诱导,抑制近端tcr依赖的信号事件,如酪氨酸磷酸酶和泛素连接酶159。有趣的是,不同Th2细胞因子对NFAT:AP-1复合物形成的要求不同。例如,依赖于NFAT的IL-4启动子的转激活需要完整的AP-1相互作用域,而不能结合AP-1的NFAT突变体仍然可以转激活IL-13启动子160。这与t细胞无能有关的生理后果目前尚不清楚,但其中一个暗示是IL-4和IL-13基因表达的信号需求将有所不同。
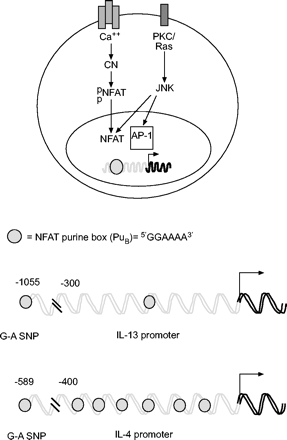
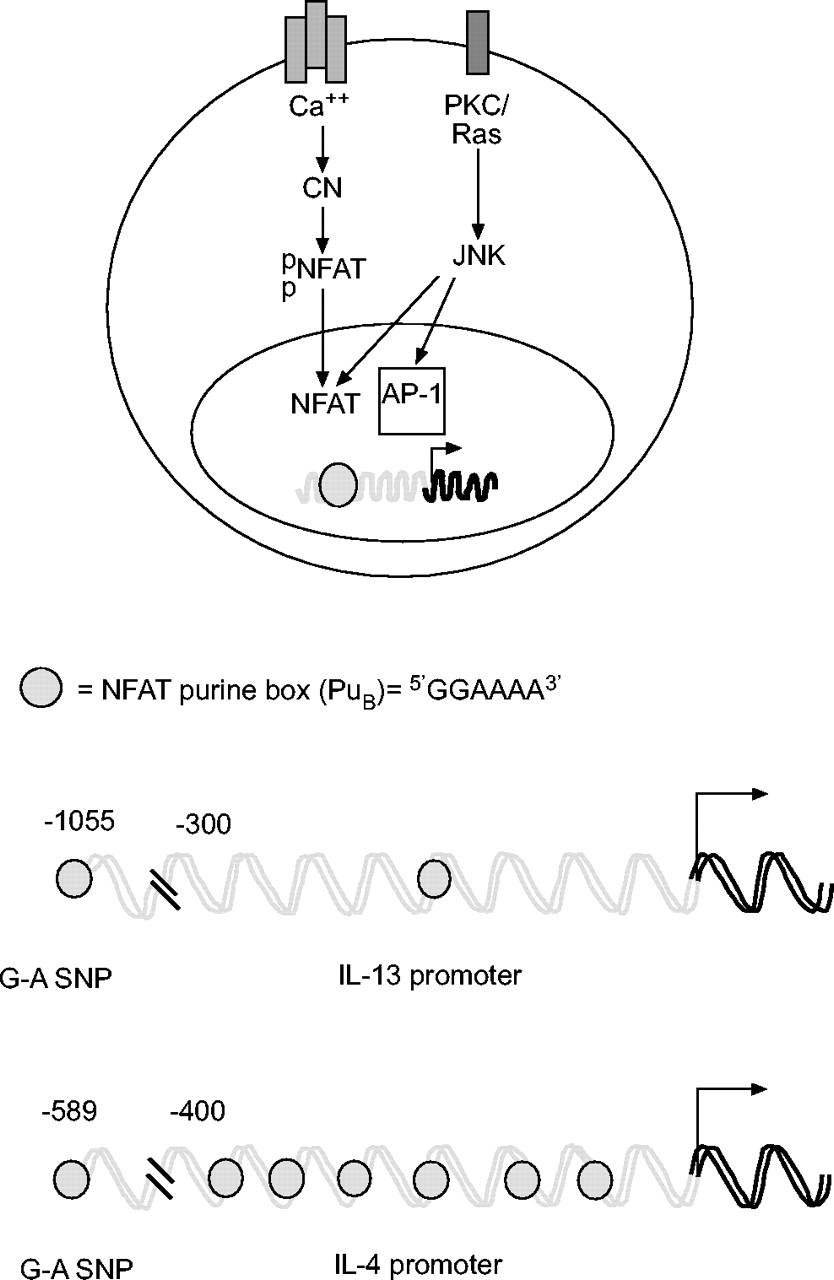
活化t细胞的核因子(NFAT)包括一个钙活化转录因子家族,与白细胞介素(IL)-4和IL-13启动子结合。细胞质NFAT被磷酸酶钙调神经磷酸酶(CN)去磷酸化,促进其核进入。核NFAT可以与激活蛋白1 (AP-1)家族的成员相互作用,形成转录活性复合物。AP-1蛋白被c-Jun n端激酶(JNK)磷酸化激活,它也可以再磷酸化NFAT并促进其核输出(未显示)。单核苷酸多态性(SNPs)位于IL-13和IL-4启动子区域的5′,影响NFAT的结合,并与不同种类的哮喘和/或慢性阻塞性肺疾病表型相关。PKC:蛋白激酶。
IL-4和IL-13启动子区域都含有与不同人群哮喘或哮喘严重程度相关的snp(表1)⇑)。有趣的是,这两种多态性都影响NFAT的结合。IL-4启动子多态性位于相对于转录起始位点的-589位,在非裔美国人和白种人中等位基因频率分别为~ 0.5和0.27。C-T SNP与哮喘严重程度相关,其重新创造了更高的亲和NFAT位点(即。与野生型受试者相比,一秒钟用力呼气量减少4.5%),尤其是白人受试者7。IL-13启动子SNP(位于-1055位)也能重建一个更高亲和力的NFAT位点,并且被认为通过降低尚不清楚的CD2和其他信号对IL-13基因表达的抑制作用以一种复杂的方式发挥作用9。因此,NFAT可能导致哮喘Th2偏倚的一种方式是由于Th2细胞因子启动子中的多态性结合位点。
NFAT与位于IL-5和IL-13启动子内的其他嘌呤盒以及远端增强子元件结合161,162。使用DNase超敏分析发现了IL-4基因3 ' -的一个有趣的NFAT位点163。这个地点,称为V一个通过染色质免疫沉淀(ChIP)试验确定,该蛋白在报告结构中具有增强子活性,并可以支持NFAT和GATA3以th2依赖的方式结合(图6)⇓)164。因此,即使NFAT蛋白在Th1中没有差异表达与Th2细胞(见下文),V一个调控元件仅限于Th2谱系。在后续的一系列实验中,Solymaret al。164使用基因靶向删除V一个并观察到IL-4在骨髓来源的肥大细胞(bmcs)和小鼠中的表达显著降低在体外分化Th2细胞。有趣的是,V细胞中IL-13的表达也部分降低一个-null小鼠,特别是在BMMCs。这些结果证实了V一个是IL-4和Th2细胞因子基因表达的关键增强子,并强调了许多转录因子结合位点位于编码区3′的概念165。Avniet al。166表明NFAT与GATA3在V处有功能合作一个在瞬时转染实验中增强IL-4启动子活性。再加上最近观察到NFAT还与T-bet合作调节IFN-γ基因表达167这些数据表明NFAT可与谱系限制因子相互作用,诱导Th1和Th2细胞中细胞因子基因的表达。
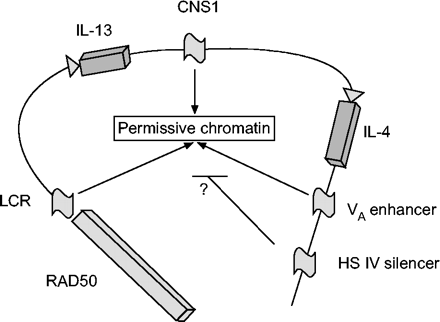
使用DNase超敏分析和/或跨物种序列比对确定了t辅助细胞(Th) 2型基因簇中的保守调控元件。RAD50基因3 ' -区域的基因座控制区(LCR)最近通过转基因小鼠被发现。保守的核苷酸序列-1 (CNS-1)位于白介素(IL)-4和IL-13基因之间,与Th2谱系承诺有关。IL-4基因的两个邻近元件具有相反的功能。V一个增强子是最大化IL-4基因表达和Th2分化所必需的,而超敏感位点IV沉默子抑制naïve、Th1和Th2细胞中的IL-4。中枢神经系统:保守的核苷酸序列;海关:高度敏感。
目前已知的NFAT家族成员有5个,它们的组织分布和调节不同。NFAT1 (NFATp或NFATc2), NFAT2 (NFATc或NFATc2)和NFAT4 (NFTAx或NFATc3)在免疫细胞中占主导地位153。尽管名字叫NFAT,但它的表达并不局限于t细胞,也可以在b细胞、肌肉细胞、肥大细胞、嗜碱性粒细胞和嗜酸性粒细胞中检测到78,168。迄今为止的证据表明NFAT蛋白不应该在Th分化中起主导作用。这一结论是基于以下三点观察得出的。首先,不同NFAT蛋白的共识结合位点几乎相同(反映了保守的dna结合域),这意味着不同NFAT蛋白的靶基因应该非常相似。其次,在比较高度极化的Th1和Th2细胞时,NFAT的表达没有显著差异。第三,NFAT与谱系限制因素相互作用(如。GATA-3和T-bet)激活IL-4和IFN-γ基因表达。然而,使用基因靶向和嵌合小鼠的实验发现,NFAT2在Th2极化中发挥了令人惊讶的重要作用,而不是NFAT1或NFAT4169,170。事实上,NFAT1的缺失(特别是与NFAT4结合)导致了过度活跃免疫的表型,其特征是IL-4基因表达升高和嗜酸性粒细胞招募到肺169,171,172。由于NFAT1和NFAT2在瞬时转染实验中都能激活IL-4或IL-13启动子173,在NFAT1缺失小鼠中IL-4基因表达的增强可能并不反映NFAT1对Th2基因表达的直接抑制作用。相反,这些小鼠的Th2偏倚可能是免疫过度增殖的结果,这是由于抑制了nfat1依赖基因的调节,这些基因抑制了Th2淋巴细胞增殖和/或细胞周期进程(如。CDK4和ROG174,175)。因此,NFAT蛋白可以直接或间接地影响Th细胞因子基因表达模式,特定NFAT家族成员的作用取决于激活强度和持续时间的差异,并可能与独特的辅因子相互作用。
目前的作者在t细胞系中证明,糖皮质激素干扰nfat2驱动的IL-4启动子活性通过糖皮质激素受体和NFAT2之间的一种新的蛋白质-蛋白质相互作用176这可能有助于解释GC对IL-4基因表达的一些抑制作用。新的钙调神经磷酸酶拮抗剂正在开发中,可能也被证明是过敏性哮喘的有用药物,特别是如果与糖皮质激素联合使用。有趣的是,有一些证据表明NFAT蛋白的激活与特异反应和哮喘的易感性相关。比如,陈et al。177研究表明,使用非特异性药物激动剂,特应性皮炎患者外周血单个核细胞中NFAT优先被激活。此外,Keenet al。178研究表明,在“哮喘易感”A/J小鼠(与耐药C3H/HeJ小鼠相比)中,NFAT2激活与IL-4基因优先表达相关。这些差异的生化基础目前尚不清楚,但值得进一步研究。
激活蛋白-1家族(包括junB)和c-Maf
Ap-1由二聚体转录因子c-Fos和c-Jun家族的成员组成通过它们的亮氨酸拉链和调节多种肺细胞类型中不同基因组的表达179。如上所述,AP-1在IL-4基因调控中与NFAT相互作用,IL-5启动子调控中也有类似的相互作用161。目前尚不清楚AP-1蛋白是否参与IL-13基因调控:初步证据表明NFAT:AP-1相互作用对于IL-13启动子活性是不可或缺的161。一些报道表明,不同的AP-1家族成员对Th2细胞中IL-4基因的表达很重要。例如,李et al。180结果表明,junB在Th2细胞中优先表达,瞬时转染实验激活IL-4启动子,增强转基因动物IL-4基因表达。这一结果令人惊讶,因为junB最初被发现是一个激酶不活跃的突变体,可以抑制c-Jun的交易功能181。在最近的一篇论文中,Hartensteinet al。186在泛素启动子控制下生成表达junB的转基因小鼠,以挽救junB-null小鼠的胚胎致死率。从这些嵌合动物中分离出的淋巴细胞很少或不表达junB, IL-4水平显著降低。此外,在junB-null嵌合小鼠中,卵白蛋白诱导的气道炎症也随之减少。目前缺乏将junB激活和/或表达与哮喘联系起来的临床研究。
在Th2细胞中,另一个含有IL-4转录激活的重要转录因子是c-Maf。自最初由何描述et al。183c-Maf在Th2克隆中优先表达,在b细胞系中过表达时可诱导IL-4基因表达,一些研究已经完善了该因子在Th2基因调控中的作用。目前的想法是c-Maf直接激活IL-4而不是IL-13基因表达,并且可以与其他因素协同作用,包括NIP-45、NFAT和junB180,184,185。此外,c-Maf可以直接抑制IFN-γ的产生,独立于IL-4186。因此,这些研究表明c-Maf在Th2极化中起着重要作用。在这方面,使用c-Maf基因靶向小鼠的研究应该是有趣的。有趣的是,Li-Weberet al。187发现Maf响应元件对于人IL-4启动子的完全激活是可有可无的,这可能反映了该位点下游的微妙序列差异。因此,c-Maf在人IL-4基因转录调控中的作用有待进一步研究。有趣的是,两项研究发现哮喘患者支气管活检或诱导痰白细胞中c-Maf的表达高于对照组31,188。c-Maf在这些细胞中的功能,以及这种表达的增加是否反映了哮喘患者Th2细胞的倾斜或其他因素,仍有待确定。
除了JunB和cMaf外,其他AP-1蛋白在凝胶转移实验中已被证明与IL-4启动子结合,但迄今为止,没有一种蛋白在Th亚群中有选择性表达。然而,调节c-Jun活性的c-Jun n端激酶JNK1和JNK2在Th2分化过程中发挥着复杂的作用。使用基因靶向小鼠或转基因小鼠的实验发现,JNK1令人惊讶地拮抗Th2分化,特别是在naïve t细胞激活的早期阶段。这归因于AP-1之外的其他靶标的磷酸化,如NFAT2。相反,JNK活性是IL-4在效应t细胞中完全表达所必需的。这些研究强调了naïve t细胞中的Th分化和再刺激效应t细胞中随后的细胞因子基因表达是具有不同中间体的独特生化事件。这一概念也适用于考虑其他转录因子在Th2基因调控中的作用,包括Stat6和GATA3(见下文)。
ccaat增强子结合蛋白
CCAAT-EBP家族的成员也是IL-4基因的有效transactivator。C/EBP-β尤其与IL-4启动子中的多个位点结合,其中一些位点以th2特异性的方式结合,突变分析表明这些位点对IL-4的转录调控至关重要147,187。C/EBP也激活IL-5启动子189。t细胞中激活C/EBP的信号级联是复杂的,似乎与激活NFAT的信号级联不同。例如,C/EBP在t细胞中以依赖pkc、不依赖钙调神经磷酸酶的方式被激活。一种可能是IL-6受体下游的信号(已知在其他细胞类型中激活C/EBP)参与了这一过程。然而,虽然IL-6可以增强IL-4基因表达和Th2分化190最近,这被归因于IL-6对NFAT2表达的一种新的增强作用191。IL-6和相关细胞因子对t细胞中C/EBP激活的影响的进一步研究应该有助于解决这个问题。在小鼠淋巴细胞中使用反转录病毒介导的C/EBP-β过表达的研究提供了强有力的证实,该因子增强了原代细胞中IL-4(并抑制IFN-γ)的表达192。在未来的研究中,C/EBP-β的水平可以在小鼠中被基因操纵,这将有助于解决其在过敏原驱动的Th2肺免疫反应中的作用。有趣的是,最近在哮喘受试者的支气管平滑肌细胞(而不是循环淋巴细胞)中发现了C/EBPα的缺陷表达193。C/EBPα的这种缺陷赋予了糖皮质激素对平滑肌细胞增殖抑制的抗性,这种抗性通过过表达C/EBPα而恢复。这项研究表明,C/EBP-β可以促进th2介导的炎症,C/EBP-α的表达缺陷也导致哮喘平滑肌功能障碍194。这对于理解如何将C/EBP定位为潜在的治疗靶点具有重要意义。理想的策略是拮抗淋巴细胞中的C/EBP-β,同时恢复支气管平滑肌细胞中的C/EBPα,这将是一项具有挑战性的任务。
信号换能器和转录激活子-6
IL-4和IL-13受体的连接导致潜在的细胞质Stat6的磷酸化和激活,目前的观点是Stat-6介导了IL-4/IL-13信号通路下游的大多数转录程序。上面讨论了IL-4在Th2极化中的重要作用,使用基因靶向小鼠的研究表明,这需要Stat6195。Stat6激活Th2细胞因子基因转录的分子机制是间接的。目前的作者和其他人在IL-4启动子中检测到Stat6结合位点,这表明IL-4诱导的Stat6仅仅增强了IL-4基因的转录激活的初始模型。然而,在瞬时转染实验中,Stat6通过取代NFAT来抑制IL-4启动子活性148。为了支持这一模型,多拉多最近的一项研究et al。196表明Stat6在Th2细胞激活的后期下调IL-4的表达。Fulkerson最近证实,Stat6可以下调过敏性肺中的基因表达(包括趋化因子)et al。197。Stat6是否直接调控其他Th2细胞因子的转录还有待进一步研究。Stat6可能间接促进Th2表达的一种方式是在染色质重塑水平上;最近的数据表明,Stat6结合到5号染色体Th2基因簇中开放染色质结构所需的基因座控制区域(见下文)。Stat6可能间接调节Th2反应的第三种方式是通过增强其他Th2转录因子的表达,如GATA3和c-Maf(图7)⇓)198。虽然已经确定了两个GATA3启动子区域199,目前缺乏Stat6激活GATA3启动子的直接证据。Stat6的另一个间接作用是促进Th2细胞的存活。在两项关键研究中,朱教授和同事200,201表明Stat6对Th2的扩张是必要和充分的,部分是通过诱导生长因子的表达(如。Gfi-1)促进Th2细胞增殖并防止其凋亡。最近的研究表明,在Th2分化过程中对Stat6的需求并不是绝对的。例如,在缺乏抑制分子CTLA-4的基因靶向小鼠中,Th2分化可以在缺乏Stat6的情况下进行202。这项研究表明,在某些情况下,无对抗的TCR信号可以替代Stat6(可能是由于最近描述的IL-2和Stat5驱动Th2分化的能力)。
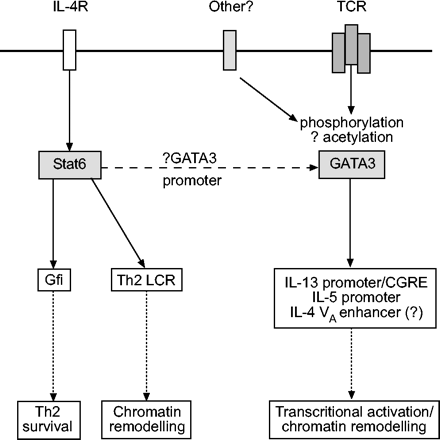
信号换能器和转录激活因子(Stat)6和GATA3对t辅助细胞2型(Th2)分化的调控白介素(IL)-4激活的Stat6可以通过诱导促进Th2存活的基因表达来增强Th2反应(如。Gfi),并与位于Th2基因簇的基因座控制区(LCR)结合。虚线表示Stat6增强GATA3表达的潜在能力。在再刺激的Th2细胞中激活GATA3的信号通路需要进一步研究。GATA3可与Th2基因簇中的多个位点结合,诱导naïve Th细胞染色质重构,并在重新刺激的Th2细胞中激活IL-13和IL-5的转录。TCR: t细胞受体;CGRE:保守GATA响应元素;Gfi:生长因子独立-1。
使用基因靶向小鼠的研究也证明了Stat6在过敏性气道炎症中的重要作用203。Stat6在肺中诱导的分子数量众多,包括th2诱导的趋化因子,如eotaxin (CCL11)和可能的粘液基因。Stat6在多种肺细胞类型中普遍表达,并调节IL-4和il -13诱导的基因表达。库珀曼et al。46在这些小鼠中,在Stat6缺失的背景下,气道上皮细胞中Stat6的表达得以恢复。粘液产生和AHR是哮喘的两个主要特征,在这些表达stat6的小鼠中被诱导,而不是对照组,以il -13依赖的方式46。这与一小部分基因的表达有关204。这些研究表明,IL-13/Stat6在哮喘中的关键靶点是一种结构性肺细胞。然而,使用野生型和Stat6缺陷t细胞的过继转移实验发现,淋巴细胞(而不是受体肺)中Stat6的表达对AHR至关重要205。在蠕虫诱导的肺嗜酸性粒细胞模型中,最近的研究表明,骨髓来源细胞中Stat6的表达对于嗜酸性粒细胞向肺募集至关重要75。在未来的研究中,Stat6的表达可以在离散的细胞谱系中被遗传操纵,这将有助于在哮喘模型中识别关键的Th2靶细胞。有趣的是,与非特应性哮喘患者或对照组相比,在特应性哮喘患者的支气管粘膜活检中发现Stat6过表达31。此外,过敏原刺激诱导Stat6在鼻黏膜的表达,可被局部类固醇阻断206。因为大多数研究使用纯化细胞体外表明Stat6在Th1和Th2细胞中表达相等,这些研究表明气道微环境促进了Stat6的表达:其生化基础仍有待确定。Stat6中的单核苷酸多态性与哮喘有关,但其功能意义目前尚不清楚97。
CP-2和阴阳一号
在详细的缺失分析中,作者发现了IL-4启动子中其他因子的结合位点,包括CP-2和阴阳-1 (y -1)。149,150。新出现的数据表明,这些因素在IL-4基因调控中起着以前被低估的作用。CP-2(也称为LBP1)最初被认为是α-珠蛋白表达所需的转录因子。目前已知CP-2在多种细胞类型中表达,并能调节许多基因的表达。通过瞬时转染检测,作者发现IL-4启动子CP-2位点的完整性对于构成性和诱导性启动子活性都是至关重要的。此外,显性阴性CP-2结构(LBP-1d)的表达抑制了D10 Th2系中IL-4基因的表达149。导致t细胞中CP-2激活的信号通路目前正在研究中,涉及MAPK家族的成员。有趣的是,在目前正在进行的研究中,作者发现CP-2的表达在分化人类Th2细胞中也上调。因此,CP-2是IL-4的一种新的转录调节因子。
使用类似的方法,目前的作者在近端IL-4启动子中发现了多效因子YY-1的四个结合位点。这些位点(称为Y元素,Y0-Y4)与上述富含嘌呤的NFAT位点相邻或重叠。作者使用一种谨慎的突变策略来保持邻近NFAT位点的完整性,发现Y0突变对瞬时转染检测中IL-4启动子活性有特别剧烈的影响150。令人惊讶的是,在共转染实验中,作者没有观察到y -1和NFAT蛋白在驱动IL-4启动子方面的任何正向相互作用。因此,尽管y -1和NFAT在dna模板上密切相关,但它们似乎通过不同的机制增强IL-4的转录活性。y -1可以与多种其他辅助因子相互作用,包括组蛋白修饰酶207一种可能是y -1将这些酶招募到IL-4位点。y -1已被证明在t细胞系中抑制IL-5启动子208,也能与IFN-γ启动子结合209。YY-1调控IFN-γ启动子活性的能力存在争议,在不同的实验中表现出增强和抑制作用209,210。y -1是否在t细胞极化中起作用目前尚不清楚。y -1最近被证明可以调节MCP-4的表达通过MCP-4启动子中的多态位点211。由于YY1缺失导致早期胚胎致死,因此有必要在淋巴细胞发育的不同阶段控制YY1的表达,以阐明其在t细胞分化和过敏性气道反应中的作用。虽然已知y -1是一种组成性核磷酸化蛋白,但新数据表明y -1的表达和/或功能可以通过翻译后修饰改变。比如Bovolentaet al。212显示y -1在il -2处理的外周血单个核细胞中被蛋白水解降解,而Yaoet al。213表明y -1的事务功能受赖氨酸乙酰化的调控。旨在解剖调节y -1在肺中的表达和功能的信号级联的研究正在进行中。
GATA3
自原来由张描述et al。214在Th2细胞中GATA3表达上调,而在Th1分化过程中GATA3表达下调,多个实验室的研究证明该因子在协调诱导Th2细胞因子基因表达和分化中起关键作用151。GATA3在Th2细胞中的优先表达涉及Stat6和核因子-κB提供的信号41,72,198,以及一个稳定GATA3表达的自动激活正反馈循环215。在中性或Th1条件下,t细胞激活后熄灭GATA3表达的信号不太清楚。GATA3的另一个重要特性是其抑制IFN-γ基因表达的能力,从而增强Th2表型216。有趣的是,与抑制IFN-γ相比,GATA3的不同结构域似乎是激活Th2细胞因子所必需的217。最近,Zhu提供了关于GATA3在Th2分化中的重要作用的确切证据et al。218。利用淋巴细胞中GATA3的条件缺失,Zhuet al。表明GATA3对于启动Th2反应和维持Th2存活都是必不可少的在活的有机体内。此外,在缺乏Th1诱导因子的情况下,GATA3的缺失导致Th1分化(如。IL-12),表明GATA3是Th1/Th2反应的主要开关218。GATA3在Th2位点的精确结合位点正在研究中。高亲和力GATA3结合位点位于IL-5和IL-13基因的上游,但有趣的是不在IL-4启动子中163,214,219。在朱的报道中et al。218在已经感染的Th2细胞中,有条件地删除GATA3抑制了IL-5和IL-13的表达,但仅轻微地阻止了IL-4的产生(细胞内染色和流式细胞术显示,IL-4阳性细胞的数量变化不大,但平均荧光强度降低了50%)。这与在Th2细胞中的一个模型是一致的,在该模型中,GATA3在转录水平上主要增强启动子驱动的IL-5和IL-13的表达,对IL-4的影响较小,尽管这是一个有争议的领域,需要更多的研究。GATA3启动Th2分化的能力可能独立于其促进转录激活的能力;本综述的结论部分讨论了GATA3在Th2基因簇染色质重塑中的潜在作用。
一些研究已经在小鼠模型和人体内直接将GATA3与过敏性气道炎症联系起来。例如,Finottoet al。220结果表明,吸入GATA3反义寡核苷酸可抑制小鼠Th2细胞因子表达和AHR。张et al。40在t细胞中诱导表达GATA3显性阴性的转基因小鼠,并为GATA3在过敏原驱动的Th2细胞因子产生和气道炎症中的作用提供了明确的证据。通过免疫组织化学分析GATA3表达的研究发现,与对照组相比,哮喘患者气道活检中表达GATA3的细胞更多,并且在过敏原刺激后,GATA3阳性细胞显著增加29,221。如果GATA3表达被用作Th2作用的替代标记,那么这些研究表明,在过敏原作用后,Th2细胞显著富集。最近的一项研究发现,GATA3基因中的三个单倍型与哮喘和特异反应相关的表型相关98。这项研究支持了过敏个体中Th2偏斜有遗传基础的观点。
基于Th2染色质的基因调控
除了上面所述的急性转录事件(发生在TCR和NFAT和其他因素的共刺激分子依赖性激活之后),现在有大量证据表明naïve Th细胞向Th2效应体的转变也在染色质重塑水平上受到调控。在这个模型中,静息状态naïve t细胞中Th2位点的染色质结构通常是抑制转录的,因为组蛋白与其他蛋白质和DNA模板之间的紧密相互作用。在th2偏置条件下(如。强IL-4受体),染色质被重塑成一种更允许的形式,允许更容易获得NFAT和其他转录因子所需的持续,高水平的IL-4基因表达。染色质的重塑状态在有丝分裂后传递给子细胞,并代表一种表观遗传特征222。Th2细胞染色质重塑的精确生化基础正在研究中,包括组蛋白的翻译后乙酰化和甲基化以及DNA的被动去甲基化166,223,224。在Th2细胞中同样重要的是IFN-γ基因的沉默,这也部分发生在表观遗传水平。
调控染色质重塑的DNA元件正在积极研究中。一个元素在染色质水平起作用的明确证据需要使用基因靶向小鼠或转基因小鼠进行实验,其中感兴趣的元素被用于驱动报告基因的表达。当连接报告基因时,赋予拷贝数依赖和整合位点独立的基因表达模式的元素在操作上被定义为基因座控制区域(LCRs)。225。LCRs在少数遗传位点上已被鉴定,并被认为在数千个DNA碱基对上散布开放的染色质结构域,从而促进基因簇的表达。在标准转染检测中,LCRs可以或不可以作为经典增强子。最近在RAD50基因的3’区域发现了一个候选LCR(图6)⇑)224。几个研究小组正在研究调节LCR的DNA结合因子,似乎包括Stat6,但有趣的是不包括GATA3226。除了LCR外,IL-4基因周围的几个元件在染色质水平上协同作用,限制IL-4基因在Th2细胞中的表达227。
LCR和其他调控元件是在DNase超敏分析的基础上发现的。另一种识别参与基因调控的重要非编码元件的方法是跨物种序列比较。如果一个内含子或非编码元素在多个物种中是保守的,这就认为它可能在基因调控中发挥功能作用。在一篇开创性的论文中,Lootset al。228他们在Th2基因座中发现了几个高度保守的区域,大部分位于非编码区域,长度为几百个碱基对。其中一个长度约为400个碱基对,位于IL-4和IL-13基因之间(保守的核苷酸序列-1 (CNS-1),图6⇑)228。小鼠CNS-1的缺失导致IL-4阳性细胞的频率降低,并且每个细胞IL-4的产生受到适度损害,反映了与染色质重塑影响一致的谱系承诺的减少229。cns -1缺失小鼠对吸入蠕虫刺激的反应减弱(如。降低AHR和免疫球蛋白E),表明尽管IL-4总体表达有微小变化,但减少的谱系承诺可对肺部Th2免疫反应产生实质性影响229。结合DNase超敏性分析和跨种序列比对来鉴定其他对IL-4基因表达重要的调控元件,V一个增强剂和超敏(HS) IV消声器。这两个位点都位于IL-4基因的3′位点,并且在具有有趣表型的小鼠种系中被删除(图6)⇑)。V一个增强子,结合NFAT和GATA3,是t细胞和肥大细胞中IL-4完全表达所必需的。最近报道了HS IV基因靶向小鼠的表型230。naïve HS IV-null t细胞中IL-4和IL-13表达增强,同时Th2偏斜。此外,IL-4在Th1细胞中异常表达,并损害Th1免疫应答利什曼虫主要。目前,V一个增强剂和HS IV消声器对染色质重塑的影响尚不清楚。发现与Th2基因簇中假定的LCR、CNS-1和其他非编码调控区域结合的交易因子的身份将是有趣的。研究这些因子在哮喘Th2免疫应答中的表达和调控是值得的。掠夺者et al。231最近开发了一种用于高通量发现保守DNA序列的计算工具,称为rVista。rVista结合了预测转录因子结合位点的聚类和种间序列守恒分析,以最大限度地识别功能元素。这个基于网络的工具应该有助于许多对基因调控感兴趣的研究者。
老鼠和人的区别?
细胞因子库的极化是CD4+和CD8+ t细胞的一个特征。虽然最初定义在小鼠t辅助克隆232在包括猫、狗、牛、马、猴子和人类在内的许多物种中都观察到Th2极化。由于它们易于生长和基因操纵,迄今为止绝大多数研究都是使用小鼠细胞进行的。人类的t细胞也有类似的行为,并且可以极化在体外通过刺激通过TCR、共刺激受体和外源性IL-4233,234。因此,Th2分化代表了一个协调的基因表达程序,在进化中是保守的。罗根et al。235最近使用一种新的转基因小鼠模型显示,人类Th2细胞因子基因在Th2条件下被协调诱导,与小鼠相似。因此,即使它们在基因上不相同,人类和小鼠的Th2基因簇都受到协调调节在活的有机体内。然而,人和小鼠在Th分化程序上存在一些明显的物种特异性差异。例如,Th2极化存在学科间的可变性,通常需要使用长时间的极化条件来使用人类t细胞(如。长达数周234)。这可能反映了近亲繁殖人群的遗传变异,以及获得真正naïve前体t细胞进行研究的困难。解决后一个问题的方法是使用脐带血来源的t细胞233。此外,极化转录因子在人类中的表达和作用也存在差异与小鼠Th2细胞开始出现。例如,尽管细胞因子库的极化,GATA3和T-bet分别在Th1和Th2条件下的消失,通常不像使用人类Th细胞那样戏剧性(De Fanis和Casolaro,约翰霍普金斯哮喘和过敏中心,巴尔的摩,美国;个人沟通)。最近还发现,T-bet不能像在小鼠Th2细胞中那样有效地消灭人类Th2细胞中的IL-2或IL-4236。此外,c-Maf在人类Th细胞中并不是th2限制因子234,而人IL-4启动子c-Maf响应元件对于完全启动子活性是可有可无的187。这些观察结果的生理意义目前尚不确定,但一种可能性是人类t细胞中Th2细胞因子基因表达存在替代途径(即。c-Maf-和/或gata3独立?);这应该是未来研究的一个有趣领域。
总结
对免疫反应的细胞和分子调节的理解已经以惊人的速度增加。目前,人们对参与Th2基因调控的分子因素和生化过程有了前所未有的了解。使用基因靶向或转基因小鼠品系已经获得了许多关键的见解,未来的挑战将是将这种理解转化为对哮喘病理生理学和治疗的临床有意义的见解。
- 收到了2005年1月14日
- 接受2005年3月21日。
- ©ERS期刊有限公司
参考文献
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}