





文摘
肺动脉平滑肌细胞(PA-SMC)增殖和炎症肺动脉高血压(多环芳烃)的关键组件。白介素(IL) 1β与IL - 1受体结合(R) 1,从而招募分子适配器骨髓分化主要响应蛋白88 (MyD88)(参与IL-1R1和toll样受体信号转导)和诱导IL - 1、IL - 6,通过核factor-κB激活肿瘤坏死factor-α合成。
我们调查了IL-1R1 / MyD88通路在肺动脉高压的发病机制。
标记IL-1R1和MyD88表达式主要PA-SMC疣状被发现在特发性肺动脉高压患者的肺,小鼠低氧诱导肺动脉高压和SM22-5-HTT+老鼠。海拔在肺IL-1βIL-1R1 MyD88和il - 6之前在小鼠缺氧肺动脉高压。IL-1R1−−/,MyD88−−/和控制老鼠的IL-1R1拮抗剂anakinra是保护同样对缺氧性肺动脉高压和血管周的巨噬细胞招聘。在SM22-5-HTT Anakinra逆转肺动脉高压部分+在monocrotaline-treated明显老鼠和老鼠。IL-1β-mediated鼠标PA-SMC增长的刺激是被anakinra IL-1R1缺席−−/和MyD88−−/老鼠。基因缺失局限于骨髓血统(M。lys-Cre MyD88fl / fl老鼠)肺动脉高压程度下降与控制,表明IL-1β-mediated PA-SMCs和巨噬细胞的影响。媒体的促生长效果受制于M1和M2巨噬细胞从M。lys-Cre MyD88fl / fl老鼠是衰减的。
在肺动脉高压肺血管重塑和炎症需要IL-1R1 / MyD88信号。针对IL-1β/ IL-1R1通路可能持有承诺治疗人类的多环芳烃。
文摘
IL-1R1 / MyD88通路是肺动脉高血压治疗的目标http://ow.ly/1Fpe3008RLs
介绍
慢性炎症被认为是各种形式的肺动脉高压的特征,包括人类肺动脉高血压(PAH) [1- - - - - -4]。然而,肺动脉高压和炎症之间的关系知之甚少。炎症可能发生二次事件过程中肺动脉高压,导致肺血管细胞增殖的能力分泌炎症介质,产生炎性微环境5- - - - - -7]。在肺动脉高压的动物模型,炎症先于血管重塑,这表明改变免疫可能是一个主要事件的开发过程中肺动脉高压(8]。复杂的过程,开始在非感染性炎症条件涉及到模式识别受体细胞质核苷酸等寡聚化域(点头)同受体,组装成高分子量分子平台称为inflammasomes和膜结合toll样受体(通常)[9]。TLR信号的结果之一是激活的转录因子核转录因子(NF) -κB,触发生产的白介素(IL) 1β,肿瘤坏死factor-α,IL - 6 (10]。还存在Inflammasomes,通过激活促炎症,特别是caspase-1,导致的成熟和分泌IL-1β和地震11]。作为一个主要玩家在先天免疫反应所涉及的复杂的过程,IL-1β被贴上“炎症的看门人”(12,13]。
il - 1受体配体结合细胞组成的复杂的il - 1受体1 (IL-1R1)和IL-1R辅助蛋白。自然被捕获IL-1R1分子拮抗剂IL-1Ra行为(14,15]。IL-1R1与最常见的信号通路的tlr招聘骨髓分化主要响应蛋白88 (MyD88) [16,17),这是一个关键的适配器在先天免疫信号转导蛋白,导致NF-κB激活(18,19]。这种效应解释说,IL-1β可以诱导自己的合成通过IL-1R1 / MyD88-mediated NF-κB激活。因此,IL-1βIL-1R1和MyD88关键行动者可能发生在肺动脉高压的先天免疫反应,导致炎症和肺血管重塑。然而,的潜在作用IL-1R1 / MyD88通路在肺动脉高压没有专门检查。在以前我们和其他人的研究(20.- - - - - -22),IL-1β显示作为有效的促有丝分裂的因素培养肺动脉平滑肌细胞(PA-SMCs)。此外,有证据表明,老鼠地缺乏保护对肺动脉高压的发展(23),血小板地激活导致肺动脉高压(24]。在大鼠暴露于野百合碱,导致肺部炎症和PAH,重组IL-1R拮抗剂anakinra降低肺动脉高压程度(25]。最后,提高多环芳烃被报道在病人anakinra治疗成人耳氏病(26]。
在这里,我们调查了在几个gene-deficient IL-1R1 / MyD88通路在肺动脉高压小鼠模型。鉴于我们的发现表明IL-1R1和MyD88强烈表达改建船舶从特发性患者多环芳烃(iPAH),我们IL-1R1调查−−/和MyD88−−/小鼠暴露于低氧,我们评估了anakinra对野生型的影响(WT)小鼠暴露于低氧;以及SM22-5HTT+小鼠和大鼠野百合碱处理。确定肺动脉高压感应IL-1R1或MyD88涉及直接影响PA-SMCs或巨噬细胞激活,需要我们研究培养PA-SMCs和评估肺动脉高压程度在转基因小鼠MyD88基因缺失局限于淋系巨噬细胞(M。lys-Cre MyD88fl / fl老鼠)。最后,我们研究了条件的影响,媒体从差异化M1和M2巨噬细胞WT和M。lys-Cre MyD88fl / fl老鼠PA-SMCs扩散。
方法
人类组织样本的收集
肺组织获得6 iPAH患者接受肺移植在大学医疗Ziekenhuizen(比利时鲁汶)。控制肺组织收集了八个病人肺切除手术局部肺肿瘤研究所Mutualiste Montsouris(法国巴黎)。每个机构的伦理委员会批准协议的集合。
动物
所有动物实验机构的动物保健和使用委员会批准的法国国家健康与医学研究。转基因小鼠持续删除IL-1R1 (IL-1R1−−/)或MyD88 (MyD88−−/)获得从理查德Flavell(免疫生物学、医学院的耶鲁大学纽黑文,CT,美国)和Shizuo彰(实验室的宿主防御,WPI免疫学前沿研究中心(IFReC)、大阪大学,大阪,日本)(27,28]。老鼠MyD88基因缺失仅限于myeloid-cell血统(M。lys-Cre MyD88fl / fl老鼠)生成BL6胚胎干细胞在国家科学研究中心(CNRS, UMR7355、奥尔良、法国)。转基因小鼠5 -羟色胺转运体(http)过度smc (SM22-5HTT+)生产和饲养如前所述29日]。WT C57BL / 6 j小鼠和Wistar鼠获得Janvier (Le Genest-Saint-Isle、法国)。
在线补充材料提供了动物实验的细节,anakinra治疗,巨噬细胞隔离和两极分化,PA-SMC扩散实验,分析IL-1β/ IL-1R1 MyD88 inflammasome通路和统计分析。
结果
增加表达IL-1R1和MyD88 iPAH患者的肺与肺动脉高压和老鼠
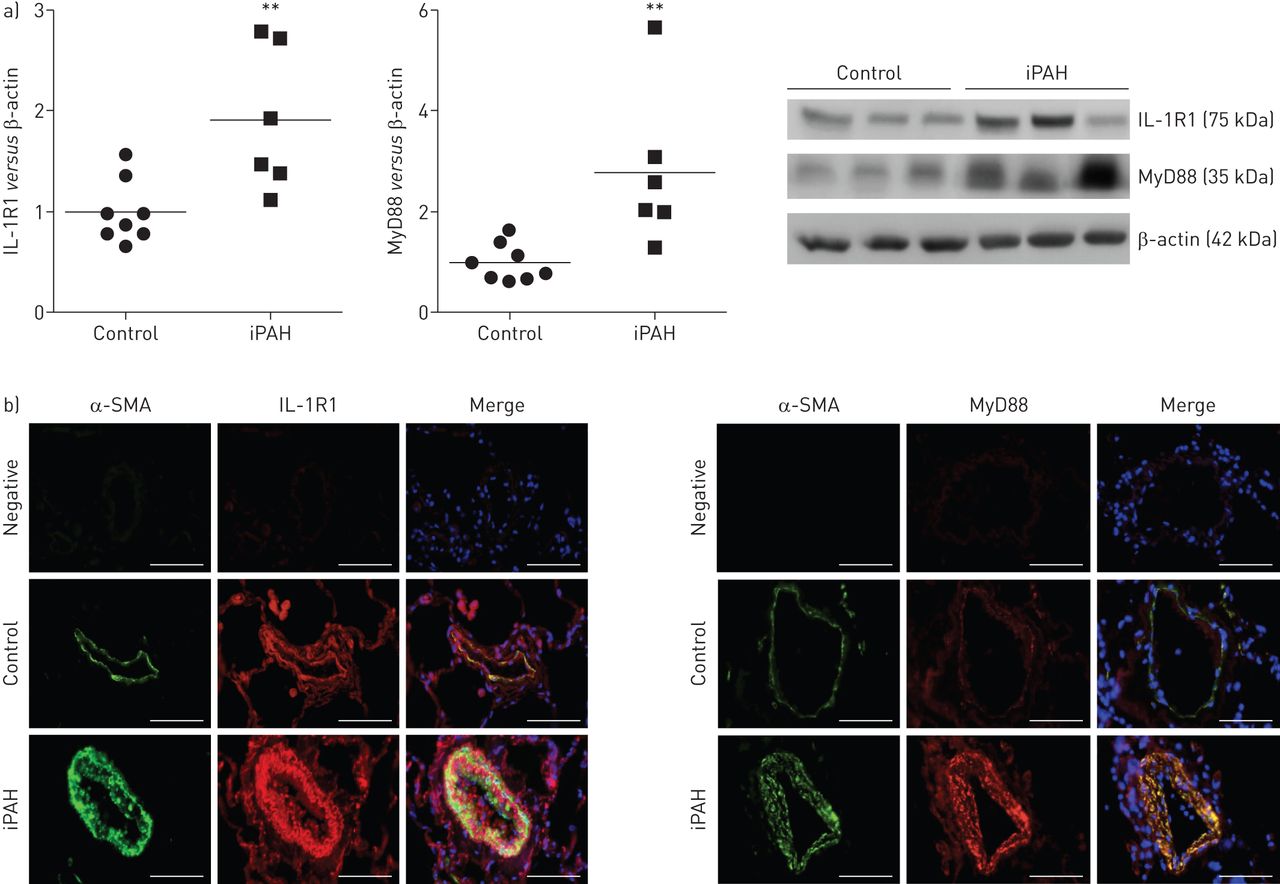
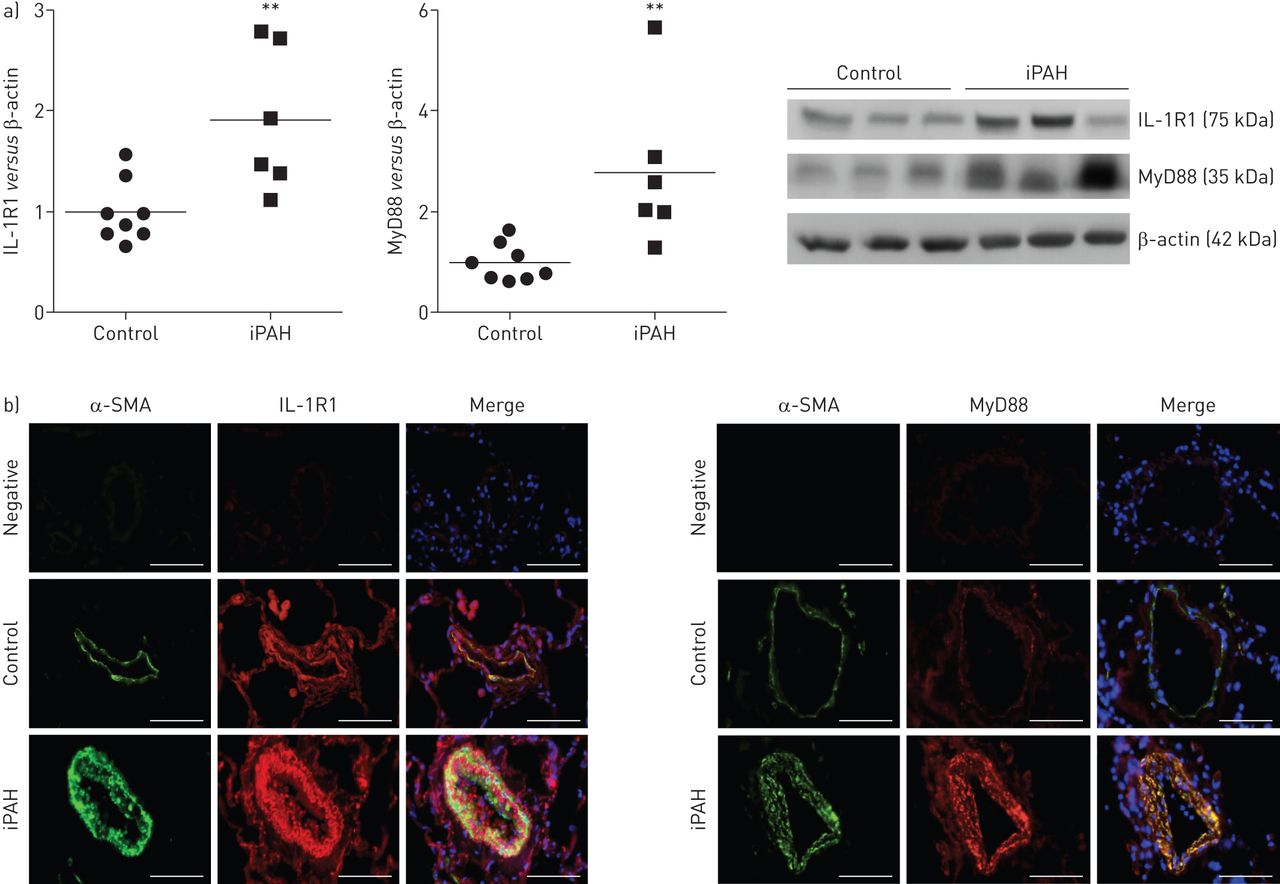
iPAH患者肺组织中,我们发现显著增加IL-1R1和MyD88蛋白水平相比,控制肺组织(图1一个)。免疫荧光染色IL-1R1和MyD88主导PA-SMCs媒体过分生长的肺血管、如图所示为α-smooth double-immunofluorescence染色肌肉肌动蛋白和IL-1R1或MyD88 (图1 b)。
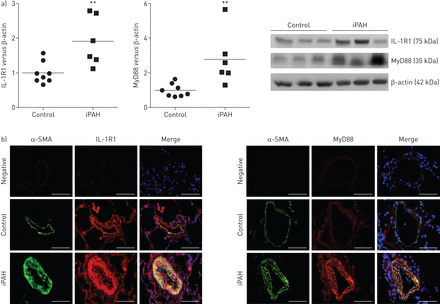
增加表达的受体1 (IL-1R)及骨髓分化主要响应蛋白88 (MyD88)在人类肺动脉高血压(PAH)。)IL-1R1 MyD88蛋白水平衡量蛋白质印迹总共lung-protein提取物6特发性(i) PAH患者和8岁,sex-matched控制。* *:p < 0.01。b) IL-1R1和MyD88 co-localise平滑肌细胞。代表病人的肺组织显微图和控制。IL-1R1或MyD88(红色),α-smooth肌肉肌动蛋白(SMA)(绿色)为平滑肌细胞染色,或赫斯特染细胞核染色(蓝色)。没有检测到阳性免疫反应性部分孵化与适当的控制免疫球蛋白其次是二级anti-rabbit和anti-mouse抗体。酒吧= 50μm规模。
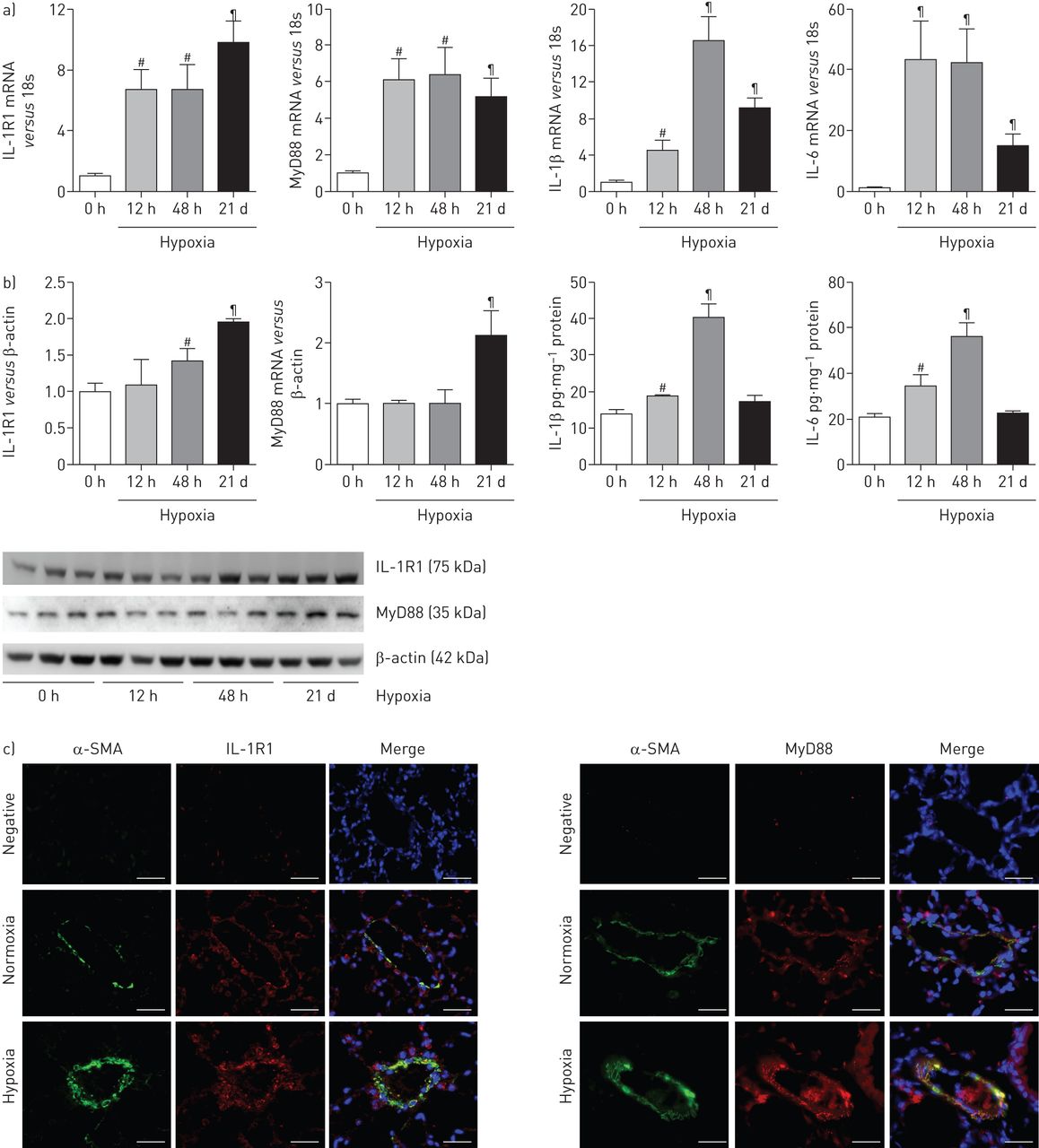
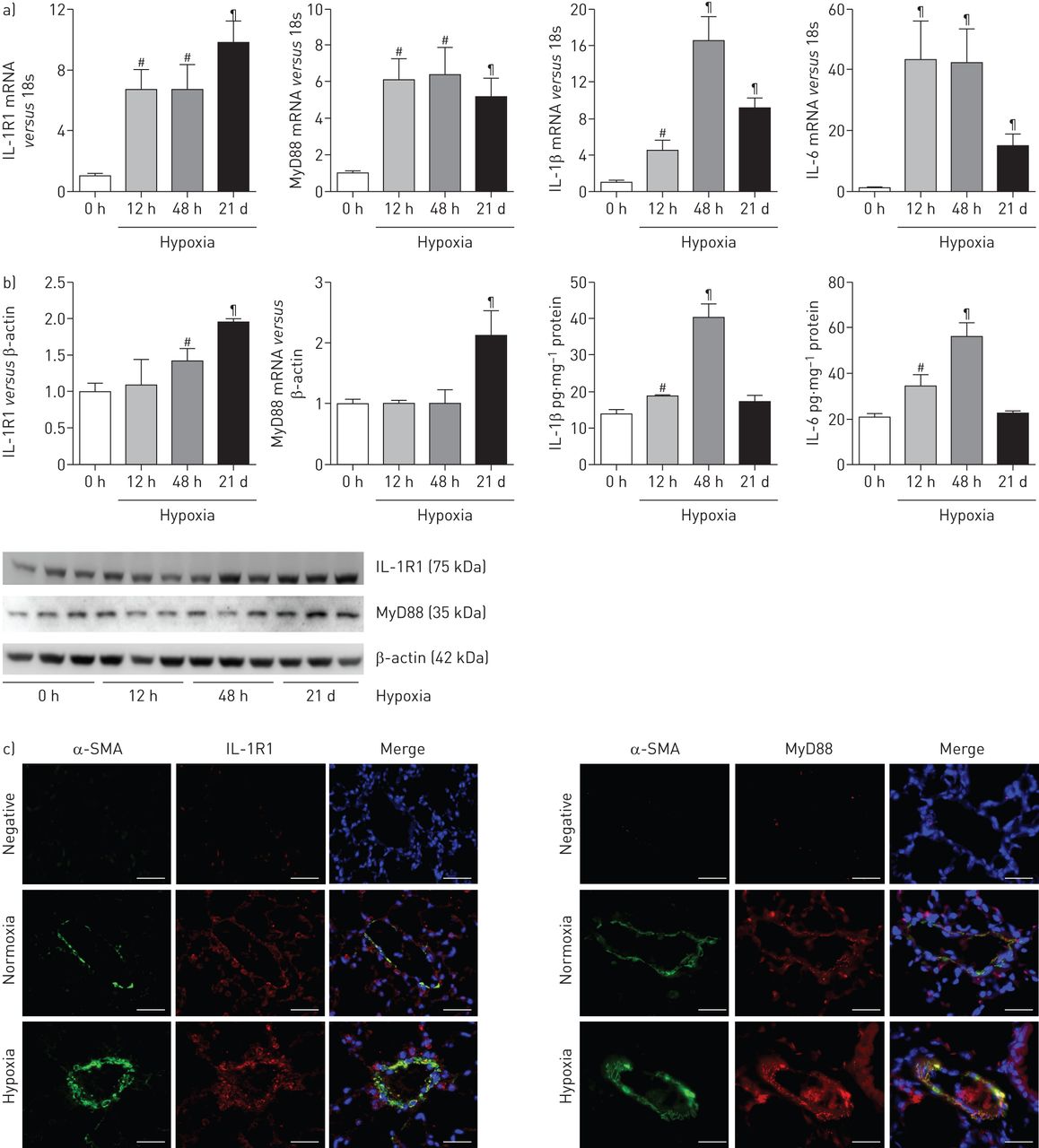
在缺氧小鼠暴露于21天,我们发现大量增加肺IL-1R1和MyD88 mRNA和蛋白水平与常氧小鼠相比图2一个),优势IL-1R1和MyD88疣状改建船舶PA-SMC层(图2 c)。我们没有发现疣状从IL-1R1 IL-1R1或MyD88肺−−/或MyD88−−/小鼠,分别(在线补充图S1)。有趣的是,肺IL-1R1和MyD88 mRNA增加早期缺氧暴露期间,在肺动脉高压的发展。肺IL-1β信使rna和蛋白质含量达到48小时然后下降,与蛋白质回到控制白天21(常氧水平图2 b)。il - 6水平平行IL-1β水平。
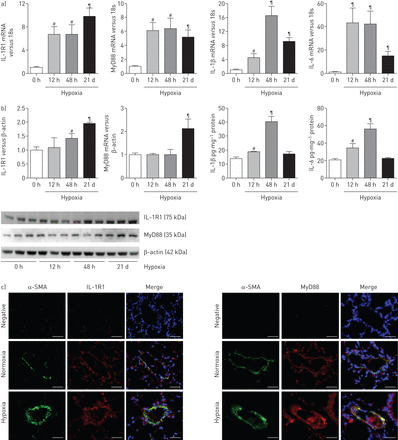
interleukin-1受体表达增加(IL-1R) 1 /骨髓分化主要响应蛋白88 (MyD88)途径在小鼠低氧诱导肺动脉高血压(PAH)。)IL-1R1、MyD88 IL-1β和il - 6 mRNA水平测量使用定量实时聚合酶链反应从小鼠肺部缺氧暴露后不同时间(12 h, 48 h或21天(d))。b)肺IL-1R1和MyD88蛋白水平用免疫印迹和肺IL-1β和il - 6蛋白水平使用ELISA缺氧暴露后不同时间测定。数据均值±扫描电镜六个动物。#:p < 0.016;¶:p < 0.0033,而对照小鼠暴露于normoxia值。c)代表从小鼠肺组织的显微图21 d后在normoxia和缺氧。IL-1R1或MyD88(红色),α-smooth肌肉肌动蛋白(SMA)(绿色)为平滑肌细胞染色,或赫斯特细胞核染色(蓝色)。没有检测到免疫反应性与兔免疫球蛋白部分孵化控制和二次anti-rabbit抗体。酒吧= 30μm规模。
同样,SM22-5HTT+小鼠自发肺动脉高压肺IL-1R1增加和展出MyD88 mRNA水平控制WT老鼠相比,尽管在常氧条件下研究(在线补充图S2A)。
影响IL-1R1或MyD88基因缺失和anakinra治疗小鼠缺氧肺动脉高压
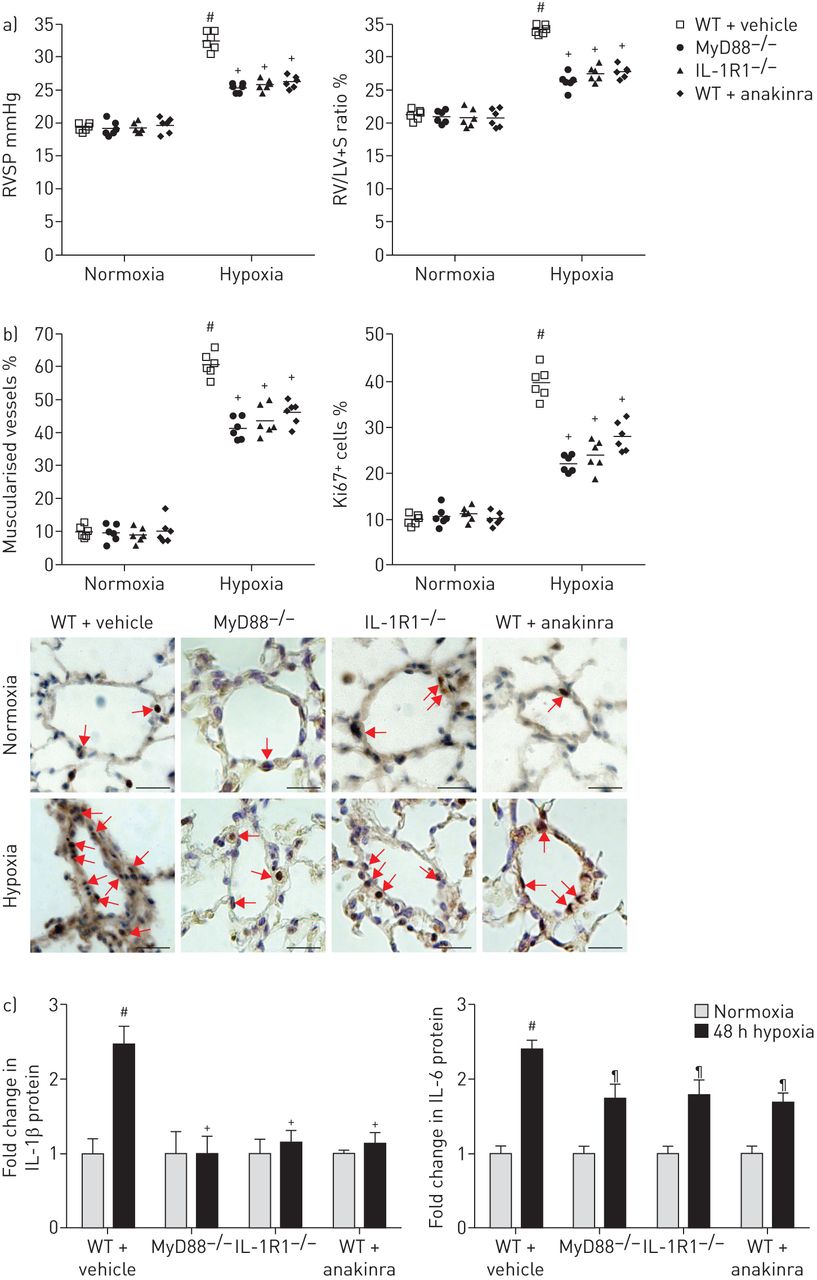
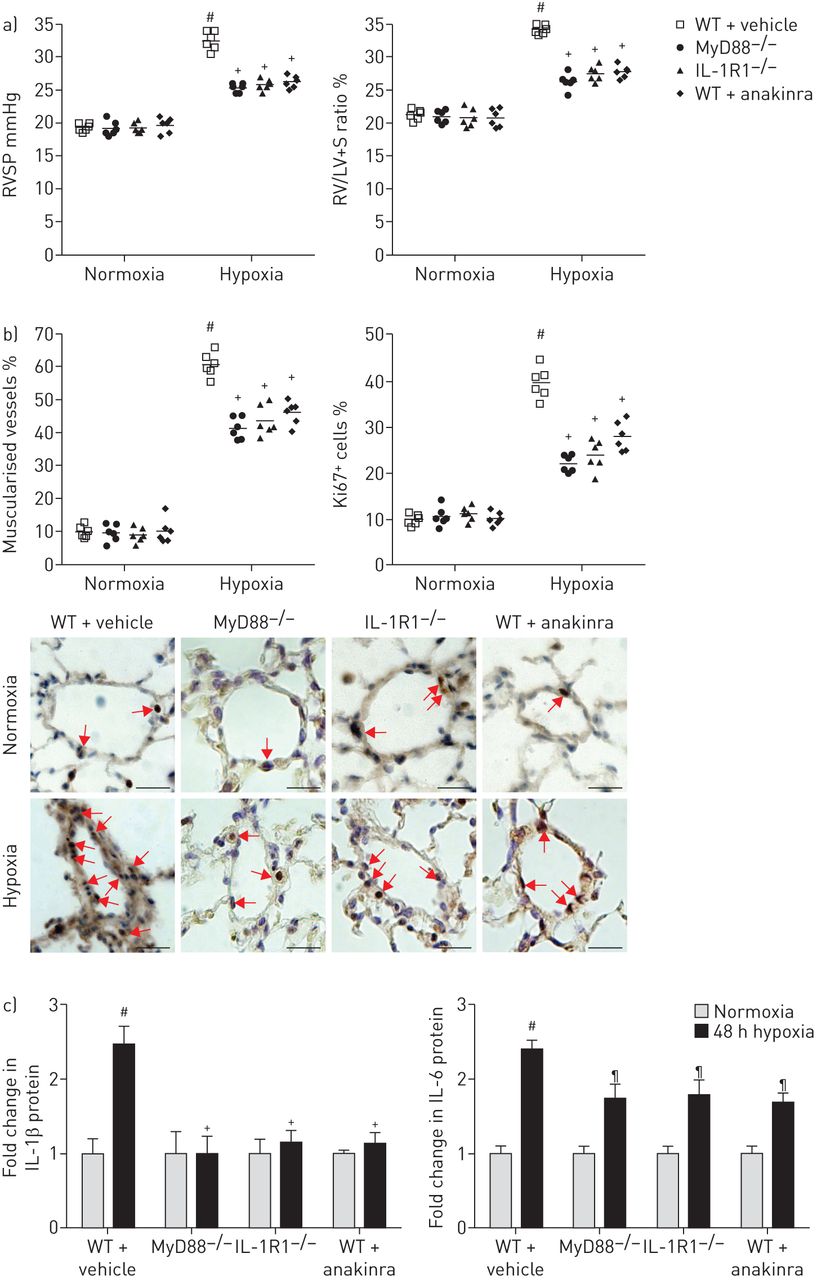
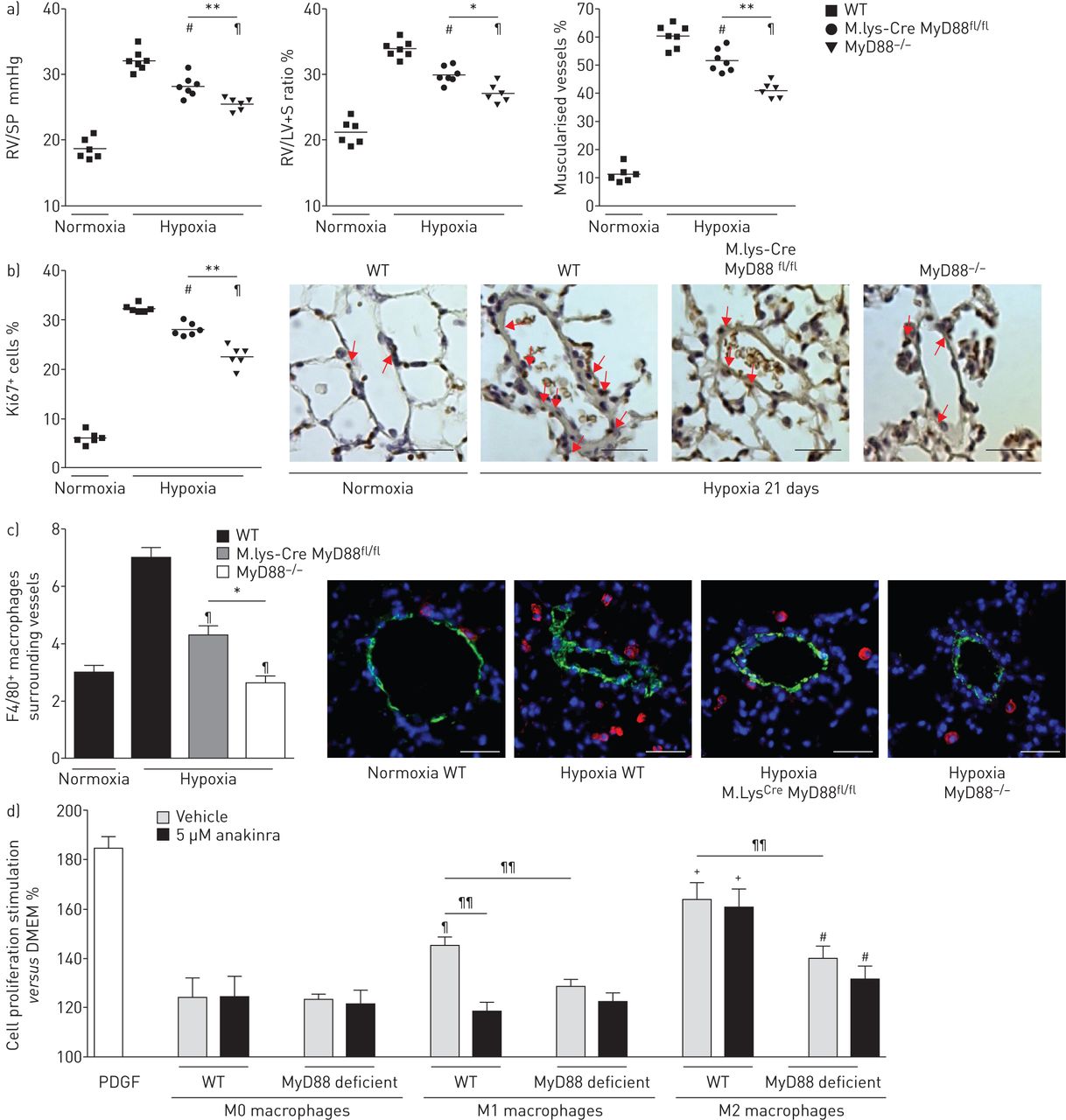
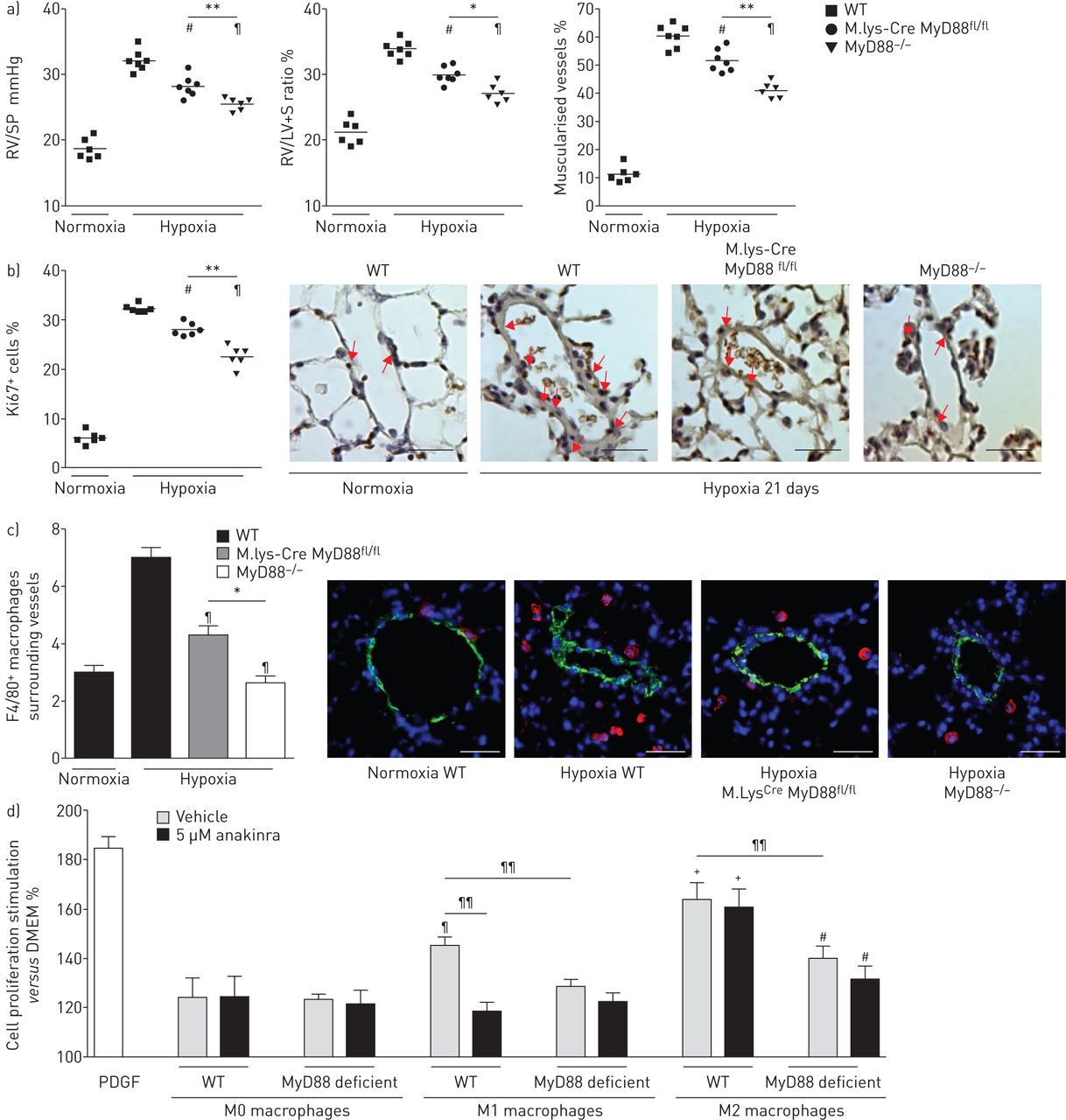
21天的缺氧暴露之后,右心室(RV)收缩压明显降低和房车肥大IL-1R1那么严重−−/和MyD88−−/老鼠比控制WT老鼠(图3)。此外,远端肺血管不突变小鼠的肌肉比WT老鼠,小分裂Ki67的百分比+肺血管细胞(图3 b)。严重肺动脉高压IL-1R1之间没有明显不同−−/和MyD88−−/老鼠。缺氧WT小鼠每日anakinra(20毫克·公斤−1)表现出类似的对肺动脉高压程度的保护,IL-1R1一样−−/和MyD88−−/老鼠(图3和b)。肺IL-1β高峰出现在抑制突变小鼠和WT老鼠anakinra处理,降低局部肺il - 6水平(图3 c)。因此,肺的水平磷酸化NF-κB p65,这显著增加在WT小鼠缺氧,在IL-1R1减少−−/和MyD88−−/老鼠,以及WT小鼠3周后接受anakinra缺氧(在线补充图S3)。相比之下,水平的提高下游inflammasome组件,包括对asc(凋亡speck-like蛋白质包含一张卡片),地震和caspase-1,仍然在IL-1R1升高−−/和MyD88−−/老鼠老鼠相比WT(在线补充图S3A和B)。
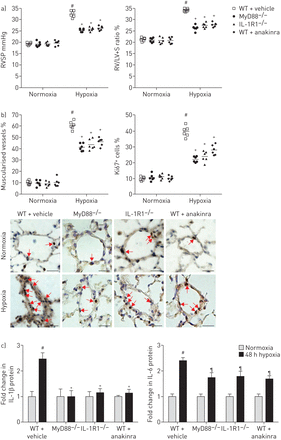
MyD88(骨髓分化主要响应蛋白88)基因缺陷,IL-1R1(受体1)基因缺陷或anakinra-induced IL-1R1抑制同样废除缺氧肺动脉高血压的发展(多环芳烃)的老鼠。a、b)右心室收缩压(RVSP)和右心室肥大指数(右心室(RV) /左心室(LV) +隔重量(S)),肺血管muscularisation分裂Ki67+野生型细胞(WT)小鼠每日车辆,MyD88−−/老鼠,IL-1R1−−/老鼠和WT小鼠每日anakinra (20 mg·公斤−1),在normoxia和缺氧(21天)。代表显微图Ki67的肺血管染色。红色箭头显示Ki67+细胞核。没有检测到免疫反应性与兔免疫球蛋白部分孵化控制和二次anti-rabbit抗体。酒吧= 40μm规模。c)肺IL-1β和il - 6蛋白水平测量使用ELISA 48 h后缺氧暴露。数据意味着±扫描电镜六个动物。#:p < 0.005相比值控制小鼠暴露于normoxia;¶:p < 0.0166,+:p < 0.0033相比值控制小鼠暴露于低氧。
IL-1β和anakinra对增长的影响从IL-1R1 PA-SMCs−−/和MyD88−−/老鼠相比,控制WT老鼠
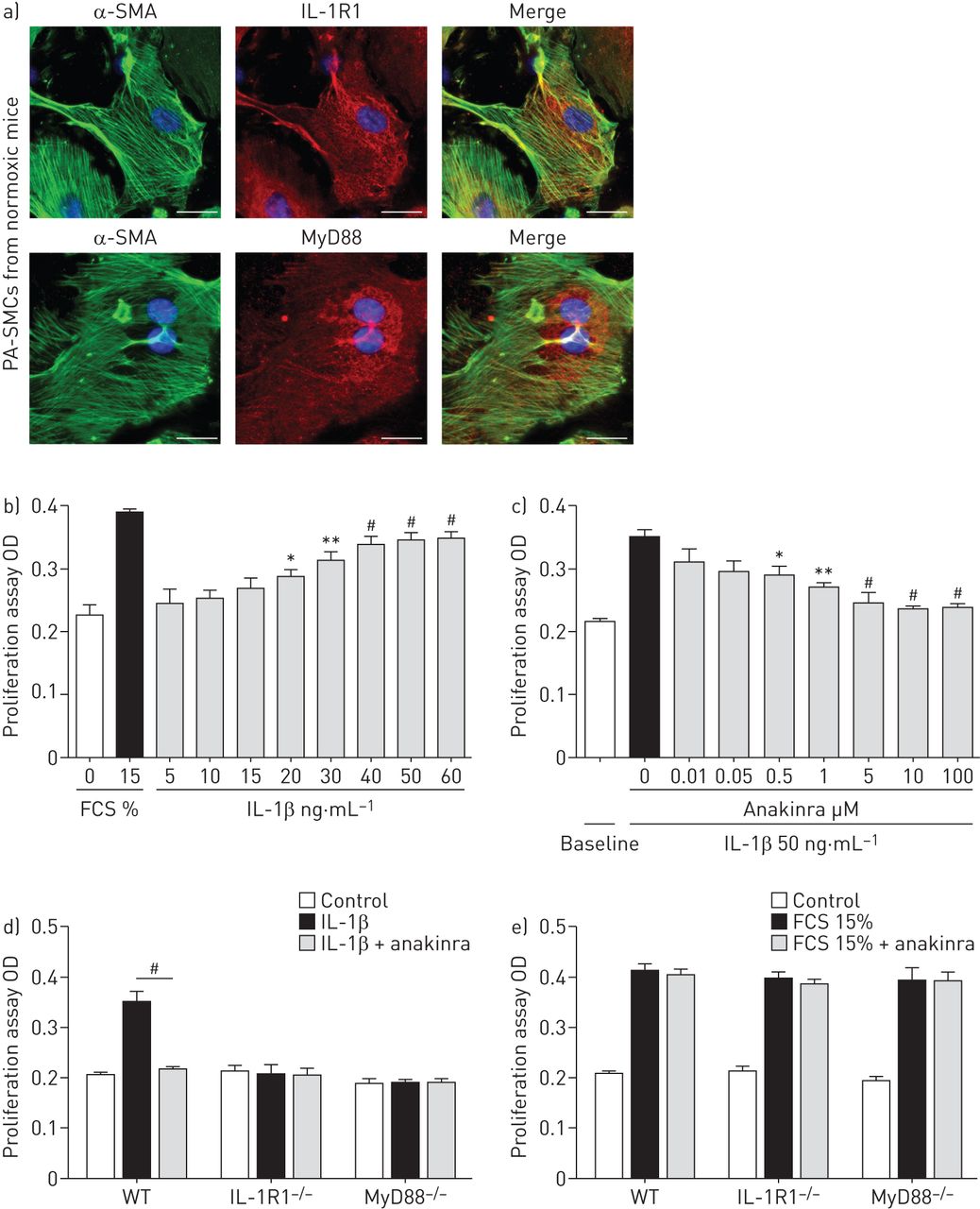
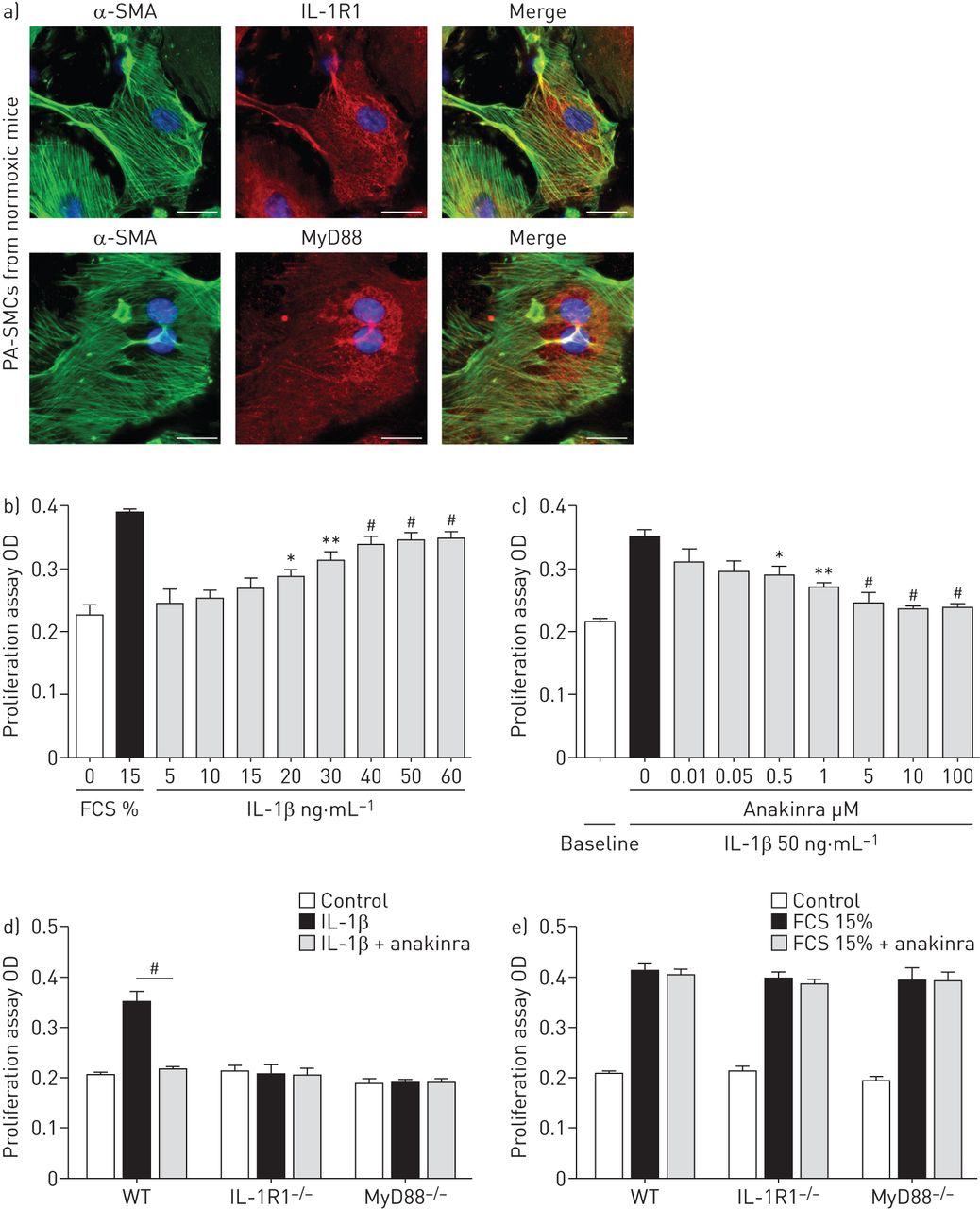
从常氧培养PA-SMCs WT老鼠表现出强烈的免疫染色IL-1R1和MyD88 (图4)。IL-1β治疗PA-SMCs WT老鼠刺激细胞生长的剂量依赖性的方式(图4 b),这种效应被anakinra抑制(图4摄氏度和在线补充图S4)。与人类相似的结果观察PA-SMCs(在线补充图S5A)。从IL-1R1 IL-1βPA-SMCs没有影响−−/或MyD88−−/老鼠和anakinra(5µM)没有产生额外的抑制(图4 d)。血小板源生长因子的刺激作用在从IL-1R1 PA-SMCs不变−−/或MyD88−−/老鼠和影响anakinra (图4 d和e)。
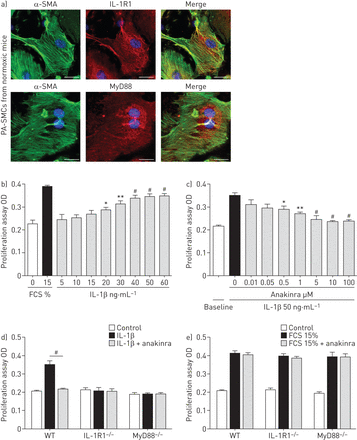
影响interleukin-1受体(IL-1R) 1激活IL-1β刺激对大鼠肺动脉平滑肌细胞增殖(PA-SMCs)。)代表的显微图PA-SMCs控制常氧老鼠。IL-1R1或MyD88(红色),α-smooth肌肉肌动蛋白(SMA)(绿色)和赫斯特对细胞核染色(蓝色)。没有检测到免疫反应性与兔免疫球蛋白部分孵化控制和二次anti-rabbit抗体。酒吧= 50μm规模。b)剂量反应曲线的影响IL-1β鼠标PA-SMC扩散(5-60 ng·毫升−1)。数据意味着±扫描电镜12到16值至少两个不同的实验。OD:光密度;FCS:胎牛血清。*:p < 0.05;* *:p < 0.01;#:non-stimulated细胞相比,p < 0.005。c)剂量反应曲线的影响anakinra(0.01 -100μM)鼠标PA-SMC扩散引起IL-1βng(50毫升−1)。数据意味着±扫描电镜12到16值至少两个不同的实验。*:p < 0.05;* *:p < 0.01;#:p < 0.005相比,细胞刺激与IL-1β孤单。d, e)扩散PA-SMCs从野生型(WT) IL-1R1−−/和MyD88−−/老鼠刺激IL-1βng(50毫升−1)或15% FCS有或没有anakinra(5μM)。数据意味着±扫描电镜在18到22岁的值至少三个不同的实验。#控制细胞相比:p < 0.005。
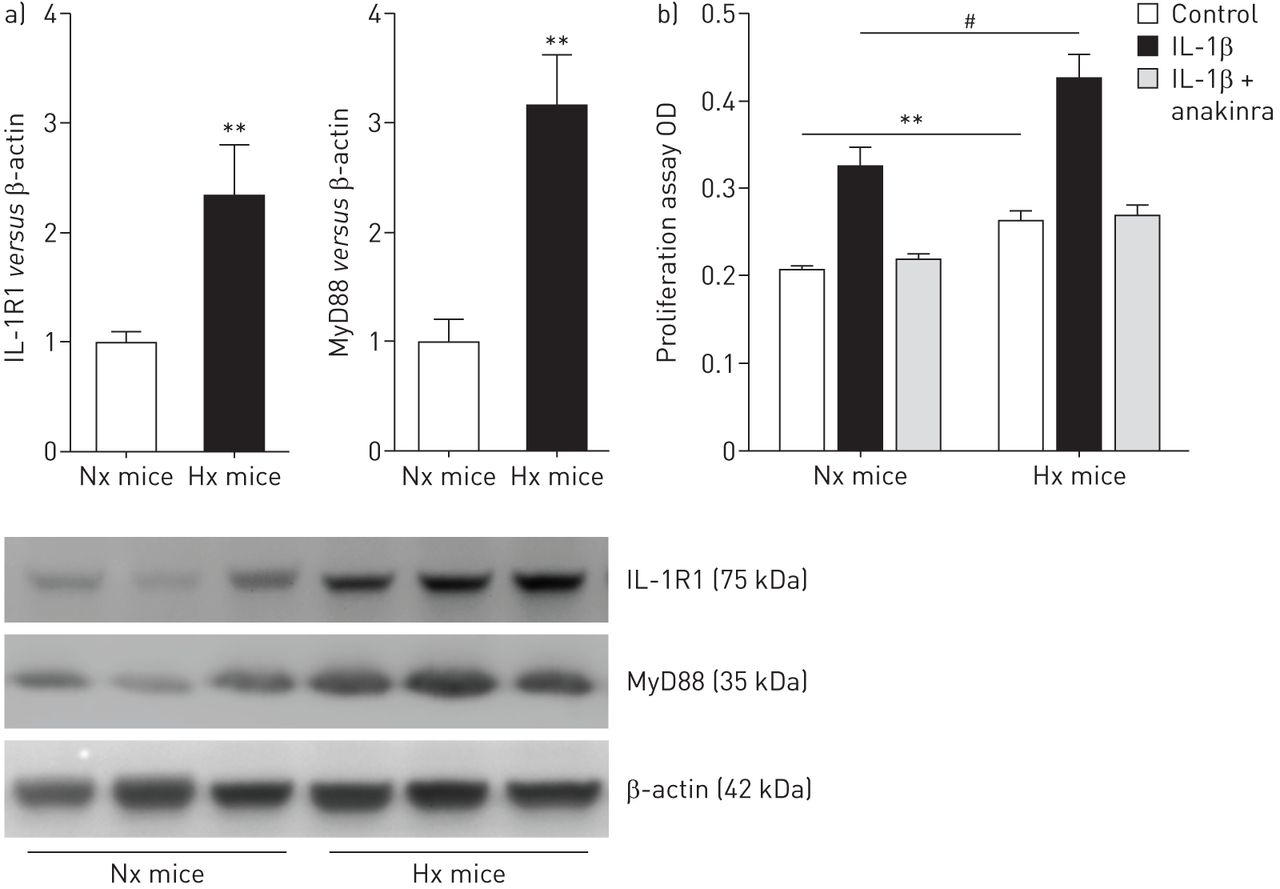
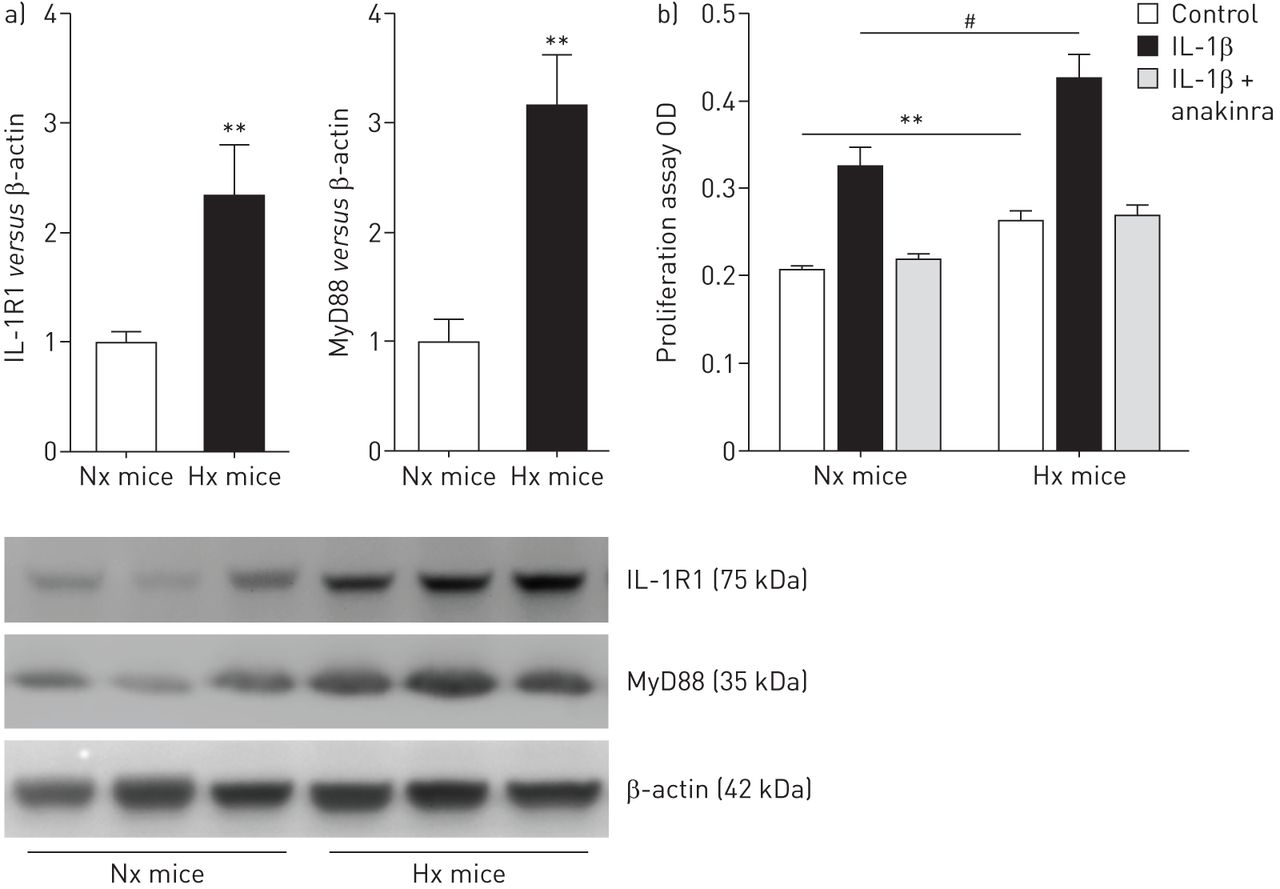
IL-1R1 / MyD88途径激活坚持PA-SMCs从小鼠暴露于慢性缺氧(图5)。因此,慢性缺氧小鼠PA-SMCs显示增加的增长来自控制老鼠相比,但这不是废除增加anakinra (图5 b)。正如所料,anakinra抑制NF-κB的激活与IL-1βPA-SMC治疗引起的。没有变化在ASC,地震和caspase-1蛋白质含量(在线补充图S6)。
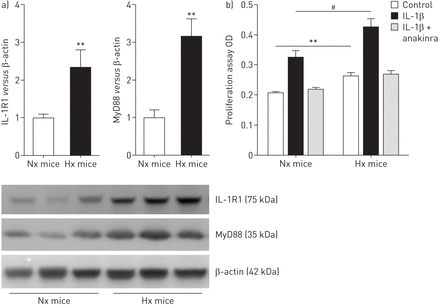
interleukin-1受体表达增加(IL-1R) 1 /骨髓分化主要响应蛋白88 (MyD88)通路和肺动脉平滑肌细胞增殖(PA-SMCs)小鼠暴露于低氧。)IL-1R1 MyD88蛋白水平测量使用西方墨点法在常氧PA-SMCs (Nx)小鼠或缺氧(Hx)老鼠。数据意味着±扫描电镜蛋白质含量在PA-SMCs七只老鼠。b)扩散PA-SMCs常氧或缺氧小鼠,刺激IL-1βng(50毫升−1)有或没有anakinra(5μM)。数据意味着±扫描电镜在18到22岁的值至少三个不同的实验。OD:光密度。* *:p < 0.01;#:p < 0.005。
贡献的巨噬细胞通过IL-1R1 / MyD88肺动脉高压的发展途径
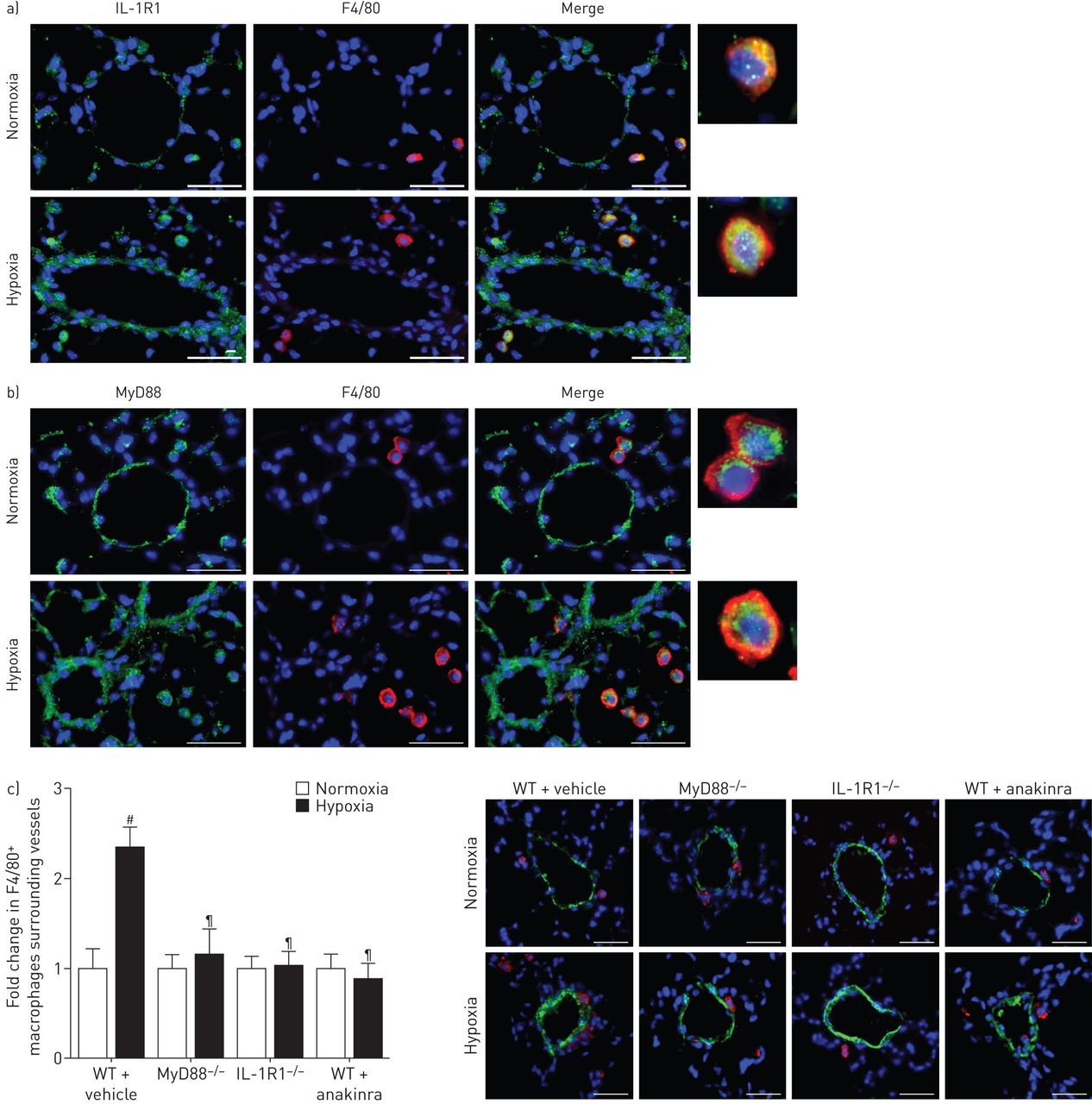
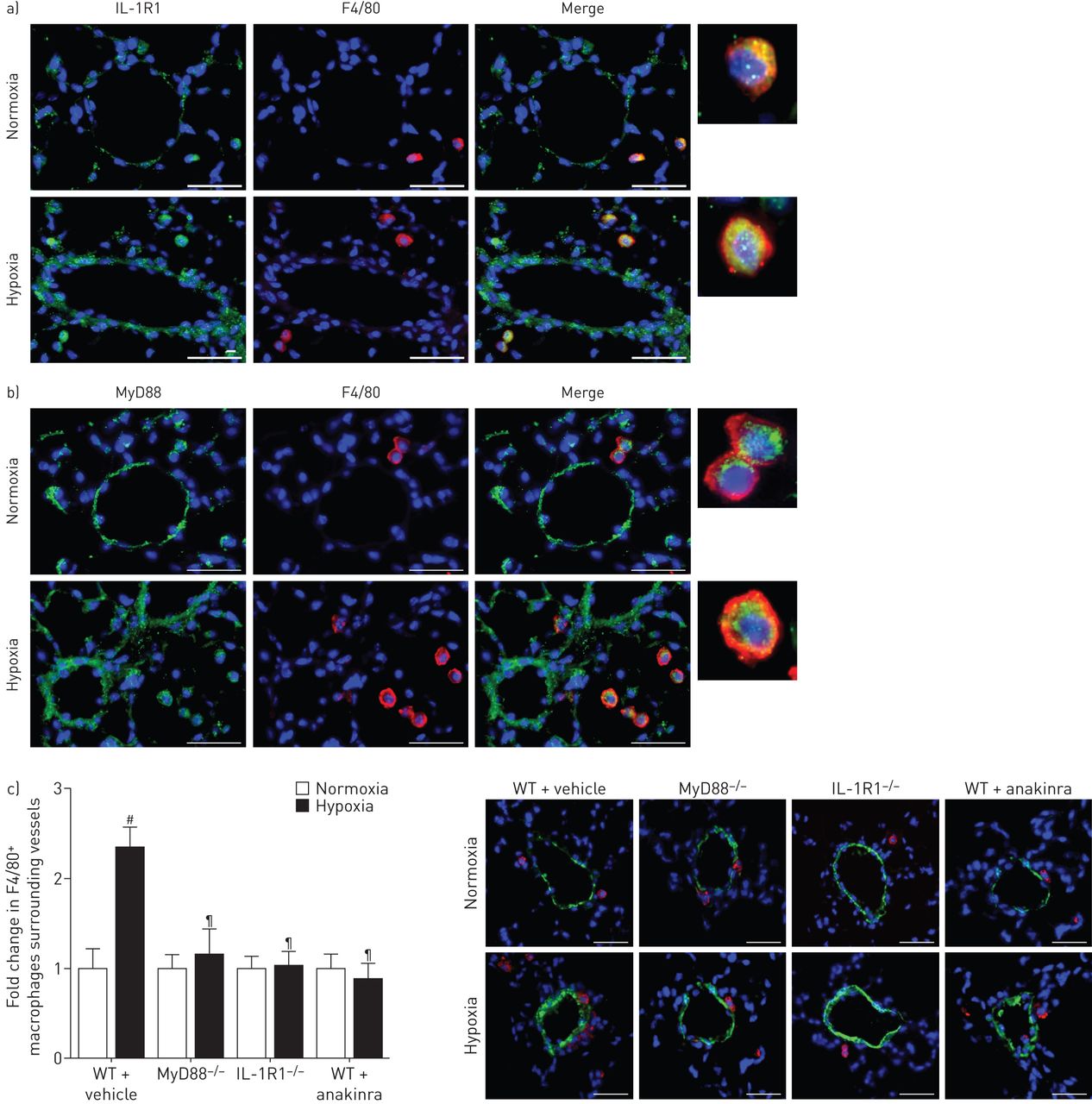
双重免疫荧光染色和F4/80 IL-1R1或MyD88显示血管周的IL-1R1 MyD88-stained巨噬细胞缺氧小鼠的肺,而在常氧小鼠染色很微弱(图6和b)。血管周的巨噬细胞的数量没有增加从normoxia在缺氧IL-1R1缺氧−−/或MyD88−−/老鼠老鼠或anakinra-treated WT (图6 c)。
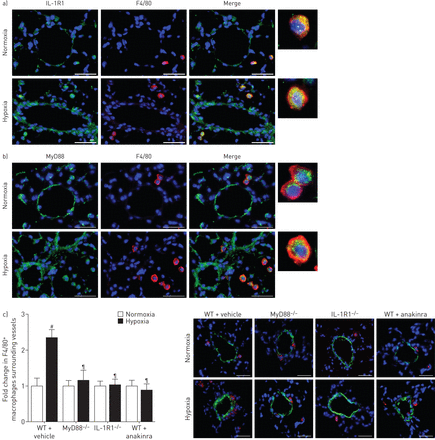
MyD88(骨髓分化主要响应蛋白88)基因缺陷,IL-1R1(受体1)基因缺陷或anakinra-induced IL-1R1抑制同样降低了血管周的巨噬细胞积累观察小鼠低氧诱导肺动脉高血压。a、b) IL-1R1 MyD88表达与F4/80巨噬细胞染色。肺部分从野生型小鼠(WT)下normoxia经过21天的缺氧是彩色IL-1R1或MyD88(绿色)和F4/80(红色)。核与赫斯特染色染料(蓝色)。c)肺小血管周围巨噬细胞计数。船只使用α-smooth肌肉肌动蛋白(SMA)染色法检测。图描绘了血管周围F4/80褶皱变化+巨噬细胞之间normoxia和缺氧。巨噬细胞的显微图显示代表染色周围肺血管在WT小鼠每日车辆,MyD88−−/老鼠,IL-1R1−−/老鼠和WT小鼠每日anakinra (20 mg·公斤−1),在21天的缺氧。没有检测到阳性免疫反应性与控制部分孵化免疫球蛋白辅助anti-rabbit或anti-rat抗体紧随其后。酒吧= 30µm规模。#:p < 0.005 WT常氧控制老鼠相比;¶:控制WT缺氧小鼠相比p < 0.0033。
进一步探索IL-1R1 / MyD88信号在巨噬细胞的作用,我们在转基因小鼠诱导缺氧肺动脉高压MyD88基因缺失局限于骨髓血统(M。lys-Cre MyD88fl / fl老鼠),MyD88相比−−/和WT老鼠。21天缺氧暴露后,肺动脉高压程度降低了M。lys-Cre MyD88fl / fl老鼠控制WT老鼠相比,但在MyD88不一样的程度−−/老鼠(图7和b)。此外,缺氧暴露后巨噬细胞周围的肺血管的数量在M较小。lys-Cre MyD88fl / fl老鼠比WT老鼠,但高于MyD88−−/老鼠(图7 c)。
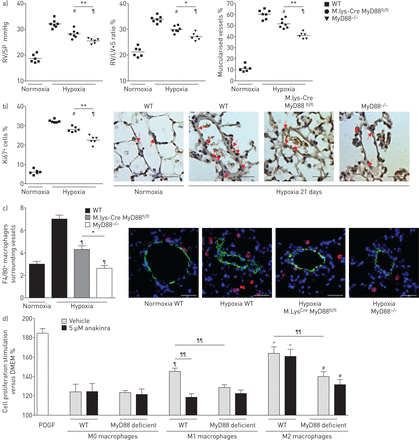
巨噬细胞肺高血压发展的贡献通过interleukin-1受体(IL-1R) 1 /骨髓分化主要响应蛋白88 (MyD88)通路。a、b)图的右心室收缩压(RVSP)和右心室肥大指数(右心室(RV) /左心室(LV) +隔重量(S)),肺血管muscularisation分裂Ki67+在野生型小鼠(WT)细胞,M。lys-Cre MyD88fl / fl老鼠和MyD88−−/老鼠在normoxia和缺氧(21天)。代表显微图Ki67的肺血管染色。箭头显示Ki67+细胞。酒吧= 40µm规模。c)巨噬细胞计数肺小血管周围WT老鼠,M。lys-Cre MyD88fl / fl老鼠和MyD88−−/老鼠在normoxia和缺氧(21天)。显微图显示巨噬细胞周围的肺血管染色:代表F4/80(红色);α-smooth肌肉肌动蛋白(绿色);赫斯特染色(核)(蓝色)。没有检测到阳性免疫反应性与控制部分孵化免疫球蛋白辅助anti-rabbit或anti-rat抗体紧随其后。酒吧= 40µm规模。#:p < 0.025,¶:p < 0.005相比值控制小鼠暴露于低氧。*:p < 0.05;* *:p < 0.01。d)影响肺动脉平滑肌细胞(PA-SMC)扩散M0、M1、M2 macrophage-conditioned媒体WT或者M。lys-Cre MyD88fl / fl老鼠。PA-SMCs来自WT老鼠和与anakinra预处理(5µM)或车辆。数据均值±扫描电镜10 - 15的值从至少两个不同的实验。#:p < 0.025,¶:p < 0.005,+:p < 0.0025而引起的扩散M0 WT macrophage-conditioned媒体;¶¶:p < 0.005。
巨噬细胞诱导扩散通过IL-1βPA-SMCs / IL-1R1 / MyD88通路
我们评估的影响从骨骨髓来源培养巨噬细胞条件培养液PA-SMC增长。的媒体从M0巨噬细胞诱导轻微PA-SMC增殖,细胞WT和M没有区别。lys-Cre MyD88fl / fl老鼠和没有anakinra的影响(图7 d)。条件媒体M1巨噬细胞诱导大PA-SMC增长比媒体从M0刺激巨噬细胞,这种效应被anakinra部分抑制或衰减时使用巨噬细胞从M。lys-Cre MyD88fl / fl老鼠。最后,M2 macrophage-conditioned媒体WT强PA-SMC扩散诱发小鼠相比,媒体受制于M0、M1巨噬细胞。这不是由anakinra抑制刺激影响,但当条件是减少媒体M。lys-Cre MyD88fl / fl老鼠使用(图7 d)。
anakinra对monocrotaline-induced肺动脉高压大鼠的进展
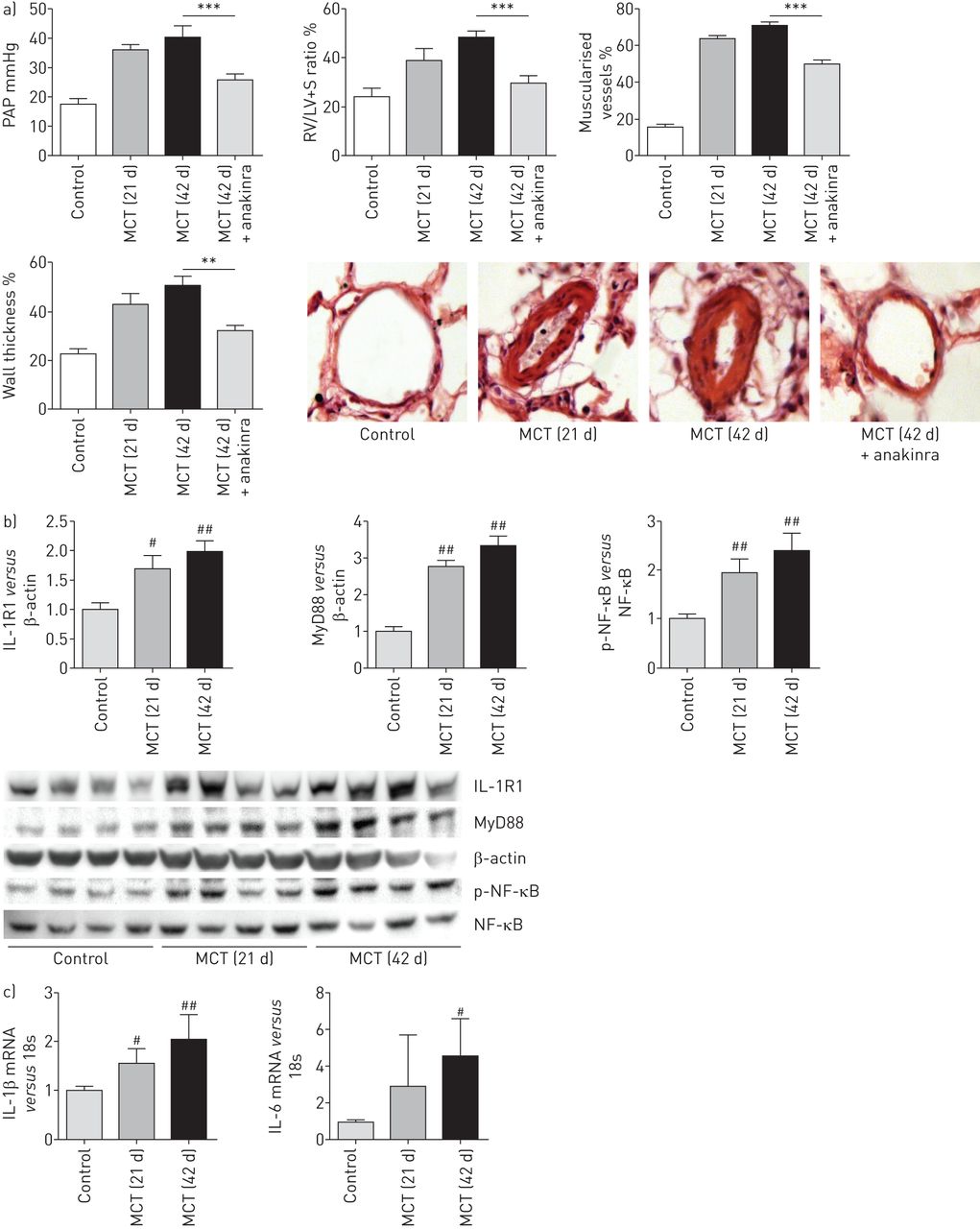
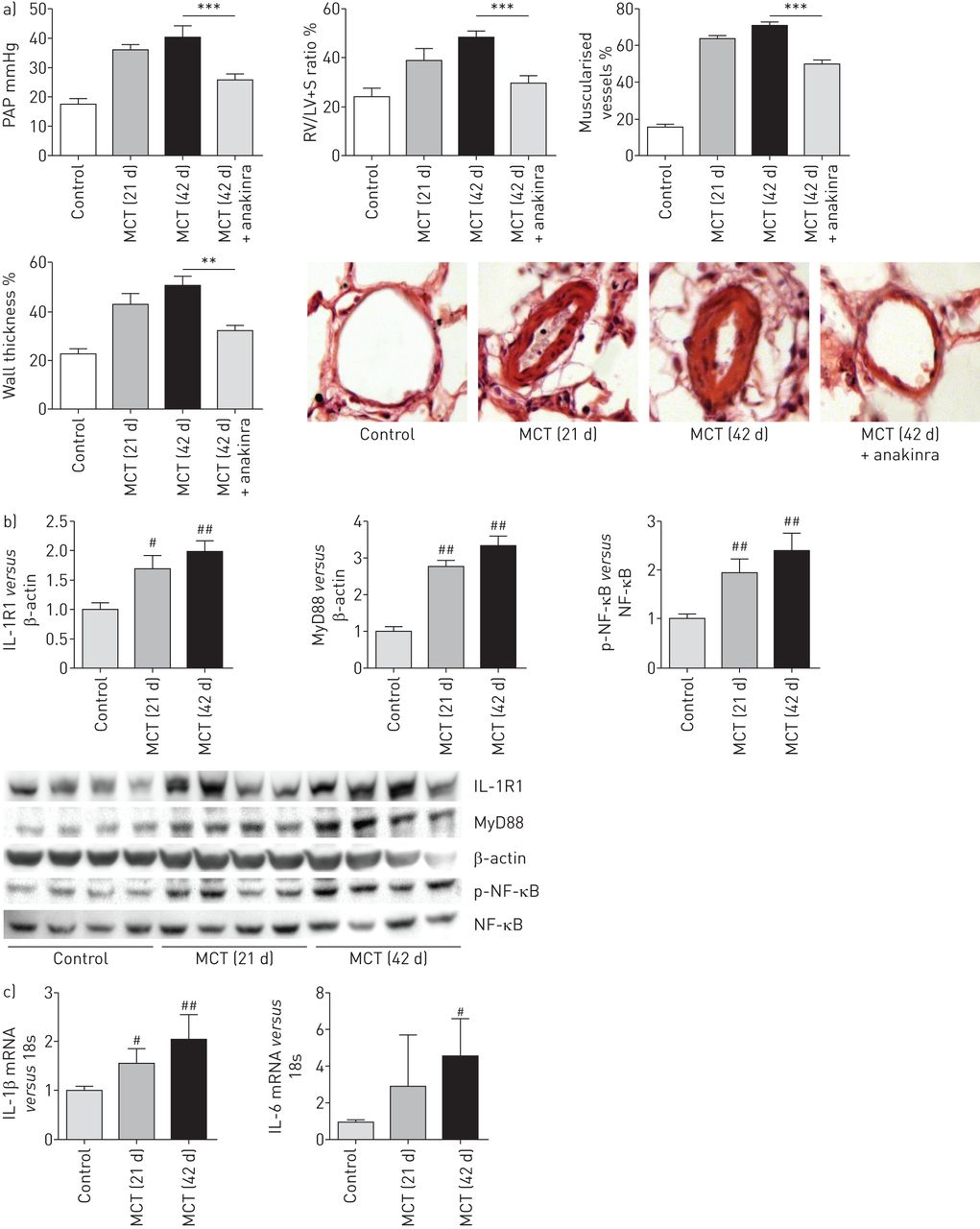
我们评估的潜在疗效anakinra与monocrotaline-induced肺动脉高压大鼠。老鼠检查后21天野百合碱政府严重肺动脉高压与肺动脉压(PAP)显著增加,房车肥大和肺动脉muscularisation相比,控制大鼠给予生理盐水代替野百合碱(图8)。Monocrotaline-treated老鼠给车辆从21天42天显示,人民行动党进一步增加,房车肥大和肺血管壁厚度。相比之下,那些每日anakinra治疗(20毫克·kg−1)从21岁到42天显示,人民行动党明显减少,房车肥大、肌肺血管和肺动脉壁厚与vehicle-treated野百合碱大鼠(图8)。Monocrotaline-induced肺动脉高压与时间的增加肺IL-1R1 MyD88蛋白水平,加上NF-κB激活(图8 b)和并行肺IL-1β和il - 6的变化mRNA水平(图8 c)。值得注意的是,anakinra治疗减少肺NF-κB活化,随后,肺IL-1β和il - 6(在线补充图S7) mRNA水平。
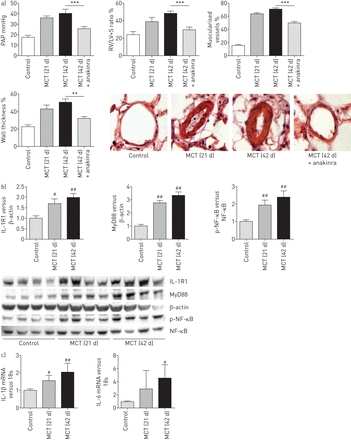
Anakinra逆转肺动脉高压的发展由注射野百合碱诱导大鼠。)肺动脉压(PAP)和右心室肥大指数(右心室(RV) /左心室(LV) +隔重量(S))在大鼠研究21天(d)或42 d经过政府的野百合碱(MCT)或生理盐水(控制)。Anakinra (20 mg·公斤−1每天)或车辆有腹腔内从21岁到42天。据肺血管、肺血管重塑:muscularisation non-muscularised的百分比或完全肌远端血管和肺动脉壁厚从老鼠在21和42天盐水(控制)或其特定管理。代表的显微图显示肺血管。每个值是平均值±扫描电镜10个独立的决定。数据意味着±扫描电镜7或8的动物。* *:p < 0.01;* * *:p < 0.001。b)肺interleukin-1受体(IL-1R) 1蛋白质的水平,骨髓分化主要响应蛋白88 (MyD88)蛋白质及磷酸化的比例核转录因子(NF) -κB p65 Ser536 (p-NF-κB) NF-κB p65蛋白,用西方墨点法测量之前和在不同时间点后未经中华人民共和国交通部管理。#:p < 0.025,# #价值观:p < 0.005,而控制。c)肺IL-1β和il - 6水平mRNA在同一时间点。数据意味着±扫描电镜六个动物。#:p < 0.025,# #价值观:p < 0.005,而控制。
讨论
目前的结果表明,肺动脉高压的肺血管重塑和炎症特点发展需要IL-1R1 / MyD88信号。IL-1R1和MyD88都明显在肺血管从iPAH患者。在小鼠暴露于慢性缺氧,生产IL-1βIL-1R1的表达和MyD88之前肺动脉高压的发展。我们发现IL-1R1−−/和MyD88−−/老鼠同样对肺动脉高压保护发展确定IL-1R1作为主要参与者在肺动脉高压的发病机制影响PA-SMC扩散和巨噬细胞的招募。唯一的部分保护。lys-Cre MyD88fl / fl对肺动脉高压表明IL-1β/ IL-1R1影响肺动脉高压部分介导巨噬细胞激活。一起发现IL-1R拮抗剂anakinra也部分逆转肺动脉高压,这些结果表明,药理干预措施针对IL-1β/ IL-1R1通路可能持有承诺治疗人类的多环芳烃。
我们关注的潜在作用IL-1R1 / MyD88通路在肺动脉高压,基于其强大的参与先天免疫(15]。IL-1β是一个主要的炎性细胞因子的表达,成熟和分泌依赖于先天免疫的复杂的过程9,11]。我们发现早期缺氧暴露IL-1β水平上升,增加IL-1R1一起MyD88 NF-κB和il - 6的分泌。早期和瞬态海拔IL-1βNF-κB和il - 6与全球一致的激活先天免疫系统,负责启动炎症在缺氧暴露的早期阶段8]。的一个主要发现是,过度IL-1R1和MyD88紧密与肺血管重塑和肺动脉高压,在iPAH患者和我们两个鼠肺动脉高压模型,即。小鼠暴露于低氧和SM22-5HTT+老鼠。此外,IL-1R1和MyD88强烈表达了PA-SMCs以及肺巨噬细胞。这些结果证实这两个主要转导元素表达的肺癌,不仅由骨髓细胞谱系,也通过本构血管细胞的增殖是肺血管重塑过程。
我们发现IL-1R1或MyD88基因缺失和/或药物失活IL-1R1保护小鼠对缺氧肺动脉高压和部分在SM22-5HTT逆转肺动脉高压+老鼠。值得注意的是,保护缺氧肺动脉高压是MyD88类似的大小−−/老鼠,IL-1R1−−/老鼠和老鼠WT anakinra对待。这表明提供的预防肺动脉高压MyD88删除主要是由于失活IL-1R1 / MyD88通路,没有任何的TLR / MyD88通路。与先前的报道,这一发现并没有冲突地删除老鼠带来一些预防肺动脉高压(23,24]。事实上,MyD88作为适配器蛋白质不是强制性的地信号转导发生(10]。此外,我们发现高机动组box-1-induced地刺激PA-SMCs不是与一个强大的对细胞生长的影响,如前所报道(23]。另一个有趣的一点是,早期IL-1β增加缺氧反应在MyD88完全压制−−/老鼠,IL-1R1−−/老鼠和老鼠WT与IL-1R1抑制剂治疗,也明显降低肺NF-κB展出。这一发现符合IL-1β诱导自己的综合的能力通过IL-1R1 / NF-κB MyD88-mediated激活(15,30.]。相比之下,没有观察到变化在MyD88 inflammasome的下游分子组件−−/老鼠或IL-1R1−−/老鼠。
防止在远端肺动脉高压与减少PA muscularisation和血管周的巨噬细胞数量减少。我们评估的潜在影响IL-1βPA-SMC扩散。按照之前我们小组报告的结果和其他人(20.- - - - - -22ng), IL-1β(50毫升−1)大力加强培养PA-SMCs的增长。正如所料,这种刺激效应抑制了IL-1βanakinra和IL-1R1 PA-SMCs从没有被观察到−−/或MyD88−−/老鼠。因此,这些在体外结果符合直接影响PA-SMCs由IL-1R1 / MyD88信号。此外,IL-1R1和MyD88表达增加PA-SMCs从长期缺氧小鼠,与更强的刺激效应的IL-1βPA-SMCs从慢性缺氧小鼠比常氧控制。然而,anakinra未能完全逆转细胞长期增长率的增加小鼠缺氧。
在我们的研究中,血管周的巨噬细胞数量减少在缺氧IL-1R1 anakinra-treated缺氧小鼠和−−/或MyD88−−/老鼠,而各自的控制。巨噬细胞计数下降的机制可能涉及的具体失活IL-1R1巨噬细胞,或在其他细胞类型包括内皮细胞,表达对单核细胞粘附分子(31日,32]。我们因此而缺氧MyD88−−/老鼠老鼠的MyD88只在巨噬细胞(M基因被删除。lys-Cre MyD88fl / fl老鼠)。我们发现M。lys-Cre MyD88fl / fl老鼠对肺动脉高压保护,比MyD88但程度不一样−−/老鼠,还显示小血管周的巨噬细胞数量减少。这些结果表明,影响IL-1R1失活有关的血管周的巨噬细胞。全球删除被更高效的肺动脉高压比macrophage-specific删除MyD88符合IL-1R1的双重效应,在巨噬细胞迁移和活化,以及PA-SMC增殖。进一步描绘角色IL-1β在PA-SMC增殖调节巨噬细胞的影响,我们对媒体的影响条件由WT巨噬细胞和M。lys-Cre MyD88fl / fl老鼠,与文化条件下产生M0、M1和M2巨噬细胞。正如之前报道的(33,34),我们发现从任何这三个巨噬细胞条件培养液类型PA-SMC刺激增长,M2与M1巨噬细胞相比,具有更大的影响和M1 M0巨噬细胞。影响PA-SMC增长macrophage-derived IL-1β显然与M1显示巨噬细胞,巨噬细胞的刺激作用介质以来减少了anakinra。没有发现anakinra效果使用从M2巨噬细胞条件培养基。然而,MyD88缺乏减少对PA-SMC增长M2巨噬细胞的刺激作用,支持一个重要的角色的MyD88巨噬细胞激活和随后mitogenic-factor释放。综上所述,这些研究支持一个角色IL-1R1 / MyD88通路在调节巨噬细胞招聘肺血管周围和巨噬细胞激活负责释放PA-SMC有丝分裂原,包括IL-1βM1巨噬细胞释放出来的因素之一。
通过关注IL-1R1 / MyD88通路,我们研究一个主要机制参与先天免疫反应发生在肺动脉高压发展的早期阶段。基因失活或药理IL-1R1 / MyD88通路可能预防肺动脉高压的发展是一致的一个关键贡献炎症引发的肺动脉高压。此外,我们的研究结果表明,药理干预针对这个途径建立阶段的肺动脉高压可以部分逆转肺动脉高压。在大鼠建立monocrotaline-induced肺动脉高压,每日anakinra治疗开始后3周注射野百合碱PAP的大幅减少,房车肥大、肌肺血管和肺动脉壁厚与monocrotaline-injected老鼠给车辆代替anakinra。因此,anakinra逆转肺动脉高压的发展由野百合碱诱导大鼠注射在某种程度上,在SM22-5HTT大于显现+老鼠。此外,增加IL1R1的表达式和Myd88被发现在肺部monocrotaline-treated老鼠,加上IL-1β表达的增加,也在先前的研究报告(35]。这些发现,再加上强烈的anakinra NF-κB激活和抑制作用IL-1β和il - 6表达,符合一般的概念主要炎症组件monocrotaline-induced肺动脉高压(25),并建立的关键重要性IL-1R1 / MyD88通路在肺动脉高压发展,即使在先进的肺动脉高压的阶段。我们观察突出MyD88过度PA-SMCs和巨噬细胞来自患者iPAH强烈支持这样一个概念,即针对IL-1R1 / MyD88途径可能为PAH患者提供临床益处。符合这种可能性,多环芳烃中最近报道的改善病人收到anakinra疗法治疗成人耳氏病(26]。因此需要进一步的研究来评估是否针对IL-1R1 / MyD88通路构成PAH患者有效的治疗选择。
确认
我们感谢理查德•Souktani Xavier Decrouy和Christelle Micheli IMRB平台设施(INSERM U955、完成、法国);和佛罗伦萨萨维尼(UMR7355、奥尔良、法国)的技术援助。
脚注
编辑评论:欧元和J2016;48:305 - 307。
可以从本文的补充材料www.qdcxjkg.com
支持声明:本研究支持由INSERM Ministere de la矫揉造作的,巴黎Chancellerie des的大学。UMR7355收到支持组织的生成MyD88-deficient老鼠从区域中心,纳(pour la医学基金会)。
利益冲突:没有宣布。
- 收到了2015年8月31日。
- 接受5月3日,2016年。
- 版权©2016人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}