





摘要
HIF1和HIF2可以起到互补,相对或在调节不同的细胞类型,以低氧气水平的响应无关的作用。这项研究表明HIF2在介导PH的发展在应对环境缺氧主导作用。http://bit.ly/2Cy46sR
肺动脉高压(PH)是血管疾病的多样集合原因重塑的肺小动脉,导致肺血管阻力和肺动脉压增加。世界卫生组织这些疾病划分为五组。组III包括与缺氧肺部疾病如慢性阻塞性肺病相关的肺动脉高压。虽然不是每个人都与COPD发展肺动脉高压,谁做更可能经历急性加重,住院治疗和预后较差。由于肺泡缺氧是推动这一反应的关键因素,研究人员已经研究如何缺氧慢性有助于PH的发展。
细胞缺氧通过表达的基因,防止氧气剥夺的后果响应,增强件/恢复组织氧气输送,和移代谢朝向保存细胞稳态的途径。驱动这些反应中最重要的转录因子是缺氧诱导因子1和2(HIF1和HIF2)1]。这些主要是通过调节它们各自的亚单位的丰度来控制的,根据细胞的氧水平。在常氧条件下,HIF1αHIF2α迅速退化,而在缺氧大量增加,允许他们heterodimerise HIF1β单元,把原子核,激活转录反应(2,3]。
研究HIF1和HIF2的pH值的作用,研究人员已采用遗传或药理学操作来抑制或激活他们的答复。不足为奇的是,HIF1α的生殖细胞缺失是胚胎致死,而HIF2α的缺失在一定的遗传背景和其他严重的表型产生的杀伤力。为了避免这个,Shimoda等。[4]研究了小鼠是杂合的HIF1α,发现到慢性缺氧肺响应显著衰减。其他人在成年小鼠中,使用平滑肌特异性HIF1α删除其衰减(但不废除)pH值和血管重塑应对环境缺氧[五]。总的来说,这些研究和其他人[6,7]认为HIF1激活有助于血管重塑响应和PH。然而,其他人已报道,HIF1活性降低通过降低肌球蛋白轻链磷酸化,其主张的HIF PH针对具有保护作用肺血管紧张[8]。
了解在PH HIF的作用,需要考虑的HIF在不同细胞类型不同的影响(图1)。例如,HIF在内皮细胞中可帮助驱动增殖和存活,并因素如内皮素-1促进细胞的增殖和收缩在肺动脉的中间层的表达。此外,全身低氧血症触发促红细胞生成素的表达,从而导致血细胞比容增加(其增强肺血管阻力)。缺氧在右心室还可能导致在重构,从那些引起的肺动脉压增加独立地发生HIF依赖性影响。我们还知道,HIF1和HIF2调节重叠但不同的基因,使得每一个激活(或使其失活)取决于细胞类型不同的基因组[9]。这些观察得出结论,理解和HIF1的HIF2的角色,以慢性缺氧肺血管反应需要了解的细胞环境中。
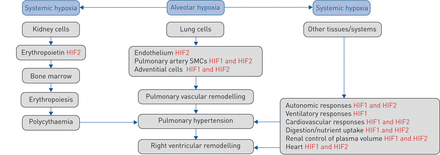
缺氧诱导因子1和2(HIF1和HIF2)在调控影响肺动脉高压的发展转录反应中发挥重要作用。肺泡缺氧直接作用于肺上皮细胞,内皮细胞,血管平滑肌和血管外膜细胞,其中编排的肺高血压发展细胞特异性HIF1和HIF2驱动响应。肺泡缺氧还导致全身性低氧血症,从而激活了影响肺动脉高压的程度HIF-依赖性反应。例如,肾缺氧驱动促红细胞生成素通过HIF2激活;血细胞比容的增加所得增强肺动脉压。在颈动脉体HIF依赖性转录影响缺氧的通气反应,从而影响肺泡和动脉血氧分压。全身低氧血症降低右心室动脉氧张力,潜在地激活影响心脏重塑HIF依赖性应答。重要的是,HIF1和HIF2控制所特有的不同的细胞类型不同的转录概况。校董会:平滑肌细胞。
认识到这一点,Hü等。[10]着手了解PH HIF1和HIF2的角色。据报道中的这个问题,欧洲呼吸杂志他们首先使用UBC-Cre的ERT2,这会导致细胞和组织中普遍缺失删除HIF1α全身在成年小鼠。出人意料的是,在与以前的工作中,他们发现了慢性低压缺氧后无右心室压力系统性的衰减(右室收缩压,肺动脉压力的替代)或右心室肥大。Using the same approach to delete HIF2α, they found that the mice all died after ∼3 weeks of hypoxia, possibly due to lack of a sufficient autonomic sympathetic response. Mice with systemic deletion of only one HIF2α allele survived in hypoxia and exhibited an attenuation of right ventricle remodelling and right ventricle weight, but no decrease in RVSP. Next, they turned to antisense oligonucleotides to suppress HIF2α, and found that survival was preserved while haematocrit, RVSP, Fulton's index and catecholamine responses during chronic hypoxia were attenuated. Antisense nucleotides against HIF1α had no effect on the development of PH, underscoring their Ubc-Cre-ERT2-HIF1α findings. Collectively, their studies identify an important role for HIF2, but not HIF1, in the pulmonary vascular response to chronic hypoxia. Finally, they utilised a HIF2-specific inhibitor, PT2567, in rats and found that mean pulmonary artery pressures, right ventricle remodelling and pulmonary artery wall remodelling responses were attenuated while the abundance of inflammatory cells, proliferating cells and expression of tenascin c (a glycoprotein lined to extracellular matrix remodelling and proliferation) were decreased. These findings were consistent with their murine studies and support the importance of HIF2 in diverse cell types in the pulmonary artery wall.
值得赞扬的是,Hü等。[10]加强了他们体内通过评估在常氧下培养血管细胞HIF依赖性基因表达模式的分析与低氧条件。这些结果,连同siRNA敲除研究和条件培养基实验,强调了内皮细胞中hif2依赖基因反应在控制缺氧反应中的重要性。但是哪些细胞负责驱动hif2依赖的PH表型呢?为了回答这个问题,他们使用HIF2α的内皮细胞基因敲除小鼠,并发现这实质上废除了肺血管慢性缺氧反应,随着RVSP反应和右心室重塑。这些研究证实了HIF2在内皮细胞中驱动肺动脉对慢性缺氧反应的重要性。
由于经常与有趣的研究的情况下,该报告由Hü等。[10[英语泛读材料回答一些问题的同时提出另一些问题。HIF2调节肾脏促红细胞生成素表达[11因此,系统反义抑制抑制了慢性缺氧时形成的多胱氨酸血症也就不足为奇了。这在多大程度上减轻了肺动脉压力的增加和右心室重构的减少?系统应用PT2567也应该可以消除慢性低氧诱导的多胱氨酸血症,尽管该研究中未报道红细胞压积水平。似乎hif2抑制小鼠血液粘度增加的失败可能会降低PH的发展,从而抑制右心室重构。此外,虽然他们表明与PH相关的许多基因的表达受到HIF2缺失的影响,但这些基因在介导PH对慢性缺氧反应中的作用还需要在未来的研究中确定。
有趣的是,HIF2的内皮细胞基因敲除几乎取消了PH和右心室反应,慢性缺氧,然而,这对血细胞比容的增加没有影响。以前,示出胚胎发育过程中血管内皮PHD2缺失引起含氧量正常成熟小鼠严重HIF2依赖PH响应[12]。然而,PHD2删除在成年小鼠不产生相同的表型。由H研究ü等。[10表明,在阻断对慢性缺氧的反应方面,胚胎内皮细胞HIF2敲除(与PT2567或对成年小鼠的反义寡核苷酸干预相比)具有更显著的效果。因此,HIF2可能在肺的发育中发挥重要作用,为以后的ph的发展奠定了基础。无论如何,Hü等。[10]通过识别在介导PH响应于肺泡缺氧内皮HIF2的极端重要性向前移动字段。
可共享PDF
脚注
利益冲突:G.B.Waypa有没有透露。
利益冲突:公厕Schumacker有没有透露。
支持声明:这项工作是由美国国立卫生研究院(HL109478,HL122062和HL35440)的支持。本文资金的信息已交存交叉引用出资者注册。
- 收到了2019年9月30日。
- 公认二○一九年十月三十零日。
- 版权©2019人队











{kind=link}
{kind=link}