





抽象的
结节病是一种主要影响肺部的肉芽肿疾病。假设微生物因子在疾病发病机制中的作用,但尚未在大队列中系统地进行调查。
这项横断面研究比较了71例结节病患者、15例特发性肺纤维化患者(非感染性对照)和10例健康对照(HCs)的肺微生物群。下一代16S DNA测序用于支气管肺泡灌洗液样本,以表征微生物组成,分析其多样性和指示物种。确定了13种已知结节病风险变异的宿主基因型,并与微生物参数相关。
微生物组合物在顺节病和HC样品(冗余分析ANOVA,P = 0.025)和射线照相胆汁类型之间有显着不同。Atopobium68%结节病标本中检出spp, HC标本中未检出spp。fusobacterium.spp在结节病标本中明显多于hc标本。71份结节病标本中有2份检出分枝杆菌。宿主基因型分析显示rs2076530 (BTNL2.)风险等位基因降低细菌负担(p=0.002)。
我们的结果表明在结节病BAL样本中存在Scadding类型依赖的微生物群。AtopobiumSPP。和fusobacterium.Spp.被鉴定为结节病相关细菌,这可能为疾病的发病机制和治疗提供新的见解。
抽象的
结节病肺微生物谱http:///wly/iftc30gxm2u.
介绍
结节病是一种全球流行的炎症性疾病,其特征是存在非干酪样肉芽肿[1].约90%的病例中肺受累是常见的,但其他任何器官也可受累。根据疾病表现的严重程度,1-5%的患者死于呼吸衰竭和结节病相关纤维化的后果[2,3.].胸部射线照相的五种称为膀胱阶段的三种结节病类型:0型显示胸部射线照相扫描的异常,而I型通过淋巴结病变和实质病变,III型仅为III型,仅通过实质疾病,患有肺纤维化的IV型。这些射线照相类型有助于对群组的临床和科学分层。
和其他复杂疾病一样,结节病被认为是遗传和环境因素相互作用的结果。虽然许多遗传风险变异是已知的,但环境诱因大多是未知的。已调查有机及无机因素,例如职业性接触可吸入尘埃[2,4].由于与分枝杆菌疾病的相似性,一种吸入的感染性病原体已经被假设,分枝杆菌抗原以及一些其他的候选细菌,如Chamydophilia pnemoniae和Propionimibacterium Acnes.作为潜在的触发因素[5- - - - - -10.].然而,关于可能的微生物参与疾病表现的结果是不一致的。有令人信服的证据表明,微生物群在慢性阻塞性肺病(COPD)、哮喘和特发性肺纤维化(IPF)等其他肺部疾病中发挥了作用[11.- - - - - -14.].对于结节病,迄今已有两项研究对此进行了系统研究,但两项研究均因样本量小而受到严重限制[15.,16.].在目前的研究中,我们使用下一代测序(NGS)技术,使德国患者的大型样本中的常规和培养细菌进行了检测,与健康对照(HCS)和健康对照(HC)相比,进一步了解肺肺癌的洞察力微生物群比较结节病放射学类型。患有IPF的患者被包含作为具有纤维化方面的非传染性控制,并且患有肺炎的患者被列为分析细菌负担的原则的证据。此外,我们研究了酸碱病变肺部微生物与结节病风险相关的单核苷酸多态性(SNP)的潜在相互作用。
方法
学习参与者
共纳入71例结节病患者(表格1),来自大学医院Freiburg(n = 29)和研究中心Borstel(n = 42)。所有参与者都提供了他们的书面同意,该研究得到了当地伦理委员会的批准。患有结节病的患者,39名是男性,32名是女性。年龄在24至67岁之间,中位数为43年。通过射线照相型分层,将三名患者分配到射线照相型0,17至I型,39型至II型,六型至III型,六型至IV型。没有一个患者在支气管肺灌洗(BAL)采样时接受抗生素或有任何其他肺部疾病,除了两个患者的射线照相型0患者,也具有支气管哮喘。在结节病例中,56名来自非闻名者,来自前吸烟者11人,偶尔吸烟者4。所有具有诈骗类型I,II,III或IV的受试者满足了肺或淋巴受累的要求。
10大学医院弗赖堡(N = 5)和研究中心博尔斯特尔(N = 5)的健康受试者被包括作为HCS,15名IPF作为额外的非传染性患者组患者,预期低细菌多样性[14.].作为测量高细菌负荷的对照,纳入了22例不同细菌来源的感染性肺炎患者的样本,对其进行了细菌负荷评估,但没有生成微生物谱。22例肺炎标本均以下呼吸道细菌过度生长为特征。这项研究没有确定这些微生物组的确切组成,因为这里只有细菌DNA的绝对数量是相关的。
BAL及样品制备
如前所述在患者和对照的招生网站上进行的BAL [17.].简言之,从200-300 mL无菌生理盐水(0.9% NaCl)中取25 mL等量注入肺舌部或肺叶中部,立即抽吸。将一个供体的抽吸灌洗等量物汇集并保存至−80°C下进一步制备。用标准的实验室程序提取和制备微生物DNA和RNA (补充方法).用引物27F结合454生命科学适配器B和引物338R结合454生命科学适配器A (Roche, Penzberg, Germany;引物来自Metabion, Planegg, Germany)。反向引物包含一个多重条形码标识序列(10 bp),允许对单个样品进行识别。DNA提取使用Molysis Complete 5 Kit (Molzym,不来梅,德国),该试剂盒也能提取细胞内细菌。样品按照Stratil等.[18.](补充方法详情)。为了评估总菌群和潜在活性结节病菌群之间的差异,使用MO BIO PowerMicrobiome™RNA分离试剂盒提取16S rRNA,同时进行DNA消化,通过逆转录酶PCR (RT-PCR)转录成cDNA,并准备进行类似于DNA的测序。将16S rRNA与16S rDNA进行比较,鉴定生理活性细菌。
焦磷酸测序分析
使用GS 454 FLX钛技术和化学序列仪(Roche,Penzberg,德国)测序扩增子,其中六次运行随机抽样。包括技术控制样品以检测药物的潜在污染细菌,并没有显示系统污染。序列和质量文件使用Pangea进行条形码[19.].reads长度小于200 bp,质量评分小于25的序列被拒绝。使用motherur软件进行噪音消减[20.].消除了底漆或条形码序列不同的含糊不清的序列和序列,与具有超过八个均聚物的序列和具有嵌合序列的序列。为了纠正序列的变量,输出被标准化为每个样本的1000个序列。
分类学分析:基于操作单元的分类学方法
运营分类单位(OTUS)是基于相似性聚集的序列组,允许分配属或物种。使用Mothur管道,序列以97%相似的阈值聚集。使用已建立的参数和方法进行分析微生物组合物,例如α-多样性分配的香农指数[21.],以及用海灵转换数据进行冗余分析以获得beta多样性[22.].多样性是指一个样本内细菌类型的数量和分布,而多样性是不同样本之间的相同比较。采用Kruskal-Wallis检验评估结节病、IPF和HC样品间物种分布的差异。alpha多样性和细菌负荷差异采用Wilcox秩和检验,beta多样性采用ADONIS检验。文中给出了更多细节补充方法.
基因分型和相互作用分析
使用DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany)从上述BAL样本中提取人类DNA,随后使用RPLI-g Single Cell Kit (QIAGEN)进行全基因组扩增。采用Taqman检测基因型®基因分型技术(Life Technologies,USEN,USA)以下13种SNP变体,已知与结节病有关,括号中给出的各种基因基因座:RS1049550(安慰), rs2076530和rs5007259(两者BTNL2.), rs4143332 (HLA-B),RS9277542(HLA-DPB1), rs479777 (chr11q13.1), rs1050045 (OS9),Rs10484410(ZNF451),Rs1040461(RAB23), rs12069782 (il23r.),RS4921492(IL12B),RS653178(ATXN2.)及rs223498 (nfkb1./曼巴).给出了包括参考和基因型频率的所选变体的详细描述补充表E1.采用Wilcox秩和检验对宿主基因型进行Alpha多样性和细菌负荷差异分析,并采用错误发现率(FDR)校正对多重检测结果进行校正。Beta多样性采用按基因型分层的转化冗余分析(tb-RDA)进行分析。
细菌负担
采用Taqman法测定BAL标本中细菌DNA含量,以评估细菌负荷®[23.],用引物Eub338F和Eub518R估计细菌负荷,用引物bActin_F和bActin_R测定人DNA量。
结果
经过质量检测和预处理,NGS共获得374,341条序列,并归一化为1000条序列。分类分析将这些序列划分为3413个OTUs,其中121个OTUs高于丰度阈值(>0.1%)。OTUs >0.1%的总序列之和除以整个数据集的总序列之和,数据集中90%的分类信息都表示在这个截断点上。
微生物多样性(α和β多样性)和结节病,IPF和控制中的细菌负担
平均alpha多样性在结节病(香农指数(SI))之间没有显著差异。不仅= 3.0,SD.=0.52)和HC (SI控制= 2.8,SD.=0.69)样本,而结节病和IPF样本的比较显示IPF的多样性降低(p<0.001;如果IPF= 2.4±0.94;图1一个).通过顺曲放射线照相类型歧视,不同类型之间的α多样性没有显着差异(多样性指数:0:Si0= 2.7,SD.= 1.2;I型:SI我= 3.1,SD.= 0.34;类型2:如果II= 3.0,SD.= 0.46;类型III:如果3= 3.1,SD.= 0.41;和IV型:SIIV.= 3.0,SD.= 0.82;图2一个).
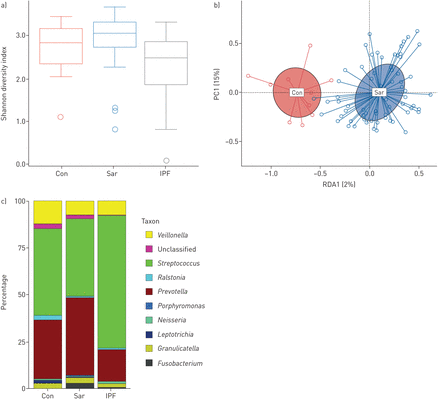
一个)Sarcoidosis(SAR)(N = 71),特发性肺纤维化(IPF)(N = 15)和健康对照(CON)(N = 10)样品的α多样性,如Shannon指数测量的。框内的水平线代表中位数。晶须代表1.5倍的间形范围内的最低值和最高值。异常值和个别样本值用圆圈表示。b) Beta多样性表现为结节病样本的hellinger -transform冗余分析与健康的控制。轴显示冗余分析(RDA1)和第一主组件(PC1)的受限轴。c)基于九个最突出的属的结节病,IPF和健康对照样品的微生物组成。
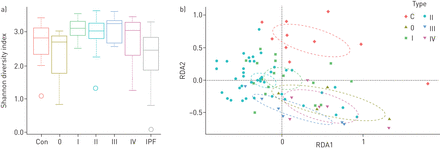
一)Alpha多样性在健康对照样本(Con) (n=10),结节病影像学类型(0型(n=3), 1型(n=17), II型(n=39, III型(n=6), IV型(n=6))和特发性肺纤维化(IPF)样本(n=15)中。方框内的水平线代表中值,须代表1.5的四分位数范围内的最低值和最高值。异常值和个别样本值用圆圈表示。b)结节病类型的聚类分析与通过基于转换的冗余分析(tb-RDA)进行健康控制,包括95%置信区间(虚线)。
肉样瘤病与HC (ADONIS)样品的微生物组成差异显著RDA, p = 0.025;图1 b)和在结节病和IPF样品之间(阿多尼斯RDA,p = 0.001)。5个OTUs在结节病、IPF和HC样本之间显著失衡:韦永氏球菌属(OTU91)在HC样品中含量较高,而链球菌(OTU1)在HC和IPF中含量较高Atopobium(OTU41)和fusobacterium.(OTU16)在结节病标本中含量较高。oribacterium.(OTU47)在IPF样本中含量低于结节病和HC样本。表2显示p值和丰度。关于结节病放射学类型的beta多样性,观察到结节病类型和HC样本具有明显的聚类(ADONIS)RDA,p = 0.011;图2 b).IPF样本与结节病类型和HC样品略微聚集(补充图E1).
人类DNA的数量与细菌载量无关(补充图E2).与HC样品相比,IPF和肺炎样品中的细菌负担分别增加了2.3和171倍,而结节病毒样品显示细菌负担减少2.5(图3.),第I类略有增加,第II、III和IV类有所减少(平均负担:对照组=1.12×10−12,i = 3.03×10−12, 2 = 2.12×10−13, 3 = 2.58×10−14,IV = 5.14×10−14).
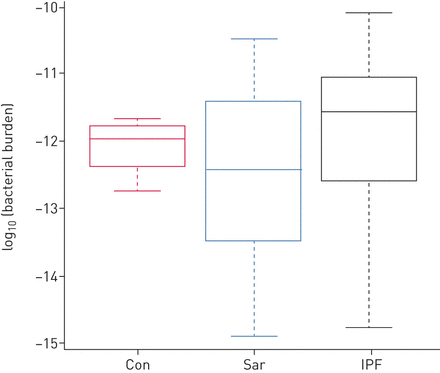
结节病(Sar)、特发性肺纤维化(IPF)和健康样本(Con)的细菌负荷方框内的水平线代表中值,须代表1.5×四分位数范围内的最低值和最低值。
对潜在混杂变量的分析显示,性别、年龄、吸烟史、招募地点或测序批次对alpha或beta多样性或细菌负担的微生物谱没有影响。
核心微生物群和指示物种
对频率阈值以上的121个otu进行定性分析,发现结节病、IPF与对照共有95个otu,而4个otu (高豚spp。Porphyromona年代。Moraxella.spp。MoryellaSPP。)在结节病样品中特别发现。核心微生物群分析表明链球菌pseudopneumoniae在71份结节病样本中,95%的样本中发现,OTUs平均丰度最高(57%)。四个链球菌(OTU1,OTU4,OTU6,OTU8),两个普氏菌(OTU2, OTU9)和1韦永氏球菌属(OTU5)在90%的rARACOITION病例中发现了物种。总细菌组合物的比较表明,这些属的代表增加了高达总细菌载荷的70%,这与HC样品的结果一致。详细的细菌物种分布在顺曲样本中显示出来图1 c.
此外,分析了微生物物种的存在或不存在以鉴定微生物标记物。在分析结节病样品中与控制,Atopobiumsp. (OTU41)在68%的结节病样本中发生,平均相对丰度为0.55%,而在HC样本中未检测到sp. (p=0.004)。Atopobium在IPF样本中也存在,但频率较低且丰富(平均值±SD.ipf: 1.22±2.16;结节病,5.51±7.31)。一份在场/缺席地图补充图E3.大量的Atopobium与HC和IPF样本相比,I型和II型结节病显著增加(P我vs..C= 0.036, PIIvs。C= 0.036, P我vs..IPF= 0.045,纠正FDR校正;图4.).fusobacterium.spp. (OTU16)在II型和III型结节病中明显多于HC (PIIvs.C= 0.040, PIIIVS.c.= 0.042;图4.),也发生在IPF样本中。一个物种(OTU91、韦永氏球菌属spp.)对hc是特异性的。
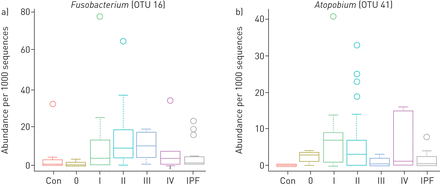
候选物种的相对丰度(%)Atopobium(OTU41)和fusobacterium.(OTU16)通过顺序(SAR)类型分层(0型(n = 3),型1(n = 17),II型(n = 39),III型(n = 6),型IV(n = 6))与对照(CON)(n = 10)和特发性肺纤维化(IPF)(n = 15)样品进行比较。方框内的水平线代表中值,须代表1.5的四分位数范围内的最低值和最高值。异常值和个别样本值用圆圈表示。
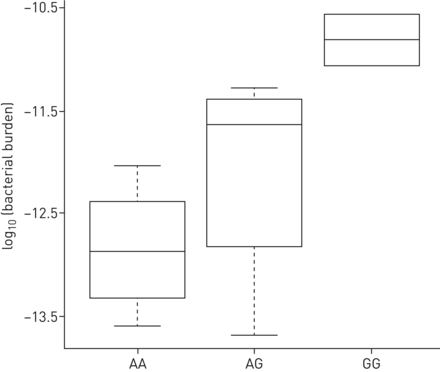
BTNL2 (rs2076530)基因型(GG (n=6), AG (n=23), AA (n=16))对结节病标本细菌负荷进行分层。方框内的水平线代表中值,须代表1.5×四分位数范围内的最低值和最低值。
水平的放线菌
作为血小霉菌类等类的成员,就像分枝杆菌和丙杆菌一样,都会影响结节病的疾病表现,propimibactium.,分枝杆菌与之密切相关的棒状杆菌属被选为候选者进行详细分析[10.,24.,25.].分枝杆菌spp在71个结节病样本中的2个(<5个观察值)和3个IPF样本中发现低水平。propimibactium.和棒状杆菌属在54%的病例和50%的HC样本中检出高频率,IPF、HC和结节病样本之间或结节病类型之间的水平无显著差异。
活性微生物群
在潜在活菌群和活性菌群(16S rRNA)中,该属的平均百分比链球菌升高(61.3%与39.1%),而脱位杆菌和Ralstonia减少了(0%与5.4%和0.2%与分别为29.2%)。以上所述的病害类型及其鉴别属,包括Atopobium和fusobacterium.,发现在活性微生物群中。
讨论
在这项研究中,我们描述了下呼吸道的微生物谱,并证明在我们的样本中,在结节病样本和对照组之间以及在结节病的x线片类型中,微生物群的组成是不同的。我们将IPF作为一种额外的表型纳入,因为这种纤维化情况可能在某种程度上类似于结节病的纤维化。结节病样本与HC和IPF样本之间的显著差异表明存在结节病特异性微生物谱。
由于支气管镜检查程序,由于支气管镜检查程序,在BAL的微生物分析中,可以完全排除样品的污染。然而,许多研究描述了人类呼吸道的各个部分中的不同的微生物谱[26.,27.].我们的研究中包含了技术控制,揭示了零星的存在propimibactium.sp.,与既往报道一致[28.].这一发现强调了propimibactium.在技术对照中未发现与结节病相关的菌种。虽然这些先证者在BAL时均未接受皮质类固醇治疗,但我们没有皮质类固醇使用情况的信息。因此,我们不能排除吸入糖皮质激素对调查样本中微生物多样性的潜在长期影响[14.,29.].
正如预期的那样,IPF和肺炎样本中的细菌负担增加,而在顺序化样本中略微下降。为了排除由于BAL样品中的总DNA不同量的技术偏差,我们测试了具有细菌负荷的人DNA量的相关性,发现无显着的相关性。此外,与HC样品相比,结节病患中的α多样性没有显着变化。以前的研究表明,αα多样性的结节病毒样品的结果相矛盾。虽然S.雪儿等.[16.与HC样本相比,发现α多样性降低了G.arzoni等.[15.]没有找到任何差异。这两项研究都受到小样本大小的限制(分别为n = 10和n = 7)。在这种较大的研究中(n = 71)中,齐全样品在α多样性上显示出薄弱的升高,但这并不统计学意义。符合当前的顺序病理学模型[8]这清楚地讨论了结节病的经典传染过程,其特征主要是特异性细菌种类的过度生长,从而降低了α多样性[30.].
调查的结节病只有最丰富的属链球菌,Fvootella.和韦永氏球菌属物种,这是根据先前对肺部微生物的研究[15.,31.].案件和控制之间五场丰度有显着不同。虽然小以前的研究发现了减少洋葱[16.]或者根本没有差异[15.与健康和患病的对照样本进行比较,我们发现了该属的成员veillonella,Oribacterium,链球菌,面食和fusobacterium.在结节病,IPF和HC样本之间不平衡。后两本身在顺序化样本中更丰富,因此可能代表了与结节病相关细菌的新候选者。作为一个重大限制,我们的研究设置不允许结论所识别的物种是否对结节病有关影响。但是,重要的疾病协会Atopobium和fusobacterium.SPP。可能表明潜在的疾病相关性。此外,观察到这些细菌活着,并且可能在患有结节病患者的肺部中繁殖的患者在结节病的病理生理学中支持他们可能的作用。
Atopobium是阴道共生菌群和口腔菌群的成员[32.],并在COPD的背景下已经描述了[33.].在目前的研究中,Atopobium在I型和II型结节病中显示出最高的丰度,因此可能是这些类型所代表的早期疾病状态的候选指标。它也可能是结节病的初始触发因素,这需要在纵向研究设计和适当的肉芽肿模型中进行研究。潜在的机械相关性,Atopobium属于同一门的细菌结核分枝杆菌。因此,两种物种很可能均为高度保守的抗原,能够诱导结节病患者的类似免疫应答[34.].第二种可能的机制是通过的自身抗原样反应Atopobium抗原,如类风湿性关节炎(RA)所述[35.].
升高的水平fusobacterium.在II型和III型放射学样本中发现sp .,两者均以实质疾病为特征。fusobacterium.是呼吸和肠道菌群的共生细菌,具有致病潜力和组织侵入能力。发现临床相关的关联对于牙周炎,伤口感染和结肠直肠癌等疾病进行了疾病[36.- - - - - -38.],炎症环境中的侵入性质增加[39.].fusobacterium.也被发现是某些肺炎型的炎症增强社区的一部分[40.],并影响四环素的反应[41.,42.].具体是否存在fusobacterium.在我们的结节病例中,样品是一种原因,或后果的辐射照相型II和III中的实质受累和慢性疾病仍有待阐明。
对我们的知识,Atopobium之前尚未与结节病有关,同时尚未描述fusobacterium.以前发现在10个结节病样品中的两种中有一个高度丰厚,并在RA的研究中进行了对照[16.].
分枝杆菌和Propionibacteria已在结节病的背景下被广泛讨论,但部分结果相互矛盾[8].我们的研究并没有为这个话题增加任何基本的证据分枝杆菌和Propionibacteria在结节病和HC BAL样品之间未发现不平衡。然而,正如我们没有调查肉芽肿活检的那样,我们也不能排除肉芽肿或疾病初始化这些细菌的分枝杆菌或丙杆菌因子的存在。相反,在我们的假设方法中,我们建议Atopobium和fusobacterium.作为具有现在需要在纵向设置和直接实验方法的不同群体中需要独立确认的合格性新的疾病相关的候选者。
此外,还有一个协会BTNL2.在我们的研究中发现了rs2076530风险等位基因减少了细菌负担。微生物群与宿主基因型的这种相关性已经显示在良好的肠道诸如复杂的疾病之类的良好的区域中[43.,44.而在肺部疾病中,只有相互作用MUC5B目前已经描述了IPF样本中的细菌负担的多态性[14.].我们研究中观察到的相关性可能是BTNL2在抑制t细胞增殖中的作用[45.].SNP rs2076530导致BTNL2蛋白功能丧失,该变体的存在可能导致t细胞过度激活[46.免疫系统普遍过度激活,导致肉芽肿形成。同时,这可能导致所观察到的细菌负担的减少。我们发现,I型结节病的细菌负荷略有增加,而II型、III型和IV型结节病的细菌负荷降低,提示随着疾病进展,细菌负荷减少。据我们所知,rs2076530的潜在scadd类型特异性关联尚未被研究。
综上所述,我们对BAL样本的发现支持结节病特异性微生物群的假设,Scadding类型显示出不同的细菌模式。我们无法确认结核分枝杆菌使用NGS,但我们发现在结节病微生物群中的不平衡,特别是AtopobiumSPP。和fusobacterium.与结节病特异性scadd类型相关。因此,我们首次描述了结节病中BTNL2风险基因型与细菌负荷的关联。这突出了进一步评估肺微生物群在结节病表现、进展和治疗中的作用以及它与遗传因素的相互作用的必要性。
补充材料
确认
我们感谢所有研究参与者和医生的贡献。我们进一步感谢临床分子生物学研究所(德国基尔)的实验室团队提供的技术援助。
作者贡献:数据分析:A. Zimmermann, H. Knecht;样本或数据集:R. Häsler, G. Zissel, ki . Gaede, J. Müller-Quernheim;研究设计:S. Hofmann, S. Schreiber, A. Fischer;原稿:A. Zimmermann, H. Knecht, R. Häsler, A. Nebel, A. Fischer;手稿修订和最终手稿:所有作者。
脚注
本文提供了补充材料www.qdcxjkg.com
支持声明:Deutsche Forschungsgemeinschaft(DFG)支持该研究通过临床局部卓越的炎症群体。BioMaterialbank North部分由Airway Research Center North(Arcn)部分资助,德国肺部研究中心(DZL)成员,是Popgen 2.0网络(P2N)的成员,由授权支持德国教育和研究部(01EY1103)。本文的资金信息已存入Crossref资助者注册表.
利益冲突:无声明。
- 收到了2016年4月13日。
- 接受2017年8月28日。
- 版权©2017人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}