





摘要
误吸与非结核分枝杆菌(NTM)肺病有关,气道生态失调与炎症增加有关。我们研究了NTM疾病是否与独特的气道微生物群和免疫特征相关。
从106名有呼吸道症状和与NTM相符的影像学异常的参与者中收集了297份口腔冲洗和诱导痰样本。在20名接受支气管镜检查的参与者中获得下气道样本。16S rRNA基因和嵌套分枝杆菌组测序方法表征了菌群组成。此外,还检查了下气道样本的炎症特征。
NTM的患病率+培养是58%。各组间口腔洗液和痰液样品的菌群特征或组成变化不大。在特种加工+样本中,27%的下气道样本富集分枝杆菌.一种分枝杆菌组方法分枝杆菌在更大比例的样本中,包括一些非致病性菌株。在特种加工+下气道样本,鉴定为口腔共生菌的分类与炎症生物标志物的增加有关。
16S rRNA基因测序方法在培养阳性的气道样本中识别NTM不敏感。然而,下气道炎症与微生物群特征之间的关联表明,这些微生物在NTM疾病的炎症过程中具有潜在作用。
摘要
16S rRNA基因测序检测不敏感分枝杆菌但可以识别与炎症相关的微生物群特征http://ow.ly/opXm30ldtQH
简介
Nontuberculosis分枝杆菌(NTM)病的发病率估计为每年每10万人1.0-7.2例,其发病率正因未知原因而不断上升[1]。尽管广泛接触这些生物体,但只有少数接触者会获得NTM,更小的亚群会发展为临床明显的疾病。重要的是,由于缺乏低毒和良好的抗菌药物,活动性NTM疾病的治疗效果有限在活的有机体内对抗有机体的活动。因此,NTM的治疗并不推荐给每个人,因为肺部疾病的临床病程是可变的[2]。
目前临床上可用的培养气道样本的方法不能代表NTM可能与存在于复杂微生物环境中的其他细菌有机体的相互作用。随着不依赖培养技术的改进,微生物-宿主免疫相互作用可以进一步详细检查[3.]。有几项研究检查了非囊性纤维化支气管扩张症患者的气道微生物群[4- - - - - -10]使用来自NTM不流行人群的痰。然而,下气道微生物组的描述一直具有挑战性,因为肺中的细菌负担大约比肠道中的细菌负担低100万倍,比上气道中的细菌负担低100倍[11,12]。声门上微生物的存在,如韦永氏球菌属或普氏菌常见于下气道[11- - - - - -18],它们与炎症增加有关[11,19],支持了微生物群变化与气道黏膜中宿主免疫表型相关的观点。因此,我们试图识别与培养阳性NTM相关的可能影响宿主免疫表型的微生物特征。在这里,我们采用16S rRNA基因高通量测序方法,同时采用改良的“分枝杆菌组”测序方法,对一组非囊性纤维化支气管扩张和高患病率NTM的大队列受试者进行测序。
材料与方法
研究对象
这是一项前瞻性观察性研究,纳入纽约大学(NYU)的非hiv感染患者(n=106),诊断为非囊性纤维化支气管扩张,纳入美国多中心支气管扩张队列(支气管扩张研究登记),为期2年。所有参与者都签署了知情同意书,该方案得到了纽约大学机构审查委员会的批准(S14-01400)。看到在线补充方法有关病人选择的详情。
程序
在入组时收集每位患者的口腔清洗和诱导痰样本,在2年的研究期间,当痰有临床指征时再次收集(在线补充表S1).此外,对于每一次诱导痰,我们都在诱导痰前收集口腔清洗样本。部分诱导痰样本送往临床实验室进行培养,根据上皮细胞计数,90.5%的诱导痰样本符合优质标准[20.]。将等份的口腔洗液和诱导痰冷冻于−80°C进行细菌DNA测序。为了研究诱导痰评估下气道微生物群和下气道粘膜炎症状态的可靠性,一组患者(n=20)进行了支气管镜检查(14例临床指征,其余6例仅用于研究目的)。采样包括设备背景对照(无菌生理盐水、Yankauer和支气管镜)、声门上(使用Yankauer采样)和两个支气管肺泡灌洗(BAL)样本:一个来自受累肺段(基于计算机断层扫描(CT)预定义),另一个来自未受累肺段。未从急性发作期间或最近使用抗生素(<1个月)的参与者中获得样本。全部BAL液在−80°C冷冻用于细菌16S rRNA基因测序和16S定量(q)PCR。
DNA测序的细节描述在在线补充方法.除了使用Illumina MiSeq (San Diego, CA, USA)进行16S rRNA基因测序外,我们还使用了一种嵌套PCR方法并行富集分枝杆菌在文库准备测序之前对16S rRNA基因进行DNA编码,以描述分枝杆菌组,如先前发表的[21]。获得的16S rRNA基因序列使用Quantitative Insights into Microbial Ecology (QIIME 1.9)软件包进行分析[22]。
对20例支气管镜亚组患者的所有BAL样本进行免疫谱分析。在活的有机体内如前所述,使用Luminex 200 (Austin, TX, USA)从脱细胞支气管镜样本中通过细胞计数差异和细胞因子评估炎症[23,24]。体外在toll样受体(TLR)4刺激过程中评估BAL细胞细胞因子的产生(在线补充方法).
统计分析
对于与离散因素的相关性,我们采用非参数检验(Mann-Whitney或Kruskal-Wallis方差分析)。我们使用ade4基于加权UniFrac距离构建主坐标分析(PCoA) [25,26]。为了将微生物群落聚类为排他的“元群落”,我们使用了带有R包的Dirichlet多项混合模型DirichletMultinomial[27,28]。为了评估16S数据组或推断宏基因组组之间的差异,我们使用线性判别分析(LDA)效应量(LEfSe) [29]。对于与连续变量的关联检验,我们使用了非参数斯皮尔曼相关检验。使用SparCC评估最丰富的细菌属(至少一个样品中的>1%相对丰度)之间的共存[30.]进行了20次迭代和500次自举重复,以消除由异常值驱动的显著性相关性,并使用Cytoscape v3.0.2进行可视化[31]。该分析仅使用了通过错误发现率校正的生物标记物[32]。所有数据均可在序列读取存档(https://www.ncbi.nlm.nih.gov/sra),注册编号为PRJNA418131。本手稿中所包含的分析所用的所有代码均可在https://github.com/segalmicrobiomelab/ntm_bronchiectasis_microbiome.
结果
参与者
表1显示106例患者的人口统计学和临床特征。所有参与者都有影像学异常。培养数据显示,106名参与者中有61人(58%)在基线时NTM痰培养呈阳性。NTM组体重指数较低+参与者(p < 0.01)。重要的是,患者的症状和影像学表现存在差异。
痰液和口腔洗涤液微生物组的比较
为了评估气道微生物群,我们使用了所有获得的口腔洗液和痰样本(n=297)。口腔洗液的α-多样性高于痰液(Shannon指数;在线补充图S1a).此外,β多样性分析显示两种样本类型之间存在显著差异(PERMANOVA p<0.001;在线补充图S1b),尽管同一受试者的样本之间的相似程度高于受试者之间的相似程度(在线补充图S1c).LEfSe分析显示痰标本中富集有普氏菌,韦永氏球菌属而且棒状杆菌属,而口腔清洗样品中富含链球菌,罗思氏菌属而且放线菌(在线补充图S1d).
NTM患者气道微生物区系的比较+与特种加工-使用痰液和口腔洗液样本
接下来,我们比较了基于NTM状态的每种样本类型的微生物群的差异。NTM培养状态以测序标本的培养结果为依据。图1而且在线补充图S2根据样品采集时NTM培养状态评估差异。在口腔洗液样品中,α-多样性差异不显著,但β-多样性差异显著−和特种加工+样本(PERMANOVA p=0.043)。在痰标本中,NTM与NTM之间的α-多样性(p=0.05)和β-多样性(p=0.08)差异均无统计学意义−和特种加工+样本。根据美国胸科学会/美国传染病学会的诊断标准比较NTM状态时,也发现了类似的阴性结果(至少两个痰样本或一个BAL样本中NTM培养阳性)[33]或仅考虑基线样本(数据未显示)。
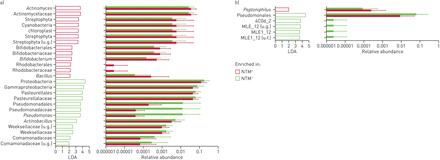
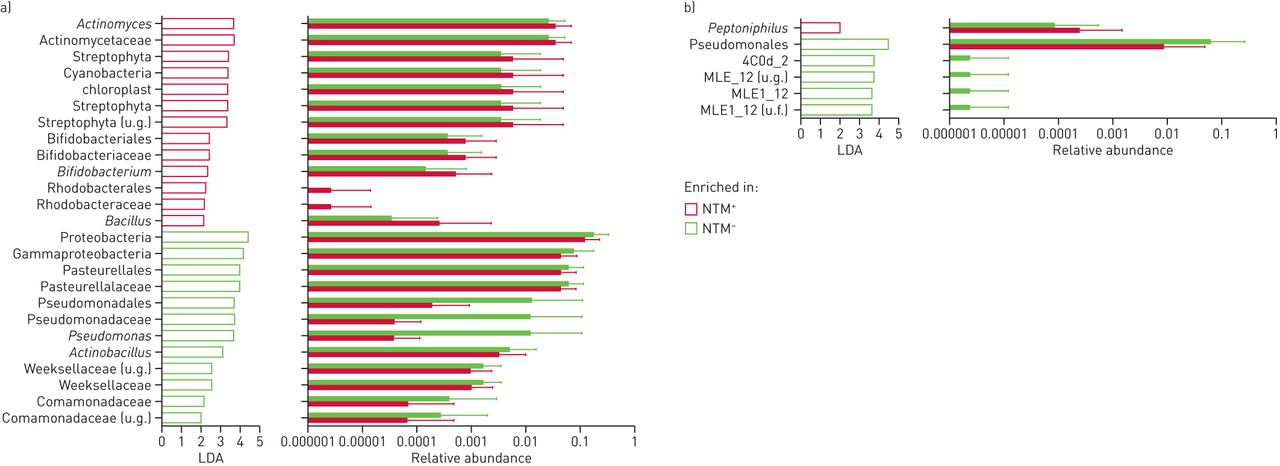
非结核分枝杆菌(NTM)口腔洗液和痰液标本的分类差异+和特种加工−组。a)对于口腔洗漱样品,线性判别分析(LDA)效应量(LEfSe)发现基于NTM状态的微生物组富集存在显著的分类差异,而与NTM状态的微生物组富集无显著差异分枝杆菌在特种加工+口腔清洗样品;b)对于痰液,LEfSe检测到的分类差异不大,与无富集分枝杆菌在特种加工+痰液样本。u.g.:未确定属;U.f:不确定的家庭。
有趣的是,分枝杆菌在NTM中未发现富集+样本。事实上,这种属只在非常小的百分比的口腔和痰样本中被发现。因此,我们研究了是否可以通过采样下气道来识别更多的微生物群差异。
使用支气管镜样本进行气道微生物区系比较
该队列中的20名参与者(40%为培养阳性NTM)接受了支气管镜检查(联机补充表S2而且图S3).首先,我们比较了痰中下气道微生物群的代表性(基于CT对受累肺段和未受累肺段进行采样)。用qPCR对16S rRNA拷贝进行定量分析,结果显示痰液中约有对数2与BAL样品相比,细菌载量较高(在线补充图S4).痰液中的高细菌负荷与口腔洗液和声门上样品中的细菌负荷相当。图2显示所有支气管镜相关样本的α-和β-多样性。β-多样性差异有统计学意义(p<0.01)。以加权UniFrac距离计算上呼吸道、痰液和BAL样品的相似度。有趣的是,与BAL相比,痰液更类似于口腔冲洗或声门上样本(受累或未受累肺段均如此;图2 c).这些数据表明,在该队列中,痰不能作为下气道的替代品来研究气道微生物群。
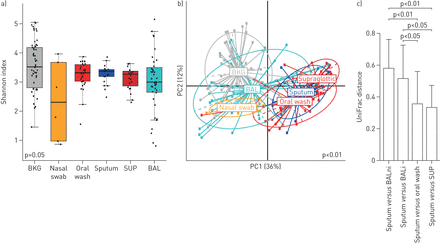
使用支气管镜样本评估下气道微生物群。支气管镜检查样本包括背景(BKG)、鼻拭子、口腔冲洗、痰液、声门上(SUP)和支气管肺泡灌洗(BAL)。a)各样品间α-多样性(Shannon多样性指数)差异显著;b) β-多样性(基于加权UniFrac)表现出基于样本类型的差异聚类(PERMANOVA p<0.01);c)基于UniFrac距离比较上下气道样本的相似度。结果显示,痰液中的微生物群与声门上和口腔清洗样品中的微生物群更相似,而与BAL样品中的微生物群(包括受累的(BALi)和未受累的(BALni))更相似。
NTM患者气道微生物区系的比较+与特种加工−使用支气管镜样本
在接受支气管镜检查的患者中,20例中有12例(60%)为NTM−8例(40%)为NTM+.NTM菌量差异无统计学意义+与特种加工−下气道样本(p=无统计学意义)。在线补充图S5根据NTM培养状态分类,BAL样品α-或β-多样性差异无统计学意义。同样,与研究支气管镜相比,作为临床指征支气管镜的一部分获得的BAL样本之间没有发现差异(数据未显示)。尽管分枝杆菌富集NTM+BAL样品(图3),该类群仅在27%的培养阳性样本中存在(中位相对丰度为0(0 - 0.014))。这些数据表明,这种测序方法无法检测到被确定为导致疾病过程的“病原体”,并且与先前文献显示的16S rRNA基因测序方法检测准确性较差的结果一致分枝杆菌[21]。
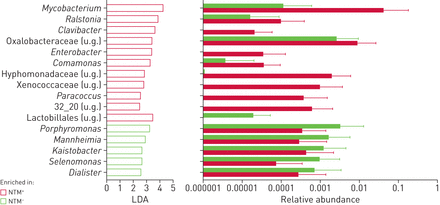
非结核分枝杆菌(NTM)分类差异+和特种加工−下气道微生物区系。线性判别分析(LDA)效应量分析显示,不同NTM状态下气道微生物群组成富集差异显著。与痰液样本不同,分枝杆菌富集NTM+支气管肺泡灌洗样本。
分枝杆菌组的评价
因此,我们使用了一种优化的协议来充实分枝杆菌利用最近发表的DNA分离方法编码16S rRNA基因[34]和嵌套PCR方法[35) (在线补充资料).首先,我们使用了一个模拟社区Mycobacteriun fortuitum而且链球菌为了建立检测的极限分枝杆菌采用这种方法(在线补充结果而且在线补充图S6有关详情)。然后,我们在所有接受支气管镜检查的参与者样本中使用这种嵌套的分枝杆菌组方法,并创建PCoA图,比较标准16S rRNA基因测序和BAL、痰液和声门上样本的嵌套分枝杆菌组方法。图4显示了大量样本的显著重叠,但确定了其他样本的成分差异。此外,直方图在图4显示不同的相对丰度分枝杆菌操作分类单位(OTUs)通过嵌套分枝杆菌组和16S rRNA基因测序方法获得。在BAL样本中,分枝杆菌在15个NTM+使用标准的16S rRNA基因测序方法,但使用嵌套的分枝杆菌组方法分枝杆菌在所有四个样本和另外三个样本中检测到NTM+BAL样本(47%)。爆炸分析表明这些序列是匹配的鸟型分支杆菌.此外,这种嵌套的方法在21个NTM中鉴定出1个(5%)−和样品分枝杆菌.对该OTU的爆破分析进行了注解分枝杆菌houstonense(一个环境分枝杆菌不知道是致病的)。使用这种嵌套的方法对背景对照样品检测到大量的分枝杆菌仅在一个对照背景样本中读取(>5%相对丰度),在19个背景设备样本中只有两个读取较少。此外,还进行了爆破分析分枝杆菌在背景设备样本中发现的读数被注释为非致病性菌株(在线补充图S7).当这种方法用于未接受支气管镜检查的受试者的口腔和痰样本时,也发现了类似的结果(在线补充资料).
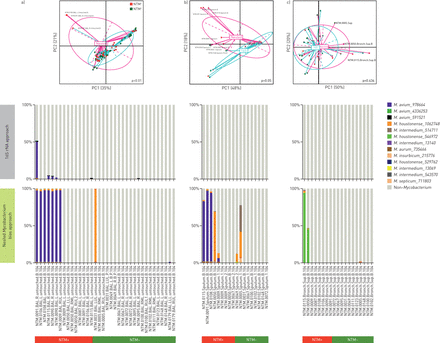
比较使用无偏16S rRNA方法和有偏分枝杆菌组方法获得的序列数据。顶部面板显示无偏16S处理的样品之间的β-多样性差异(基于加权UniFrac距离)与偏倚分枝杆菌组法。底部面板中的柱状图显示了注释的操作分类单位(OTUs)的相对丰度分枝杆菌以及它们基于BLAST在两个数据集上的注释。a)在支气管肺泡灌洗(BAL)中,经无偏置16S处理的BAL样品β-多样性有显著差异与偏倚分枝杆菌组入路(PERMANOVA p<0.01)。使用偏倚分枝杆菌组方法,鸟型分支杆菌在7种非结核分枝杆菌(NTM)中发现+培养样品,一个NTM−样本有一个分枝杆菌注释到的OTUm . houstonense(一种未知致病菌株);b)无偏置16S处理的痰液样品β-多样性差异有统计学意义与偏倚分枝杆菌组入路(PERMANOVA p<0.01)。使用偏倚分枝杆菌组方法,m . avium在三个NTM中被发现+文化样本;c)在声门上性样品中,无偏置16S处理的样品之间β-多样性无显著差异与偏倚分枝杆菌组法。在两个样本中,a分枝杆菌标注为非致病性菌株。
下气道免疫剖面
为了评估NTM疾病中微生物特征与独特的粘膜免疫表型的关系,我们检测了BAL细胞差异,在活的有机体内细胞因子水平体外细胞因子的生产。在特种加工+与未受累肺段相比,受累肺段的BAL样本中嗜中性粒细胞显著增加,巨噬细胞显著减少(表2).相反,在NTM中−受累肺段BAL的淋巴细胞明显升高。在活的有机体内在BAL中测量的细胞因子水平也显示了NTM的不同炎症特征+.在特种加工+与未受累肺段相比,受累肺段BAL中干扰素(IFN)-γ、白介素(IL)-8、IL-12p70、IFN-γ诱导t细胞α趋化剂(ITAC)、巨噬细胞炎症蛋白(MIP)1α和MIP1Bβ水平显著升高。这种模式在NTM的BAL样品中不存在−受累肺段的参与者MIP3α和IL-17A水平较低。同样地,在期间观察到明显的炎症模式体外toll样受体-4对BAL细胞的刺激作用(联机补充表S3).在特种加工+来自受累肺段的BAL细胞抑制了粒细胞-巨噬细胞集落刺激因子(GM-CSF)和IFN-γ的产生。NTM中未发现这些差异−参与者。
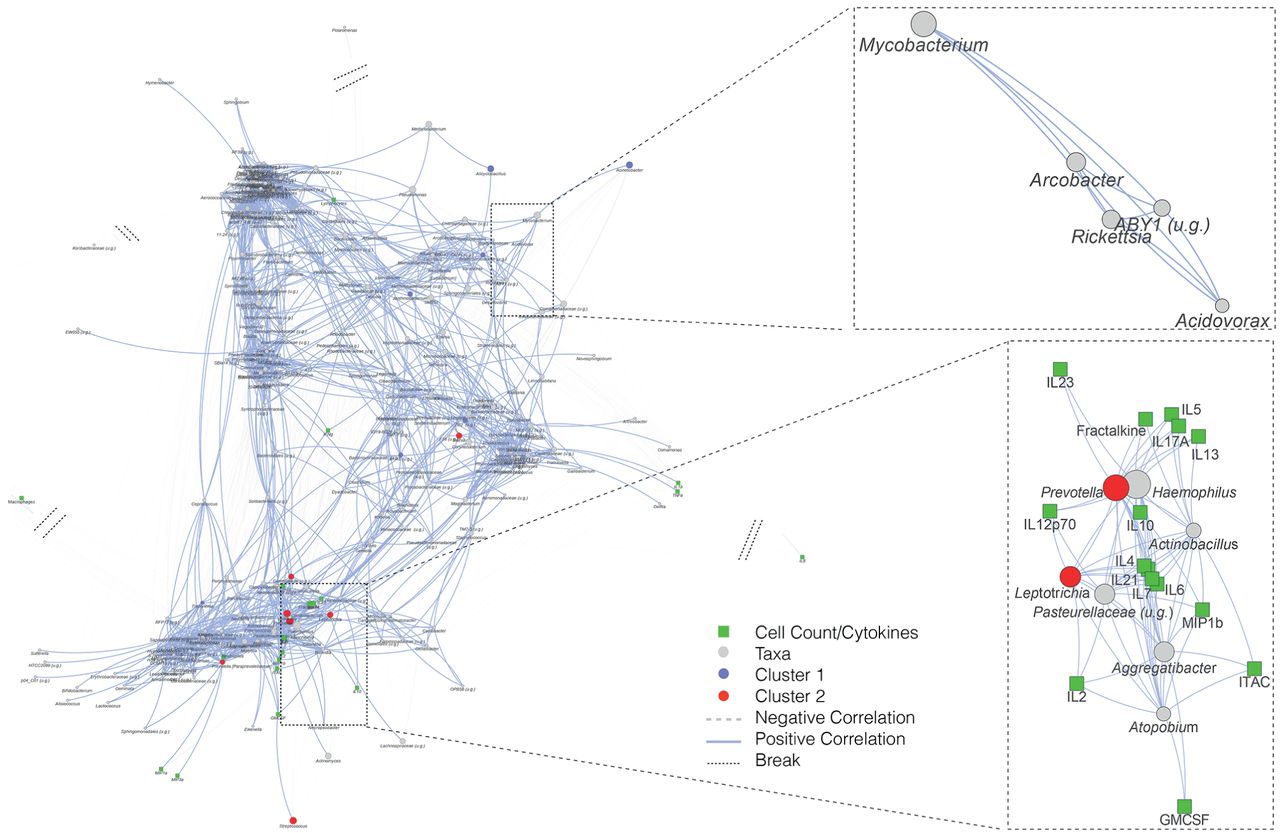
接下来,我们评估了NTM中与这些炎症生物标志物相关的微生物组特征+样品和NTM−样本。为此,我们使用网络方法评估倾向于共同出现的分类群,并基于Dirichlet多项模型聚类将其识别为不同的聚类(在线补充资料而且图S9).
在NTM的BAL+与会者,口头共议等普氏菌,韦永氏球菌属而且纤毛菌属倾向于同时发生,并与中性粒细胞和几种细胞因子包括IL-6, IL-17, IL-23和fractalkine (图5).有趣的是,分枝杆菌属于单独的共发生聚类,与炎症生物标志物无显著相关性。在NTM的BAL−下气道样本中口腔共生菌的相对丰度与细胞因子和BAL细胞相关性不显著(在线补充图S10).
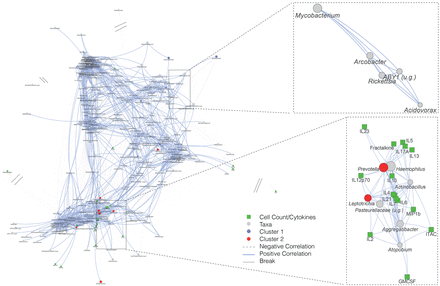
下气道非结核分枝杆菌(NTM)分类群与炎症生物标志物之间的关系+样本。使用SparCC构建网络分析,以消除微生物组数据中常见的成分和稀疏性效应,以确定属水平上分类单元与炎症生物标志物之间的相关性。每个节点代表一个属,节点的大小表示对数相对丰度从大(高)到小(低)。此外,分类群被确定为下气道微生物群簇的标记(基于DMM分析,见在线补充图S5)以颜色编码。节点之间的边表示显著相关,边的长度表示相关系数的强弱(边越短,正相关越高)。u.g.:未确定属。
讨论
本研究的目的是使用不依赖培养的技术评估疑似NTM疾病的前瞻性队列患者的气道微生物群。我们的分析表明,使用痰样本,在有和没有通过培养鉴定NTM的样本之间,可以识别出很少的微生物群组成变化。使用来自接受支气管镜检查的参与者亚组的上气道和下气道样本,我们发现诱导痰不能很好地代表该患者人群中的下气道微生物群,而更准确地反映了口腔的组成。此外,文化无关的方法没有发现分枝杆菌在很大比例的样本中。我们用a扩展了这些观察分枝杆菌-偏置嵌套序列方法确认,在NTM中占多数+参与者,与许多其他已确定的微生物相比,这种生物体的丰度很低或没有检测到。这些数据表明,这些与培养无关的检测方法的敏感性有限分枝杆菌并举例说明了目前研究低丰度病原体的通用测序方法尚未认识到的局限性。最后是NTM的下气道+参与者具有独特的免疫表型,其中几种炎症生物标志物的水平与口腔分类群微生物的相对丰度相关,而与口腔分类群微生物的相对丰度无关分枝杆菌.这些数据表明,微吸和/或未能清除吸入的口腔微生物可能有助于NTM疾病的炎症内型。
不依赖培养的技术已经证明,气道中含有复杂的微生物群,对宿主的免疫反应有重大影响[3.,11,23,36,37]。在最近一项涉及76名来自欧洲多中心队列的非囊性纤维化支气管扩张患者的研究中,流感嗜血杆菌,铜绿假单胞菌而且链球菌为痰液样本中数量最多的菌种[4]。然而,这一队列的特点是NTM患病率低。在美国,NTM是非囊性纤维化支气管扩张的常见原因,最近由aksamit等.[38]和多种NTM菌株与支气管扩张有关[39]。在我们的研究中,NTM的患病率为58%,与美国支气管扩张研究登记相似[38]。NTM疾病的诊断通常基于诱导痰。因此,我们在我们的队列中检查了诱导痰和口腔清洗中的微生物群差异。痰液和口腔清洗样品在NTM状态的多样性指标上均无显著差异。此外,在NTM+患者中,分枝杆菌在这些样品中没有富集。
为了进一步描述NTM疾病中下气道微生物群的特征,一组患者接受了支气管镜和BAL和上气道(口咽)取样。口咽菌群的差异包括富集链球菌而且罗思氏菌属在口腔清洗和浓缩用普氏菌而且韦永氏球菌属在声门上样本中。重要的是,诱导痰与上、下气道样品的比较表明,诱导痰在成分上更接近于上气道微生物群(无论是口洗液还是声门上),而不是下气道微生物群。这表明诱导痰主要受上呼吸道菌群组成的影响,而下呼吸道菌群的表现较差。与我们的发现相似,在一组哮喘患者中,使用诱导痰来评估气道微生物群提供了下气道的不完全反映,主要受口腔微生物群的影响[40]。这意味着我们对使用非侵入性样本的下气道微生物群的有限理解。在我们的队列中,BAL样本来自NTM+参与者得到了丰富的分枝杆菌BAL样本来自NTM−参与者得到了丰富的Porphyromonas.然而,类似于诱导痰,分枝杆菌在这种生物培养阳性的样本中,使用16S rRNA基因测序往往无法识别。这与之前发表的观察结果一致[21,34]。由于NTM倾向于每个基因组只有一个或两个16S rRNA基因,使用16S测序的标准方法,它们在每个细菌拥有更多16S rRNA基因的其他类群中可能被低估[33,41]。在落下帷幕,分枝杆菌仅在27%的NTM中被识别+样本使用标准16S rRNA基因测序。在这项研究中,我们试图使用一种通用的测序方法,使我们能够广泛地描述气道微生物群的细菌组成。自分枝杆菌在NTM中很少出现+使用现在广泛接受的16S rRNA基因测序的案例,我们试图通过应用嵌套扩增方法来提高识别该属的敏感性,其中第一个PCR靶向a分枝杆菌-包含16S rRNA基因V4区域的特异性区域[21,42- - - - - -44]。这是可能的分枝杆菌特异性引物在识别这种生物体时有更好的产量。尽管如此,通过这种测序方法,我们能够识别分枝杆菌在生长NTM的BAL样品中,有47%的样品中含有NTM。相比之下,分枝杆菌仅在17%的NTM中发现+痰样本,这可能也与痰下气道微生物群的代表性有关。
我们的支气管镜取样也允许我们比较NTM的炎症表型+和特种加工−参与者。先前的研究表明,在分枝杆菌感染时,通过诱导IFN-γ,激活的巨噬细胞上调促炎细胞因子的表达,以帮助预防分枝杆菌感染。这些细胞因子包括IL-6、IL-1β、IL-12、肿瘤坏死因子-α和一氧化氮[45,46]。在特种加工+从相关部位获得的BAL样本,经脂多糖(LPS)刺激的BAL细胞显示IFN-γ和GM-CSF水平显著钝化,提示先天免疫反应受损。在共现网络分析中,被确定为口腔共生植物的类群之间有显著的关联(如普氏菌。,韦永氏球菌属而且纤毛菌属)和T-helper-17细胞因子在NTM中也有表达+BAL样本。相对的丰度分枝杆菌与炎症生物标志物水平无显著相关性,提示其他微生物对NTM疾病下气道炎症张力的重要性。
这项研究有几个局限性。纳入本队列的患者均为轻度NTM。NTM患者比例增加很大鸟型分支杆菌复杂,符合美国支气管扩张研究登记[33,38]。有可能是不同的菌株分枝杆菌不同的疾病严重程度会有不同的气道微生物群和炎症特征。在本研究中,相对较小比例的患者接受了支气管镜检查,使我们能够评估下气道[47这些调查大多是出于临床原因进行的。虽然与我们较大的队列相似,但这些患者可能代表不同的疾病表型。然而,鉴于痰样本中存在的微生物群对下气道的代表性有限,有必要在更大的队列中进行进一步调查,以揭示可能与本病相关的微生物群宿主相互作用,并确定诱导痰中存在的哪些微生物群特征可用于探索下气道微生物群。此外,尽管我们在NTM中发现了下气道微生物群特征和炎症生物标志物之间的一些显著关联+小组,我们认为这些结果是探索性的和产生假设的。需要对更大的患者队列进行支气管镜取样,以证实和扩大这些发现。最后,本研究的目的是评估ntm相关性支气管扩张患者的微生物群落。因此,我们没有评估急性发作时微生物组组成的变化,也没有评估NTM治疗的效果。重要的是,本分析排除了接受NTM治疗的患者,以避免这种对NTM差异分析的潜在混杂+与特种加工−组。
总之,我们确定了当前无偏见的与培养无关的识别技术的局限性分枝杆菌在NTM培养阳性的患者中,这突出了这些方法技术改进的必要性。此外,我们还描述了NTM疾病患者如何在下气道中具有独特的炎症环境,这可能与下气道微生物群的某些成分有关,包括通常被确定为口腔共生菌的分类群。这些数据表明微吸或未能从下气道清除上呼吸道微生物可能起作用,并可能解释培养阳性NTM疾病参与者的表现和疾病进展的一些异质性。下气道微生物群对NTM疾病病理生理炎症过程的贡献值得在更大的队列中进一步研究,并可能具有潜在的治疗意义。
补充材料
脚注
这篇文章有补充资料可从www.qdcxjkg.com
作者贡献:所有列出的作者都对本手稿有贡献。I. Sulaiman和L.N. Segal参与了概念和设计。B. Scaglione, J. Wang, A. Basavaraj, Y. Li, A.S. Scott, S. Chung, K. Bantis, J. Bessich, J. Carpenito, S. Rafeq, G. Michaud, J. Donington, G. Schattner, S. Garofano, R. Condos, D. Kamelhar, D. Addrizzo-Harris, I. Sulaiman和L.N. Segal都参与了数据的获取。B.G. Wu, P. Malecha, J.C. Clemente, N. Shen, C. Naidoo, G. Theron, I. Sulaiman和L.N. Segal参与了数据的分析和解释。吴博光、李云、王杰、P. Malecha、J.C. Clemente、n.s Shen、G.塞隆、J. Bessich、K. Bantis、S. Chung、S. Rafeq、G. Michaud、J. Donington、R. Condos、C. Naidoo、A. Basavaraj、A.S. Scott、D. Kamelhar、D. Addrizzo-Harris、I. Sulaiman和L.N. Segal参与了本手稿的起草和修改。I. Sulaiman和L.N. Segal参与了这份手稿的最终批准。
支持声明:本研究由美国国立卫生研究院K23 AI102970(给L.N. Segal),美国国立卫生研究院T32 CA193111(给B.G. Wu), Helaine Lerner基金(给L.N. Segal), NTM信息与研究(给L.N. Segal),空勤人员医学研究所青年临床科学家奖(给B.G. Wu),以及石世界赫伯格基金会奖学金(给B.G. Wu)资助。本文的资助信息已存入交叉参考基金注册.
利益冲突:没有声明。
- 收到了2018年4月30日。
- 接受2018年7月29日。
- 版权所有©ERS 2018











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}