





抽象的
慢性阻塞性肺疾病(COPD)与异常上皮-间充质相互作用导致炎症和重构过程相关。我们开发了一个使用COPD、控制源性气道上皮细胞(AECs)和肺成纤维细胞的共培养模型,以了解参与COPD重构和炎症的介质。
从COPD和控制肺组织中获得的AECs和成纤维细胞与胎儿肺成纤维细胞或人支气管上皮细胞系共培养。检测炎症介质、前纤维化分子和细胞外基质(ECM)蛋白的mRNA和蛋白表达。
共培养导致肺成纤维细胞释放促炎介质白细胞介素(IL)-8/CXCL8和热休克蛋白(Hsp70),并降低ECM分子的表达(例如控制和COPD衍生的主要单元之间的胶原蛋白,Decorin)并不不同。这种促炎效应是通过上皮衍生的IL-1α介导的,并在上皮暴露于香烟烟雾提取物(CSE)时增加。当暴露于CSE时,与对照衍生的气道上皮相比,COPD衍生的AECs引发了更强的IL-1α响应,并且这与来自肺成纤维细胞的显着增强的IL-8释放相对应。
我们证明,通过IL-1α的产生,AECs诱导促炎肺成纤维细胞表型,该表型在COPD中暴露CSE后进一步增强,提示在COPD中存在异常的上皮-成纤维细胞相互作用。
抽象的
IL-1α调节上皮细胞-成纤维细胞的相互作用,并可能驱动慢性阻塞性肺病的炎症和组织重塑http://ow.ly/Rhy23008ytz
介绍
慢性阻塞性肺病(COPD)主要是由接触有害颗粒物引起的,吸烟是其中的主要危险因素[1].目前,不存在COPD治愈,目前的药理学治疗只能部分抑制症状和恶化[2].该疾病的特点是慢性炎症和组织修复缺陷,导致不可逆的慢性气流限制,由于肺的气体交换表面(肺气肿)的破坏,重构和缩小小气道[2].
当吸入时,香烟烟雾首先遇到气道上皮,它通常形成一个连续的和高度调节的结构屏障,是先天免疫防御的一部分[3.].我们以前表明香烟烟雾抑制上皮屏障功能[4].这种损伤也会导致促炎介质(白细胞介素(IL)-8/CXCL8, IL-6)的释放和被称为损伤相关分子模式(DAMPS)的危险信号,如IL-1α和热休克蛋白(Hsp70) [5].此外,气道上皮是生长因子(例如转化生长因子(TGF)-β1)可以作用于固有层的潜在间充质细胞,诱导修复[6].间充质纤维细胞、成纤维细胞和平滑肌细胞是重要的结构细胞,在肺内产生各种细胞外基质(ECM)蛋白,包括胶原蛋白和脱朊蛋白[7].我们以前证明的原发性肺成纤维细胞从严重的肺成纤维细胞产生下降(慢性阻塞性肺病(金)4)COPD患者的全球肺成纤维细胞产生与轻度(金1)COPD患者相比[8].其他研究小组也证明肺成纤维细胞对IL-1β, IL-1α有反应[9]和前列腺素PGE2 [10通过释放IL-8/CXCL8和IL-6,特别是IL-1β,也通过下调它们的ECM蛋白生产来刺激[11].上皮成纤维细胞通信已显示参与哮喘的发病机制[12,13也被认为会导致特发性肺纤维化[14].然而,关于气道上皮细胞(AECS)与COPD中的潜在肺成纤维细胞的直接相互作用的详细知识也是有限的,也相对于香烟烟雾的影响。
我们假设,通过释放介质的功能失调的上皮细胞-成纤维细胞通信在慢性炎症和慢性阻塞性肺病的重构过程中起着关键作用,而香烟烟雾暴露有助于这一异常过程。本研究的目的是建立一种共培养细胞模型,研究AECs与重度COPD患者和对照组肺成纤维细胞之间的串扰对促炎介质释放和ECM表达的影响。此外,我们还研究了香烟烟雾暴露对COPD中AECs和成纤维细胞之间的交互作用。
材料和方法
人气道上皮细胞和肺成纤维细胞
人支气管上皮16HBE14o细胞(由美国加州大学旧金山分校D.C. Gruenert博士捐赠)在Eagle最低基本培养基(EMEM)/10%胎牛血清(FCS)中培养,如前所述[15].如前所述,分离出原发性aec [16]从13例重症COPD患者接受肺移植的气管支气管组织和16例非COPD对照供体肺的剩余气管支气管组织中获得,这些患者没有进一步的信息。献血者的主题特征已列于表格1.原代AECs在含有激素的支气管上皮生长培养基(瑞士隆扎,巴塞尔)中培养,并在第3代使用,如前所述[15].
胎儿肺成纤维细胞(MRC-5;BioWhittaker, Walkersville, MD, USA)在EMEM/10% FCS中培养。原发人肺成纤维细胞(phlf)来自9名患有严重疾病的COPD患者,他们接受了肺移植,5名非COPD对照患者接受了肿瘤切除手术。如前所述,使用外植体技术从肺实质中分离成纤维细胞[8,17],在Ham's F12培养基/10% FCS(龙沙,巴塞尔,瑞士)中生长,第5代试验用。PHLF隔离的完整协议可以在在线补充材料中找到。PHLF捐赠者的受试者特征见表2..研究方案符合格罗宁根大学医学中心研究规范(www.rug.nl/umcg/onderzoek/researchcode/Index.)和民族道德和专业指南(www.federa.org).
共培养模型
16HBE14o-和MRC-5细胞最初用于建立该模型。通过以下方法重复了显著的观察结果:1)来自严重COPD患者或对照组的原发性AECs与MRC-5成纤维细胞,2)来自COPD患者或对照组的16HBE14o细胞与phlf,分别评估每种细胞类型的疾病特异性影响。简单来说,AECs被镀在0.4-μM孔6.5 mm跨孔膜上(Costar;在24孔板上播种成纤维细胞。当两层细胞融合后,将含有AECs的transwell插入物与成纤维细胞共培养,并在适当的培养基中培养72小时(见在线补充材料)。
条件培养基实验和中和抗体实验
16HBE14-细胞或原发性AECS当汇合是血清/激素 - 剥夺过夜,并用20%香烟烟雾提取物(CSE)刺激6小时。将CSE彻底洗净,在收集的无CSE条件培养基之前将细胞再次温育另外24小时。然后将已经血清剥夺过夜的成纤维细胞与已预孵育1小时的无CSE的条件培养基处理24小时,或没有4μg·mL−1IL-1α中和抗体(AB-200-NA)或IL-1β中和抗体(MAB601)(R&D Systems,Europe,Abingdon,UK)。收集无细胞上清液并通过ELISA分析,收获细胞裂解物以进行RNA和蛋白质检查。有关完整的实验协议,请参阅在线补充材料。
统计数据
数据分析使用SPSS (IBM, Armonk, NY, USA)。实验组间比较采用Mann-Whitney u检验,原代细胞组内配对比较采用Wilcoxon符号秩检验。我们对细胞系实验结果进行正态分布检验,对配对差异采用t检验。P <0.05认为差异有统计学意义。
结果
肺成纤维细胞与AECs共培养时炎症介质释放增加
当16HBE14O-和MRC-5细胞置于共培养中时,我们发现基底外侧IL-8 / CXCL8(图1一个)和hsp70(图1 b)的分泌与上皮细胞和成纤维细胞的单培养比较。IL-1β蛋白水平无法检测(数据未显示)。随后对上皮细胞和成纤维细胞的mRNA分析表明成纤维细胞是分泌IL-8/CXCL8的主要来源(图1 c).类似地,当在共同培养时,成纤维细胞中,IL-1βmRNA水平增加,但在上皮细胞中增加(图1 d).为了确定从共培养中的细胞系中获得的结果是代表原发性细胞的,我们将16HBE14 - 细胞与来自COPD和对照受试者衍生的PHLF配对,并且还与MRC-5细胞的COPD和控制供体配对原代AECS.与共培养细胞系模型一样,我们发现用16HBE14 - 细胞的PHLF的共同培养也诱导基底外侧IL-8 / CXCL8的显着增加(图1 e)和hsp70(图1 f),没有差异的IL-8 / CXCL8来自出吸烟者,从不吸烟者和当前吸烟者的PHLF的反应。此外,我们证实了IL-8 / CXCL8的mRNA(图1 g)和IL-1β(图1 h)仅在共培养的PHLFs中表达上调。此外,原代AECs与MRC-5成纤维细胞联合可导致基底外侧IL-8/CXCL8和Hsp70水平升高,IL-6蛋白水平有升高的趋势(在线补充)图1 gydF4y2Ba),虽然未检测到的粒细胞 - 巨噬细胞刺激因子和IL-33的水平。HSP70和IL-6水平强烈相关,IL-8 / CXCL8在共同培养中释放(在线补充图2 gydF4y2Ba).我们发现对照和COPD衍生的AECS和PHLF之间的介质释放没有显着差异。
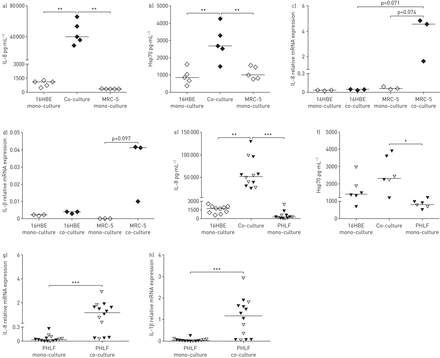
白细胞介素(IL)-8 / CXCL8,热休克蛋白(HSP70)和IL-1β水平在气道上皮细胞(AECS)和肺成纤维细胞的共培养中。单独培养16HBE14O-(16HBE)细胞,并用MRC-5细胞或衍生自对照(开放三角形)和慢性阻塞性肺病患者(填充三角形)的原发性人肺成纤维细胞(PHLF)。a)IL-8 / CXCL8浓度(用中值)和B)HSP70浓度(用中值)在共培养系统的基底外侧室的无细胞上清液(24小时)中。c)IL-8 / CXCL8和D)IL-1βmRNA表达水平(用中值)在上皮细胞和成纤维细胞(6小时)分别比较16HBE14O-和MRC-5细胞的共培养和单培养物中收获。e)IL-8 / CXCL8浓度(中值)和F)HSP70浓度(中值)在基石室的无细胞上清液中(24小时)。G)IL-8 / CXCL8和H)IL-1βmRNA表达水平(6小时)在分别比较16HBE14O-细胞和PHLF的共培养和单培养物中收获的PHLF(6小时)。mRNA水平与管家基因β相关2-microglobulin和蛋白质磷酸酶1α,并表示为2-ΔCt.*: p < 0.05;* *: p < 0.01;***:指示值之间p<0.001。
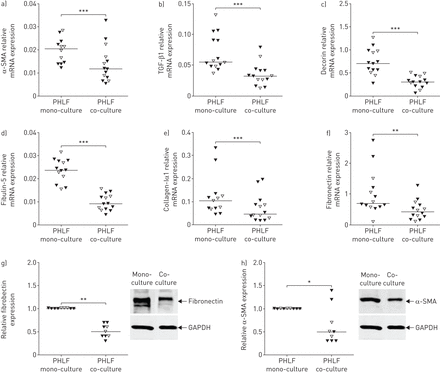
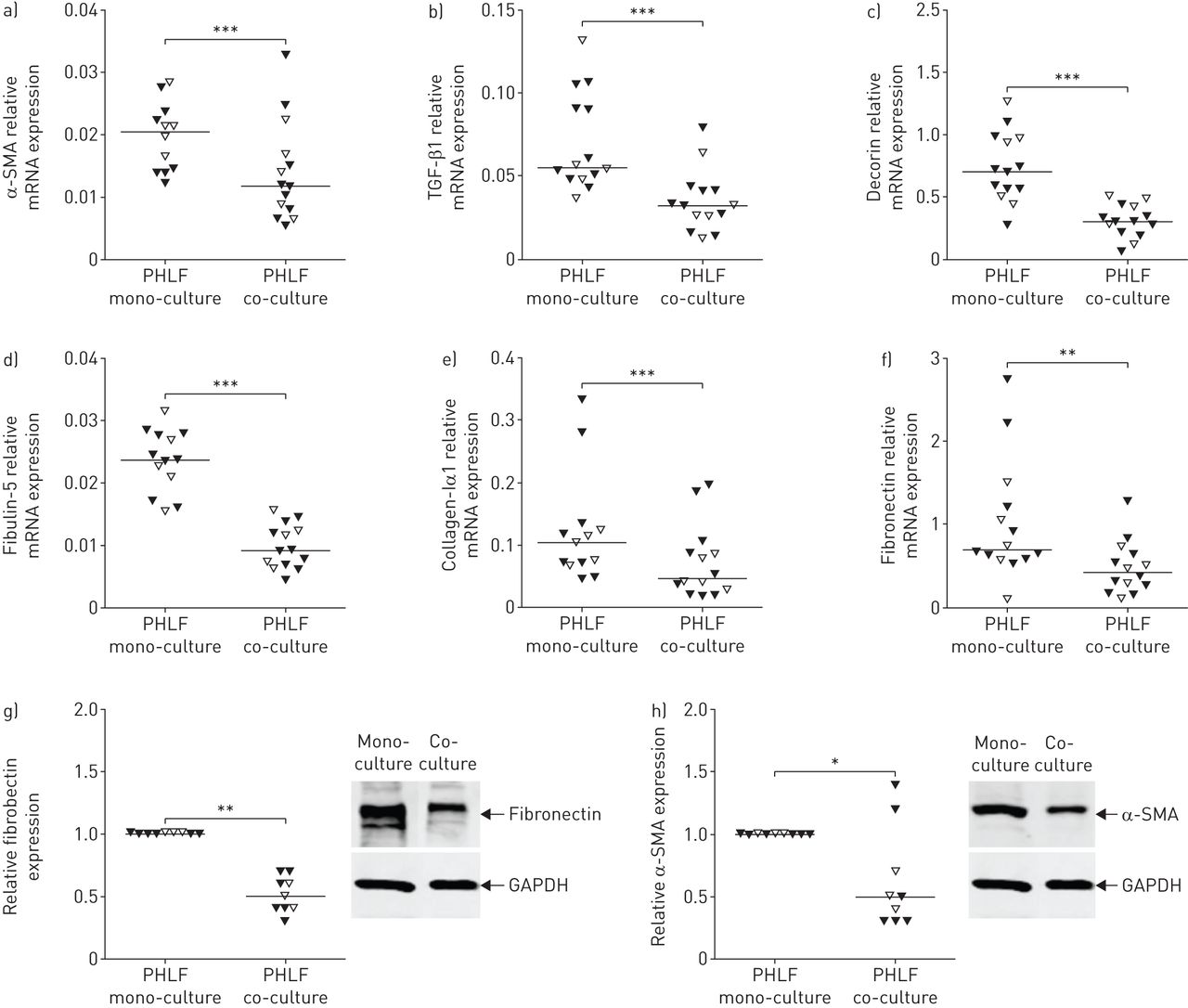
a-f)与16HBE14o-细胞共培养后,原代人肺成纤维细胞(PHLFs)细胞外基质分子和结构蛋白的表达下降。a) α-平滑肌动蛋白(α-SMA), b)转化生长因子(TGF)-β1, c)脱蛋白,d)纤蛋白-5,e)胶原α1和f)对照组供体(开放三角形)和慢性阻塞性肺疾病(COPD)患者(填充三角形)的phf中的纤维连接蛋白与16HBE14o和单培养比较。mRNA水平与管家基因β相关2-microglobulin和蛋白质磷酸酶1α,并表示为2-ΔCt.G,H)与对照供体(开放三角形)和COPD患者(填充三角形)与16HBE14O-和单声道相比,G-cultous。使用甘油醛3-磷酸脱氢酶(GAPDH)作为加载对照。*: p < 0.05;* *: p < 0.01;***:指示值之间p<0.001。
用上皮细胞的共培养中肺成纤维细胞中ECM分子和促纤维化蛋白的表达降低
与增加的促炎反应增加,来自COPD和对照受试者的PHLF的共培养与16HBE14-细胞导致α-平滑肌肌动蛋白(α-SMA)的mRNA表达中的显着下调(图2一个),TGF-β1(图2 b), ECM分子脱蛋白(图2 c), fibulin-5 (图2 d), collagen-Iα1 (图2 e)和纤维连接蛋白(图2 f)与单培养条件相比。使用Western Blotting对蛋白质水平确认纤连蛋白和α-SMA的下调(图2 g和h).这些ECM和结构蛋白在共培养时的基线表达和减少在COPD和对照来源的PHLFs之间均无显著差异。
上皮细胞来源的IL-1α在肺成纤维细胞中负责促炎症表型转换
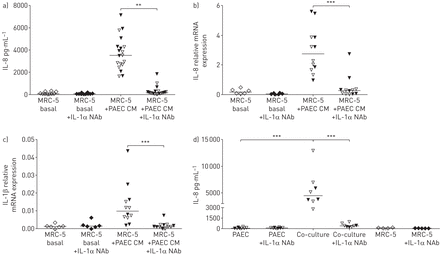
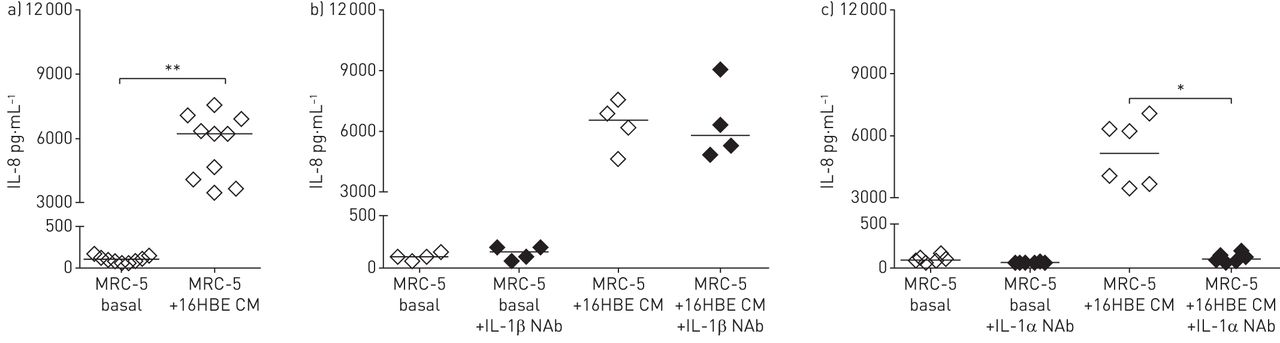
接下来,我们使用来自16HBE14o-细胞的条件培养基来研究观察到的影响是否可能是由可溶性因子引起的。事实上,我们观察到上皮条件培养基也诱导了MRC-5成纤维细胞IL-8/CXCL8分泌的增加(图3一).因为IL-1β和PGE2已经被证明在成纤维细胞中诱导类似的表型开关[9,11[我们首先研究了IL-1β中和抗体的影响(图3 b)和使用腺苷酸环化酶抑制剂MDL-12,330A盐酸盐(在线补充图4.).两者都没有防止IL-8 / CXCL8从MRC-5细胞中释放。IL-1R1受体的其它激动剂是IL-1α。有趣的是,我们发现使用UL-1α的中和抗体完全废除IL-8 / CXCL8分泌通过MRC-5成纤维细胞(图3 c).

来自16HBE14o细胞的白细胞介素(IL)-1α负责MRC-5成纤维细胞的促炎表型开关。IL-8/CXCL8浓度(含中位)从融合的MRC-5细胞中释放:a)不含(基础的MRC-5)或16HBE14o-细胞(16HBE CM)条件培养液,b) 16HBE14o条件培养液有或没有4µg·mL−1IL-1β中和抗体(NAB)和C)在存在和不存在4μg·mL的情况下用16HBE14-条件培养基中和−1il - 1α逮捕。*: p < 0.05;**:指示值之间p<0.01。
来自原代气道上皮细胞(AECs)的白细胞介素(IL)-1α负责MRC-5成纤维细胞的促炎表型开关。将MRC-5细胞培养至融合,去除血清过夜,然后在没有(基础的MRC-5)或与对照供体(开放三角形)和慢性阻塞性肺病(COPD)患者(填充三角形)原发AECs (PAEC CM)条件培养液(存在或不存在4 μg·mL)的情况下孵育−1IL-1α中和抗体(NAB)。a)IL-8 / CXCL8浓度(用中值)和B)IL-8 / CXCL8和C)IL-1βmRNA表达(用中值)在MRC-5细胞中。d)IL-8 / CXCL8浓度(带中值)在基石室的无细胞上清液(24小时)比较来自对照供体(开放三角形)和COPD患者的原发性AECS的共培养和单培养物(填充三角形)和MRC-5细胞在存在或不存在4μg·ml−1il - 1α逮捕。mRNA水平与管家基因β相关2-microglobulin和蛋白质磷酸酶1α,并表示为2-ΔCt.* *: p < 0.01;***:指示值之间p<0.001。
为了确定使用16HBE14o条件培养基获得的结果是否代表原代细胞,我们使用来自COPD和对照供体的原代AECs条件培养基对MRC-5成纤维细胞进行了相同的实验。如图所示图4.,IL-1α中和也消除了原发性AEC条件介质诱导的IL-8 / CXCL8分泌(图4一)以及IL-8 / CXCL8和IL-1βmRNA表达(图4 bc)在MRC-5成纤维细胞中。
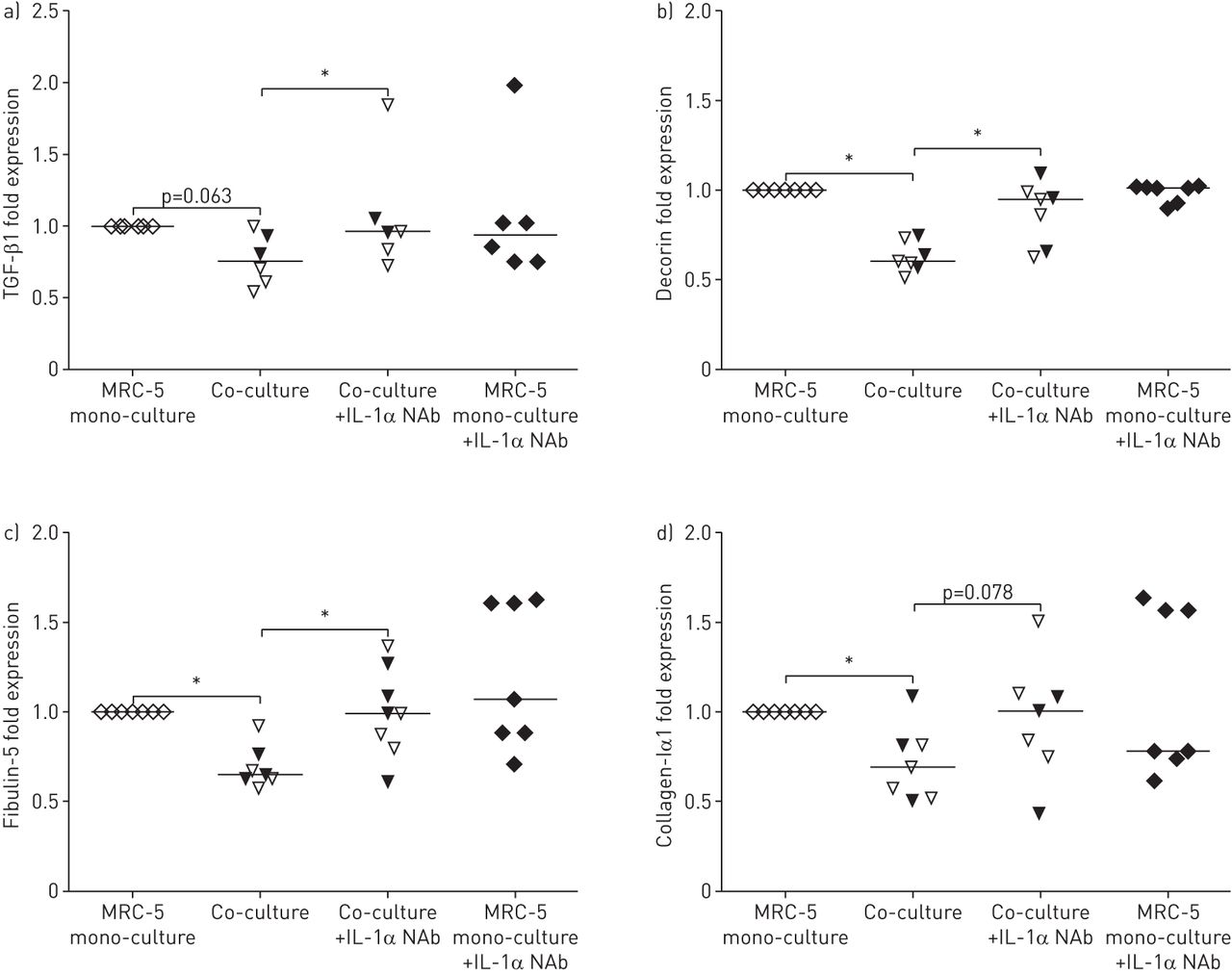
此外,我们评估了中和抗体在共培养模型本身中的作用。如图所示图4 d,在基础室添加IL-1α中和抗体,也抑制了COPD原发性AECs和MRC-5对照组的基底外侧释放IL-8/CXCL8。基底外侧IL-6的释放也有明显的下降趋势(在线补充)图1 gydF4y2Ba).此外,当与原代AECs共培养时,IL-1α在共培养中中和也能抑制MRC-5细胞中TGF-β1、脱蛋白、fibulin-5和胶原- i α1 mRNA表达的下调(图5.).
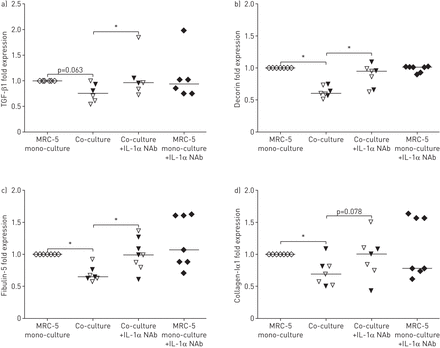
白细胞介素-1α与肺成纤维细胞与上皮细胞共培养后细胞外基质分子和结构蛋白的表达降低有关。a)转化生长因子(TGF)-β1, b)脱蛋白,c) fibulin-5和d) MRC-5细胞中胶原- i α1的含量,与对照组供者(开放三角形)和慢性阻塞性肺疾病患者(填充三角形)的原代气道上皮细胞(有和没有4µg·mL的情况下进行单培养和共培养比较−1IL-1α中和抗体(NAB)。mRNA水平与管家基因β相关2-microglobulin和蛋白质磷酸酶1α,并表示为2-ΔCt.*:指示值之间p<0.05。
香烟烟雾暴露在AECS和随后的IL-8 / CXCL8在肺成纤维细胞中产生的IL-1α表达增加
为了评估上皮细胞暴露于香烟烟雾是否会改变与肺成纤维细胞的交流,我们将成纤维细胞暴露于预处理过CSE的16HBE14o细胞条件培养基中。CSE显著增加了上皮细胞IL-1α蛋白的释放(图6)及mRNA (图6 b)与基础水平进行比较。随后将MRC-5成纤维细胞暴露于经cse处理的16HBE14o-细胞的条件培养基中,其诱导的IL-8/CXCL8的产生明显高于16HBE14o基础条件培养基(图6 c).类似地,CSE暴露显著增加了原发性AECs中IL-1α mRNA的表达(图6 d).
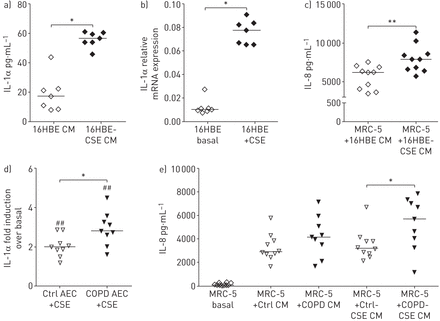
来自香烟烟雾提取物(CSE)的白细胞介素(IL)-1α - 散装气道上皮引起肺成纤维细胞中的IL-8 / CXCL8更高的释放。A-C)16HBE14-细胞预刺激,没有或20%CSE。a)IL-1α浓度(具有中值)在条件培养基(16HBE cm)和b)IL-1αmRNA表达(中位数)在16HBE14 - 细胞中。c)与16HBE14O-条件培养基孵育后,从MRC-5细胞中释放的IL-8 / CXCL8浓度(带中值)。D,E)来自对照(CTRL)供体(开放三角形)和慢性阻塞性肺病(COPD)患者(填充三角形)(填充三角形)的原发性气道上皮细胞(AECS)被预先刺激20%CSE。d)在原发性AECs中折叠IL-1α的表达。e)与原发性AECs的调节培养基孵育后,从MRC-5细胞中释放的IL-8 / CXCL8浓度。mRNA水平与管家基因β相关2-microglobulin和蛋白质磷酸酶1α,并表示为2-ΔCt*: p < 0.05;**:组间指征值p<0.01;##:组内指示值p<0.01。
有趣的是,与对照来源的原发性AECs相比,copd来源的原发性AECs在CSE暴露后IL-1α mRNA表达有更强的增加(图6 d).根据IL-1α表达的这种增加,来自CSE暴露的COPD衍生的原发性AECs的调节培养基导致来自MRD-5成纤维细胞的IL-8 / CXCL8释放的显着较强,而不是来自CSE暴露的控制原发性AECs的条件培养基(图6 e).
讨论
我们发现肺成纤维细胞直接受AECs调控,释放促炎症介质,下调ECM合成和促纤维化反应。我们的数据表明,这种调节是由上皮来源的IL-1α驱动的,因为在上皮条件培养基中和IL-1α完全逆转肺成纤维细胞释放的炎症介质。此外,我们发现香烟烟雾暴露诱导AECs中IL-1α水平升高,特别是在严重COPD患者的上皮细胞中。此外,香烟烟雾暴露可能有助于COPD上皮细胞和潜在的成纤维细胞之间的异常交叉,因为我们观察到,与cse暴露对照组来源的上皮细胞相比,来自COPD患者的cse暴露上皮细胞诱导肺成纤维细胞IL-8/CXCL8分泌更强。
在肺中,成纤维细胞位于肺间质内,靠近气道上皮,因此在正常修复和疾病状态下很容易受到上皮释放的几种因素的影响[7].然而,在COPD中,AECs和潜在的成纤维细胞之间的相互作用可能增加,细胞可能通过观察到的黏膜基膜碎裂而更紧密地接触[18,19].因此,在COPD中,上皮细胞可能对成纤维细胞发挥更强的促炎作用。肺中的成纤维细胞不仅通过肉芽组织的收缩、合成和重塑来促进修复过程,而且还通过产生细胞因子来帮助肺内正常的免疫防御机制[20.].IL-8/CXCL8是肺中性粒细胞的趋化剂[21,22],慢性中性粒细胞炎症可能有助于COPD异常组织修复、重塑和破坏[1].肺中的IL-8 / CXCL8释放增加与COPD的发病机制有关[23].因此,肺成纤维细胞在上皮接触香烟烟雾时分泌更高的IL-8/CXCL8可能在COPD的发病机制中发挥作用[1].类似地,Hsp70和IL-6的释放增加,它们都是炎症介质,在COPD中涉及慢性炎症过程[5,24].
据报道,气道上皮是介导慢性炎症和COPD结构改变的介质的来源。其中,与健康对照组相比,COPD患者肺上皮细胞上清液中IL-1β、IL-1α、PGE2、肿瘤坏死因子-α、IL-6和各种基质金属蛋白酶均升高[25].已显示PGE2和IL-1β以及IL-1α诱导IL-8 / CXCL8分泌来自成纤维细胞的分泌[9,10].我们的结果强调,IL-1α的中和完全阻止了肺成纤维细胞在上皮条件培养基刺激下产生IL-8/CXCL8。因此,我们的数据为上皮来源的IL-1α在上皮细胞和成纤维细胞之间的交互作用提供了强有力的支持,在肺部的正常免疫防御和COPD异常修复过程中。IL-1α是炎症细胞因子IL-1超家族的成员。它在正常免疫反应中起着至关重要的作用在活的有机体内并在肺上皮细胞中组成思考[26].IL-1α及其激动剂IL-1β与IL-1R1受体结合,产生类似的下游反应,随后激活转录因子,如核因子-κB和激活蛋白-1 [26].IL-1β的活性依赖于NLRP3炎症小体的激活和随后caspase-1的裂解,而IL-1α在其前形态和caspase的裂解时都是活跃的[27].
我们的研究结果表明,香烟烟雾可能增加严重COPD气道上皮细胞IL-1α的释放,随后导致纤维母细胞IL-8/CXCL8释放的额外增加,促进中性粒细胞炎症在活的有机体内.对IL-1α在COPD中的作用所做的工作相对较少,我们的数据表明有必要进一步研究IL-1α在COPD发病机制中的作用。与我们的发现一致,PAuwels.et al。[27]的研究表明,香烟诱导的小鼠炎症依赖于IL-1R1受体,IL-1α及其激动剂IL-1β均参与中性粒细胞炎症。此外,Botelhoet al。[28[表明,小鼠中的烟雾诱导的嗜嗜中性炎症依赖于IL-1α,但不是IL-1β。它们还测量了支气管活组织检查和痰中的IL-1α蛋白,并在与对照组相比,来自COPD患者的活组织检查中炎性浸润和上皮的表达增加了[28].年代uwaraet al。[9]使用了另一种慢性阻塞性肺病模型,其中人类AECs受伤在体外分别用过氧化氢和thapsigargin诱导活性氧损伤和内质网应激。他们发现,在这些细胞的条件培养基中,IL-1α和IL-1β水平增加,这导致了MRC-5成纤维细胞中IL-8/CXCL8和IL-6的释放[9].使用IL-1α中和抗体完全阻断这种效果,并且仅部分通过使用IL-1β中和抗体。这符合我们的研究,证明了对调节成纤维细胞的上皮衍生的IL-1α的关键作用,以在共同培养模型中成为促炎症在活的有机体内使适应。我们的数据还表明COPD衍生的上皮细胞更容易发生在CSE暴露时释放IL-1α。进一步研究IL-1α释放的机制是值得的,以便理解为什么COPD上皮细胞在CSE暴露时表达更多IL-1α,例如这是否涉及内质网应激或如前面所示的氧化剂/抗氧化剂失衡[9].
除了在我们的共同文化模型中生产炎症调解员外,AECS通过IL-1α还降低了促纤维化细胞因子TGF-β1、结构分子α-SMA和各种ECM分子,包括脱蛋白、fibulin-5和胶原i α1的表达。这一发现的确切意义还需要进一步研究,因为缺乏证明COPD和控制成纤维细胞之间的差异也可能是由于功率限制。然而,我们目前的发现支持了小气道可能由于组织修复缺陷而丢失的机制,这与M的发现一致cDonoughet al。[29],他在晚期(GOLD 4) COPD患者中证实小气道损失高达90%。虽然我们的上皮-成纤维细胞模型反映了在活的有机体内这种情况比单型文化更紧密,我们的模型可能无法完全反映在活的有机体内其他细胞类型也存在的情况。同样在我们的模型中,我们将第一代气管支气管组织中对照来源的原发性AECs与第三代至第五代支气管树中copd来源的原发性AECs进行了比较。尽管这些细胞并非来自肺部的同一部位,但多项研究显示,气管和支气管AECs的基因组和表观基因组具有相似之处[30.,31].我们使用从接受终级疾病移植的COPD患者源自肺组织的细胞。还需要进一步的实验来评估观察到的上皮细胞和成纤维细胞之间的异常串扰是否也存在于疾病的早期,轻度和中等阶段。如果在我们的模型中观察到的IL-8 / CXCL8分泌增加另外,将需要评估未来的工作还需要促进中性粒细胞趋化性。
总之,我们的数据表明,肺成纤维细胞受AECs的调节,以在其功能中成为促炎症。该调节由上皮衍生的IL-1α驱动,通过CSE暴露和来自严重COPD患者的上皮细胞的CSE暴露和较强的细胞的进一步增强而不是健康个体。我们的研究提供了对上皮细胞和成纤维细胞在COPD中慢性重塑和炎症发病机制中的作用的新洞察。
脚注
编辑评论:EUR RESPIR J.2016;48:301-304。
这篇文章的补充材料可从www.qdcxjkg.com
支持声明:本研究由Stichting Astma Bestrijding资助。本文的资金信息已存入乐趣.
利益冲突:可以在本文旁边找到披露www.qdcxjkg.com
- 收到了2015年11月16日。
- 接受2016年5月9日。
- 版权所有©2016











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}