





摘要
鼻咽和口咽样本通常用于指导治疗囊肿性纤维化(CF)无咳痰婴儿的下呼吸道感染。
我们的目的是通过常规培养和16S-rRNA测序研究25组鼻咽、口咽和支气管肺泡灌洗液(BAL)样本的细菌群落组成的一致性,这些样本来自17名年龄为5个月(n=13)和12个月(n=12)的CF婴儿。
聚类分析表明,BAL菌群的特征是口腔和鼻咽细菌的混合物,包括共生菌链球菌,奈瑟氏菌属,韦永氏球菌属和罗思氏菌属和潜在的病原体金黄色葡萄球菌,流感嗜血杆菌和莫拉克斯氏菌属然而,在每个个体内,上呼吸道两个生态位的微生物群和相应BAL之间的一致性程度不同。
上呼吸道和下呼吸道生态位微生物群的个体内不一致的一致性表明,CF患儿的肺部可能有自己的微生物群,似乎是上呼吸道微生物群的种子,但并不完全相同。
摘要
CF婴儿的肺部有一种微生物群,似乎是由上呼吸道微生物群播下的,但与上呼吸道微生物群并不相同http://ow.ly/1NlA306DuPv
介绍
囊性纤维化(CF)是一种导致慢性肺部感染的限制生命的遗传性疾病。在儿童早期,CF患者的肺部被一系列复杂的微生物所定植,这些微生物比那些被认为是“典型CF病原体”的细菌种类要多得多[1- - - - - -3.].儿童CF患者的核心肺微生物群似乎由属组成链球菌,罗思氏菌属,普氏菌,放线菌,韦永氏球菌属,Gemella,奈瑟氏菌属和嗜血杆菌[1],经微吸或吸入,由上呼吸道扩散至下呼吸道[4- - - - - -7].
理论上,支气管肺泡灌洗(BAL)对CF患儿进行直接肺取样[8]将最能代表LRT微生物群落中呼吸道病原体的本地存在和相对丰度。然而,1)由于BAL的侵入性,2)由于婴幼儿CF患者不会自发咳痰,3)由于这些患者缺乏获得诱导痰的实际经验[8,9],临床医生通常使用其他标本,包括鼻腔、鼻咽(NP)和口咽(OP)样本进行微生物监测[10- - - - - -15].最近的横断面研究[16,17]的研究表明,平均而言,上呼吸道和下呼吸道拥有相似的细菌群落。然而,我们认为对个体患者更相关的个体内一致性分析直到最近才出现,并且仅适用于终末期CF疾病的成人[18].这些分析表明,咽喉和(移植的)肺组织的微生物群高度不一致。
我们使用新一代测序技术和在出生后第一年连续两个时间点进行常规培养,研究了CF患儿的NP、OP和BAL微生物群谱。我们的目标是:1)评估不同呼吸生态位的个体间和个体内细菌群落的一致性;2)研究在CF患者的管理中使用基于16s - rrna的测序相对于传统培养的潜在附加价值;3)评估CF患儿肺部微生物群的时空变化。
方法
研究人群与设计
通过一项前瞻性的双中心观察性研究招募了CF患儿,该研究围绕荷兰新生儿筛查诊断为CF患儿的微生物监测项目建立[8].补充方法中详细描述了项目和研究人群[19].在目前的研究中,我们收集了出生一年内患有CF的婴儿的连续个体内NP、OP和BAL样本。在所有采样时刻,对基线特征和患者呼吸状态进行问卷调查。
道德的声明
这项研究(NL10/337)得到了乌得勒支大学医学中心和伊拉斯谟医学中心儿科医学伦理委员会的批准,并根据良好临床实践指南进行。
取样和储存方法
在全身麻醉下,根据标准的安全和支气管镜监测程序进行柔性光纤支气管镜检查。在BAL手术前和镇静后,经鼻擦拭后鼻咽[20.口咽用ESwab 482CE尼龙绒柔性无菌棉签(Copan诊断公司,Brescia,意大利)经口拭拭,并保存在Amies传输介质中[21].在BAL手术开始时(详细方案请参考补充方法),用无菌纱布擦拭瞄准镜尖端,作为细菌DNA背景噪声阴性对照[22].支气管镜通过喉罩引入;灌注1 mL·kg进行BAL−1无菌0.9%生理盐水,低压抽吸回收。为防止上呼吸道污染,在引入支气管镜时,直到鼻尖通过隆突才进行吸痰。在不同灌洗周期中,从右肺中叶、舌叶/左上叶、左下叶及其他肺段至少2个最多5个位置依次收集BAL液[23].在两个研究中心之一,由于后勤原因,来自不同叶的BAL液被汇集在每个人身上。所有BAL样本立即密封在无菌容器中,并连同单独的NP和OP拭子一起运送到两个CF参考中心的临床微生物实验室。两个上呼吸道样品的BAL液和Amies介质随后发生涡状和alialige。一个样品在−80°C冷冻8小时,直到进一步的序列分析。另一种镀液用于哥伦比亚血,巧克力,麦康基和甘露醇盐琼脂培养基和burcep板(详情见补充方法)。
Microbiomic分析
细菌DNA的分离和定量
DNA从200 μ L NP Amies培养基中分离[24]、200µL OP Amies培养基、300µL BAL液和我们的阴性对照样品(200µL裂解缓冲液(NP和OP)和300µL磷酸盐缓冲盐水,来自支气管镜纱布尖(BAL))。使用针对细菌16S-rRNA基因的引物探针组,通过定量PCR建立样品的总细菌载量[24- - - - - -26].为了避免16S-rRNA测序分析中的环境污染,我们只考虑了具有足够总细菌载量的呼吸道样本,定义为细菌密度≥0.3 pg·μL的样本−1以上为背景环境对照样品。为了证实低细菌密度NP和BAL样本不是由环境交叉污染引起的,我们同时对低细菌密度NP和BAL样本的一个子集(n=22)与背景环境对照样本(n=27)进行测序,包括DNA分离对照和术前支气管镜尖端DNA对照(即。gauze-controls)。具体方法请参见补充方法。
扩增子库制备
使用454 GS-FLX-Titanium测序仪(Roche, Hongkong, China)扩增细菌16S-rRNA基因的V5-V7高变区,生成PCR扩增文库。
数据处理
获得的原始序列使用QIIME 1.8版本进行处理[27].使用split_library.py检查序列质量,使用默认参数(补充方法)。随后,条形码和引物被修剪,嵌合序列被识别并使用嵌合层去除。接下来,利用UCLUST以97%的相似度将reads对齐并聚类到操作分类单元(OTUs)中。使用Greengenes 16S-rRNA数据库(version 13.8)对对齐的序列进行分类注释,并根据文献结果将其分类为CF的潜在病原体或一般共生菌[28- - - - - -31].数据可通过NCBI GenBank数据库获取:登录号为SRP081002。对于每个样品,在稀疏深度为1100个序列时,计算α-多样性指数(Shannon多样性、观察物种数和Chao 1多样性指数)[32,33].
统计分析
数据分析采用IBM SPSS 21版(IBM, Armonk, NY, USA)或R 3.1版软件进行。我们使用(配对)t检验或Mann-Whitney u检验来计算连续数据组间差异的统计学显著性。相关系数采用非参数斯皮尔曼相关计算。采用Bonferroni重复测量方差分析评价不同生态位间α-多样性指数差异的统计学意义事后测试。非度量多维尺度图(nMDS)和层次聚类树状图(iTol) [34]基于Bray-Curtis不相似矩阵,用于可视化不同生态位之间整体微生物群落组成的差异。采用排列方差分析(permutational ANOVA, PERMANOVA, vegan package R)计算这些差异的统计学意义[35].接下来,我们使用Kruskal-Wallis测试和Benjamini-Hochberg校正进行多次测试和随后的Nemenyi事后测试,以检测100个排名最高的OTUs在生态位(NP或OP)之间的相对丰度的显著差异与拜尔港)。前100个OTUs是基于我们75个样本(每个呼吸生态位n=25)样本集中这些OTUs的平均丰度得出的。Bray-Curtis相似性(1 - (Bray-Curtis不相似性),其中1.0表示两个样本之间的完全相似性,用于确定NP、OP和BAL的单个样本集内微生物组成的总体相似性1)(作为BAL样本的参考,我们使用舌腔灌洗(如果可用),或者另一个位置或混合样本);2)每个BAL不同肺灌洗位置之间;3)早期和后期BAL样品之间的差异。评估生态位之间最丰富的细菌种类的一致性2计算这些细菌相对丰度(+0.00001)的-转化比值(NP / BAL或OP / BAL)。
结果
研究人群的特征
在基线时进行常规择期支气管镜检查时,从17名CF患儿中收集了25组NP-OP-BAL;包括13组来自9个月以下的婴儿(“早期”,3-8个月不等)和12组来自9个月以上的婴儿(“晚期”,9 - 13个月不等)[36,37].在两个时间点对8名婴儿进行采样,两个采样时间点之间的中位间隔为8个月(范围5-10)。BAL样本平均取自3个(范围2-5)肺叶。两个年龄组婴儿的基线特征描述在表1.
密度数据和测序结果
平均而言,我们观察到OP样品的细菌密度最高,其次是NP和BAL样品。样品和环境对照样品(纱布对照和DNA分离对照)的密度见补充图S1。我们的最低密度样品和环境对照的同时测序证实,环境和我们的最低密度样品之间的交叉污染可以忽略不计(补充结果)。
使用16 - s测序,我们获得了总共590 227个序列(每个样本的平均序列数±sd: NP 5754±2084,OP 4490±1717,BAL 4705±1610),分为259个OTUs(不包括单子),代表15个分类门,分配了10个以上的归属序列。
鼻咽和口咽群落与支气管肺泡灌洗群落在人群水平上的比较
生态位之间生态多样性的变化
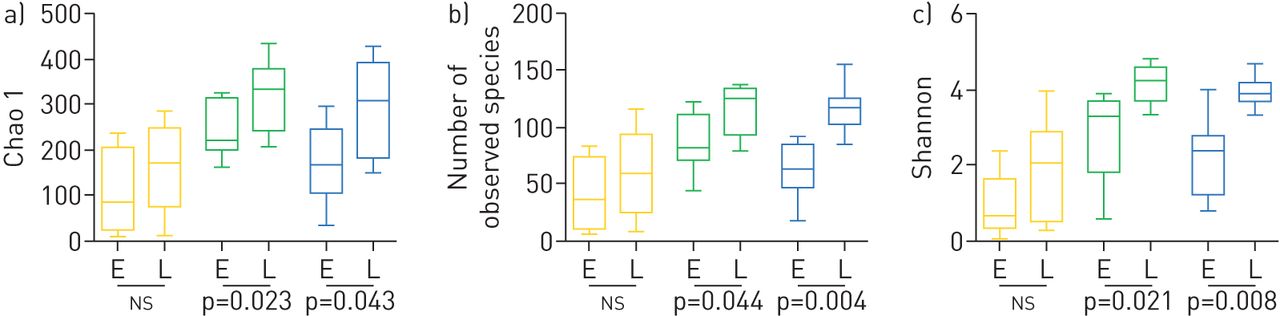
Shannon多样性指数,包括物种丰富度和均匀度,在样本点之间有显著差异(重复测量方差分析,p<0.001): NP菌群的多样性最低(平均多样性指数(DI) 1.9, 95% CI 1.4-2.5, NP的p=0.001与BAL),其次是BAL (DI 3.1, 95% CI 2.6-3.5)和OP菌群(DI 3.3, 95% CI 2.9-3.8, OP p=0.352与拜尔港)。在其他多样性测量中也观察到类似的模式(图1).
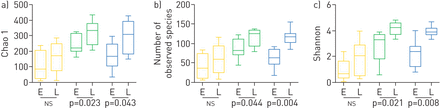
鼻咽(NP,黄色)、口咽(OP,绿色)和支气管肺泡灌洗(BAL,蓝色)样本随时间变化α-多样性分析的箱图。a) Chao 1多样性指数,b)观测物种数,c) Shannon多样性指数。研究人员对8名患有囊性纤维化的婴儿进行了两次取样,即。当3-8月龄(E)和≥9月龄(L)时。早期和后期获得的样本之间的相对丰度通过配对t检验(p值描述)评估,生态位之间的相对丰度通过Bonferroni重复方差分析评估事后测试(正文中的数据)。ns:不显著。
不同生态位间细菌群落结构的相似性
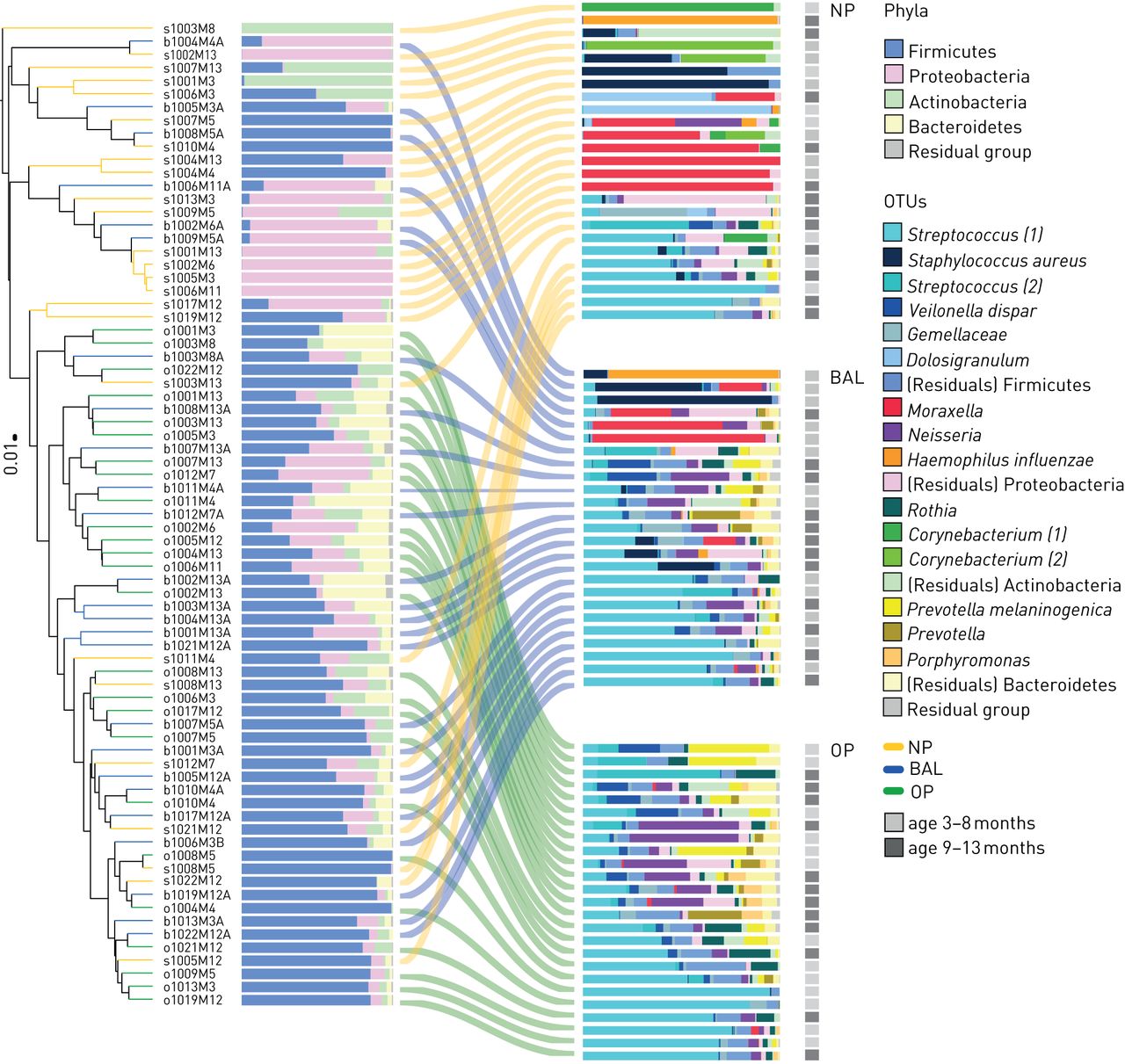
NP、OP和BAL细菌群落的门级比较见补充图S2。由于使用NP和OP样本作为BAL样本的代理[10- - - - - -14],我们评估了这两个生态位和BAL样品之间微生物群分布的总体一致性。基于Bray-Curtis不相似矩阵的nMDS图用于可视化三个生态位之间总体微生物群组成的差异(PERMANOVA未经调整R2= 0.11, p < 0.001);这说明BAL菌群一般位于NP和OP菌群之间(图2),表明BAL菌群是起源于上呼吸道两个生态位的细菌群落的混合物。平均而言,NP菌群比OP菌群和BAL菌群(NP与BAL PERMANOVA未调整R2= 0.07, p = 0.002;人事处与BAL PERMANOVA未调整R2= 0.04, p = 0.07)。与这些结果相结合,样本的层次聚类在所有三个生态位之间重叠,BAL菌群配置文件与NP和OP配置文件混合(图3).
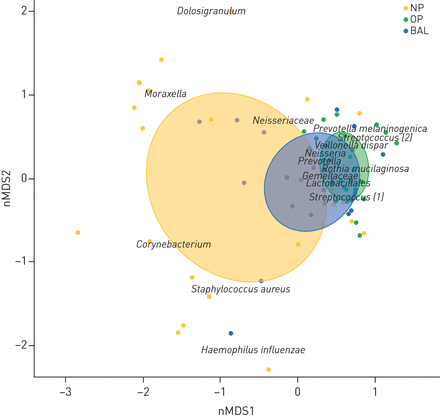
鼻咽(NP)、口咽(OP)和支气管肺泡灌洗液(BAL)样品中微生物群落组成的二维非度量多维标度(nMDS)图。基于Bray-Curtis不相似矩阵的nMDS图用于同时可视化源自NP(黄色)、OP(绿色)和BAL(蓝色)的单个样本(点);在三个生态位中15个最丰富的细菌物种的共聚类(基于n=75队列);以及按生态位(椭圆)分层的样本的几何平均值附近的标准差。β -多样性(即。微生物群的个体间变异性)在NP中最高,其次是OP和BAL样本。BAL样品的整体细菌群落组成与NP和OP生态位的群落组成重叠。此外,金黄色葡萄球菌,棒状杆菌,Dolosigranulum和莫拉克斯氏菌属与NP样本的相关性更强,而链球菌,罗氏菌,普雷沃氏菌和奈瑟氏菌属在OP和BAL样品中富集。运算分类单元(OTU)名称后面的数字表示该OTU在其属的平均相对丰度基础上的等级数量(如链球菌(2)是第二占优链球菌在群体水平观察)。

基于生态位的微生物群分布的层次聚类。树枝和边缘根据起源的生态位着色。分支的长度对应于样本(群集)之间的(平均)Bray-Curtis不相似度(标尺等于0.01所示的值)。堆叠柱状图的第一列显示了门水平上微生物群的相对丰度;第二列表示原始数据集中每个生态位(鼻咽(NP)、口咽(OP)和支气管肺泡灌洗(BAL)(各n=25)的15个总体排名最高的操作分类单位(OTUs)。早期和后期获得的样本分别用浅灰色和深灰色表示。运算分类单元(OTU)名称后面的数字表示该OTU在其属的平均相对丰度基础上的等级数量(如棒状杆菌(2)是第二占优棒状杆菌属在群体水平观察)。
属级分析表明,OP和BAL样品中共生植物的丰度相当奈瑟氏菌属,链球菌,罗思氏菌属、韦永氏球菌属,Gemella和普氏菌spp。,而NP和BAL样本含有相当丰度的潜在病原体,如金黄色葡萄球菌,流感嗜血杆菌和莫拉克斯氏菌属(Kruskal-Wallis(带Benjamini-Hochberg修正)和Nemenyi事后测试)(表2和补充图S3)。棒状杆菌属和Dolosigranulumsp .几乎只在NP样本中检测到。假单胞菌ssp。这三个生态位的丰度都很低;因此,未发现临床相关差异。
鼻咽或口咽微生物区系与支气管肺泡灌洗在个体水平上的比较
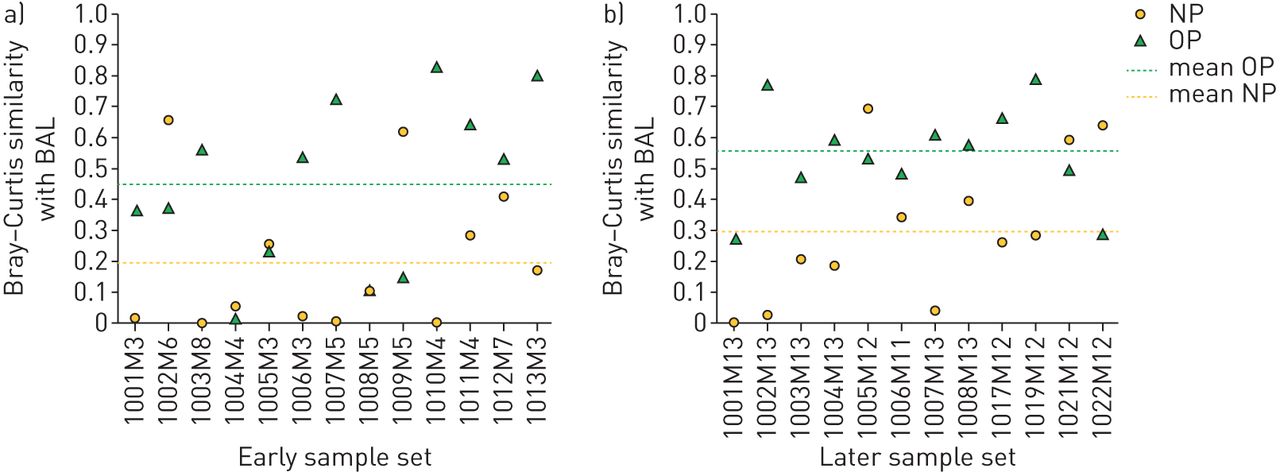
为了潜在的临床应用,我们还通过Bray-Curtis相似性度量计算了NP、OP和BAL微生物群之间的个体内一致性。由于受试者肺叶间的高度一致性,如以下部分所述,我们在这些分析中使用了25个灌洗样本中的24个样本的舌语作为肺部微生物组的代表。对于大多数患者,我们观察到NP和BAL菌群之间以及OP和BAL菌群之间的个体内一致性变化,大多数样本未能超过总体菌群相似性指数0.6 (图4).我们还观察到,从早期到后期,NP和BAL样本以及OP和BAL样本之间的总体相似指数略有增加。
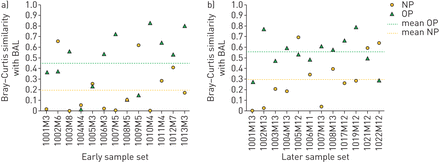
基于Bray-Curtis相似性的鼻咽(NP)和口咽(OP)微生物群谱及其配对支气管肺泡灌洗(BAL)微生物群的个体内一致性。a)“早”组,b)“晚”组。对于每个个体,我们分别计算NP和BAL样本(参考:可用语言时)(黄色)和OP和BAL样本(参考:可用语言时)(绿色)之间的个体内Bray-Curtis相似性。相似指数范围从0(两个样本之间没有相似度)到0.85(其中1.0意味着样本之间完全相似)。我们将相似度评分>0.6定义为高。均值±sd相似指数以水平虚线表示,早期OP-BAL(绿色)组为0.45±0.27,后期为0.55±0.16,NP-BAL(黄色)组为0.20±0.23,后期为0.31±0.24。
在物种水平上,我们观察到不同生态位之间的一致性存在很大差异:如。我们观察到OP和BAL样本之间的丰度在个体内具有很高的一致性链球菌(美国缓和的),奈瑟氏菌属和罗思氏菌属的丰富性之间的一致性莫拉克斯氏菌属和金黄色葡萄球菌NP和BAL样本之间的相似性更大。个人内部的和谐流感嗜血杆菌NP和BAL以及OP和BAL样本均相对较低(补充图S4)。补充结果中提供了所有参与者的所有NP、OP和BAL样本的微生物区系概况(补充图S5)。
不同叶间微生物组成的比较
根据欧洲呼吸学会的建议,在188bet官网地址两个或多个叶定期进行BAL [8],我们假设不同的肺部位置可能有不同的微生物组成。我们通过比较个体内部和个体之间不同的肺叶来研究这一点。25个BAL手术中71个BAL位置(每个BAL 2 - 5个肺区)可用于分析:同一BAL手术中不同肺叶的个体内微生物群分布的一致性较高(图5),显著高于受试者间肺部微生物群的相似性(平均相似性:受试者内0.73,受试者间0.28,p<0.001)(补充图S6)。
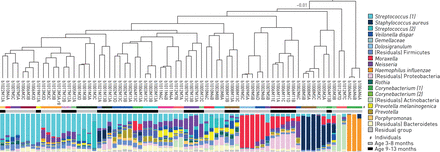
25个支气管肺泡灌洗样本的树状图显示了多个灌洗位置(总n=71)的分级聚类。聚类基于Bray-Curtis差异性和平均连锁。分支的长度对应于样本(群集)之间的(平均)Bray-Curtis不相似度(标尺等于0.01所示的值)。#不同颜色的长方形盒子分别代表不同的个体。8个个体在出生后的第一年被采样两次,分别用灰色(早期)和黑色(晚期)表示。堆叠柱状图显示了整个数据集中前15个操作分类单元(OTUs)的相对丰度。OTU名称后面的数字表示该OTU在其属的平均相对丰度基础上的等级数量(如棒状杆菌(2)是第二占优棒状杆菌属在群体水平观察)。
BAL微生物组成的时间变化
我们研究了8名婴儿出生后13个月期间BAL微生物群平均8个月的时间变化。平均而言,我们观察到随着时间的推移,微生物群多样性有所增加(Shannon指数:早期2.2,后期3.9;图1).随着时间的推移,NP和OP生态位的多样性也有相应的增加(图1).此外,我们通过计算每个个体早期和晚期BAL样本之间的Bray-Curtis相似指数,计算了微生物群分布随时间的稳定性,并将其与每个儿童样本与其他婴儿样本的相似性进行了比较。我们观察到早期和晚期BAL样本之间的个体内一致性较低(补充图S7),表明随着时间的推移,肺部微生物群发生了显著变化(补充图S6)。
传统培养与16 - s测序结果的可比性
为了与现行的实践标准进行比较,即。常规培养时,我们将测序结果与培养结果(补充方法)比较最丰富葡萄球菌,莫拉克斯氏菌属和嗜血杆菌辣子鸡,金黄色葡萄球菌,复活的和流感嗜血杆菌培养结果,确认培养结果为金黄色葡萄球菌,复活的和流感嗜血杆菌与各自有很强的相关性金黄色葡萄球菌(r年代= 0.451),复活的(r年代= 0.517)和流感嗜血杆菌辣子鸡(r年代=0.276, p均<0.001;补充图S8)。此外,测序检测潜在病原体的敏感性高于常规培养,见补充图S8。
为了测试三个呼吸生态位微生物群之间的个体内一致性,我们对常规培养结果重复了这些分析。总体上,所有培养菌的阳性预测值(PPV)和阴性预测值(NPV)均为中高水平(表3).然而,常规培养往往无法在相应的NP和OP样本中检测到肺中鉴定的完整病原体集(分别占样本集的28%和50%;补充图S9)。
讨论
我们研究中最有趣的发现是,当通过常规培养或测序技术研究时,NP或OP的微生物群落与其相应的BAL微生物群落之间的一致性在个体患者中是可变的。这表明,两种类型的上呼吸道样本在确定CF患儿的微生物定殖谱方面具有有限的诊断价值emanick等.[38]是唯一一项确定CF患儿唾液和OP样本、诱导痰和咳痰中气道微生物群的研究;他们得出结论,诱导痰可能比OP拭子更能代表肺定植。我们的研究证实了他们关于OP拭子作用有限的观点,也表明NP样本也不是合适的替代品,根据世界卫生组织(WHO), NP样本是检测普通人群中呼吸道病原体的金标准。
第二个有趣的发现是上呼吸道和BAL之间的一致性随着时间的推移而增加:这可能是抗生素使用在上呼吸道和肺生态位提供了相似的选择压力的结果。抗生素也可以解释为什么随着疾病的进展,微生物群的多样性下降,而生态位的微生物群组成开始看起来更相似。另外,上呼吸道分泌物的微吸入和胃食管反流可能会导致群落混合,尽管临床上只有四分之一的患者使用抗反流药物治疗这一问题。
然而,在种群水平上,我们的研究表明,OP微生物群的组成和多样性比NP微生物群更接近于肺部微生物群,尽管我们观察到共生菌群(OP微生物群)和呼吸道病原体(NP微生物群)的合理起源之间存在区别。OP与肺的一致性普遍较高,这与以往的研究一致。16]和学龄儿童[39]和青少年[17虽然我们不能排除OP和BAL壁龛之间的相似是BAL手术过程中污染的结果,但在两个壁龛之间观察到的低个体内一致性似乎驳斥了这种可能性。
此外,我们得出的结论是,儿童肺部微生物群在8个月的时间间隔内发生了显著变化,并伴随着多样性的增加,这表明在一岁后半段,肺部微生物群仍有动态发展。从以前的研究中我们知道,在CF患者中,多样性在儿童时期达到顶峰,然后在生命的第二个十年中下降,那时它更频繁地由铜绿假单胞菌[1,40- - - - - -42].因此,这些数据支持这样的结论,即应获得常规样本,以准确了解CF患儿LRT中细菌的动态情况。
我们还将测序结果与常规培养结果进行了比较。几十年来,大量文献试图估计口咽培养肺定植的ppv和npv。1991年Ramsey等.[43的存在金黄色葡萄球菌,铜绿假单胞菌和流感嗜血杆菌在患有CF的幼儿口咽培养中,对肺运载(高PPV)具有高度预测性。他们还指出,上呼吸道中没有这些生物并不排除它们存在于下气道(低NPV)。然而,后来的研究在这个问题上产生了相互矛盾的信息[13,14].在我们的队列中,当测量每种细菌时,我们观察到两个上呼吸道壁龛的培养结果的高ppv和npv预测了肺定植。然而,更重要的是,在任何相应的上呼吸道生态位中,都很少观察到肺中完整的病原体,这强调了上呼吸道培养对肺定植的低预测价值。
总之,我们的数据表明,CF婴儿的肺部有自己独立的微生物群,似乎是由上呼吸道微生物群播下的种子,但并不完全相同。在本研究中,我们确定了多叶抽样的附加值[8,以破译肺部是否存在不同的生态系统。我们观察到个体婴儿不同叶的微生物群分布高度一致,这与健康个体的其他研究一致[44,45]和稳定CF的成年患者[46].这一发现可能是由于这一婴儿群体的肺部仍然相当健康:因此,在表型不稳定和/或慢性感染和炎症的CF患者中,随着时间的推移,单个肺叶可能会发展出自己的微生物群落和病原体。为了能够捕捉到这种现象,BAL程序可能比诱导痰样本更有优势。然而,在婴儿中,诱导痰样本可能是首选的,因为其低侵入性和可重复性。
本研究的优势在于我们使用16s - rrna测序来回答临床相关问题。使用这种技术,我们能够描述CF患儿上呼吸道和下呼吸道中完整的微生物群落,比传统培养更详细地描述了不同细菌的组成和相对贡献。此外,我们的分析集中在个体内而不是个体间测序和培养结果的一致性上,这与个体患者的临床决策密切相关。要理解我们的研究结果,我们的研究应该考虑到几个局限性。由于研究人群年龄较小且随访时间相对较短,因此不可能评估抗生素使用的潜在影响。为此,长期随访是必要的。其次,活体肺部分泌物的取样需要支气管镜通过口咽;尽管我们在BAL和实验室过程中采取了程序性预防措施,以避免口咽污染的混淆,但不能完全排除这种可能性。此外,由于环境污染可能会对我们的发现产生影响,我们还对“阴性”对照进行了测序,并随后将“阴性”对照与我们的最低密度样本聚在一起,表明环境和样本之间的交叉污染可以忽略不计。此外,由于我们只针对16S-rRNA基因的一个小片段,物种水平的注释是基于一个不确定性水平; targeting more or longer fragments could solve this limitation. Last, our patients were asymptomatic at the time of the BAL procedure; therefore, we cannot extrapolate our findings to situations of active infection.
总之,我们已经证明了上呼吸道和下呼吸道生态位微生物群之间的个体内一致性不一致。此外,我们已经证明,不同肺叶之间的微生物群是高度和谐的。最后,我们观察到一岁后半期肺微生物群的动态发展。需要做更多的工作来设计替代的、侵入性更小的方法来监测肺定植,如。通过对不咳痰的婴儿使用诱导痰。这些数据是否可以可靠地外推到有症状性CF的婴儿需要评估。此外,为了指导CF患儿(传染性)肺部疾病的治疗,我们建议研究微生物群分析在常规培养方法之上的附加效应。
补充材料
披露的信息
补充材料
H.A. Tiddenserj - 02235 - 2016 - _tiddens
确认
作者感谢所有参与的儿童及其家庭。我们感谢研究团队的所有成员,尤其是Annelotte Visser和Eveline Nieuwhof;实验室工作人员,特别感谢Raiza Hasrat, Jody van Engelsdorp Gastelaars和Cindy Kok;感谢合作机构对项目的贡献。
作者贡献:C.K. van der Ent, D. Bogaert, G.A. Tramper-Stranders和E.A.M. Sanders设计了这项研究。G.A. Tramper-Stranders, C.K. van der Ent和D. Bogaert撰写了研究方案。S.M.P.J. Prevaes、C.K. van der Ent、K.M. de温特-德格鲁特、H.M. Janssens和D. Bogaert负责招募参与者和收集样本。M.L.J.N. Chu和D. Bogaert负责qPCR和测序。S.M.P.J. Prevaes、D. Bogaert和W.A.A. de Steenhuijsen Piters负责序列的后处理和数据分析。所有作者都参与了稿件的数据解读和起草工作,对提交的最终稿件进行了认可,并同意对所有方面的工作负责。
脚注
这篇文章有补充资料可从www.qdcxjkg.com
支持声明:威廉敏娜儿童医院基金会和荷兰囊性纤维化基金会支持这项工作。资金来源在研究设计中没有作用;在数据的收集、分析和解释方面;在报告的写作中;或者决定提交论文发表。本文的资助信息已存入交叉参考基金注册.
利益冲突:可以在本文旁边的网站上找到信息披露www.qdcxjkg.com
- 收到了2016年5月17日。
- 接受2016年11月23日。
- 版权所有©ERS 2017











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}