





摘要
呼吸道合胞病毒(RSV)是2岁以下婴儿急性细支气管炎的主要原因。坏死与呼吸道病毒感染的结局有关。我们报道RSV感染以RIPK1-, RIPK3-和mlkl依赖的方式触发小鼠原代巨噬细胞和人单核细胞的坏死。此外,坏死途径对肺泡巨噬细胞清除RSV有害。此外,Ripk3−−/小鼠免于rsv引起的体重减轻,肺部病毒载量降低。
肺泡巨噬细胞减少也保护小鼠免于体重减轻和肺部RSV病毒载量的降低。重要的是,肺泡巨噬细胞的减少消除了上调Ripk3而且MlklRSV感染对肺组织基因表达的影响。
自分泌肿瘤坏死因子(TNF)介导的RSV触发的巨噬细胞坏死和坏死途径也参与TNF的分泌,即使巨噬细胞已导致细胞死亡,这可加重RSV感染期间的肺损伤。线,Tnfr1−−/小鼠的Ripk3而且Mlkl肺中坏死的肺泡巨噬细胞的基因表达和数量急剧减少。最后,我们提供的证据表明,鼻TNF水平升高与婴儿RSV细支气管炎的疾病严重程度相关。
我们认为,靶向TNF和/或坏死机制可能是降低幼儿RSV感染引起的呼吸道疾病的有价值的治疗方法。
摘要
RSV感染引发的RIPK1-、RIPK3-和mlkl依赖的肺泡巨噬细胞坏死由自分泌TNF介导,对病毒清除有害。肺泡巨噬细胞坏死可能是RSV发病机制的驱动因素。https://bit.ly/3fS34uw
简介
呼吸道合胞病毒(RSV)是两岁以下婴儿因急性细支气管炎和病毒性肺炎住院的最常见原因[1,2]据估计,全球每年有多达20万人因此死亡[2,3.].早产婴儿或患有先天性心脏病或慢性肺病等高风险疾病的婴儿,罹患严重呼吸道合胞病毒疾病的风险增加[4].越来越多的证据表明,儿童时期的RSV感染可能导致日后反复出现喘息和哮喘[5,6].虽然世界卫生组织已将RSV列为疫苗开发的高优先目标,但到目前为止还没有临床批准的RSV疫苗。因此,了解RSV如何与宿主相互作用对于更好地描述疾病的发病机制和开发新的治疗方法至关重要。
肺泡巨噬细胞(AMs)占气道白细胞的近95%,是最先遇到肺部病原体的免疫细胞[7,8].因此,它们对感染的反应可以严重影响传染病的结果[7,9].在这种情况下,新出现的证据表明AM死亡在确定肺部病毒感染的发病机制中起着重要作用[10,11].
坏死被认为是细胞死亡的一种促炎形式,导致细胞内内容物释放,其作用可能是损伤相关分子模式(DAMPs),刺激免疫细胞[12].坏死是由受体相互作用蛋白激酶1 (RIPK1)和RIPK3组成的多蛋白复合物的组装驱动的,导致假激酶混合谱系激酶结构域(MLKL)的激活[13,14].坏死性细胞死亡可通过消除复制位点有效抑制细胞内病原体(如病毒)的传播[15,16];然而,它加重了宿主的炎症和感染后的组织损伤。因此,已有研究表明甲型H1N1流感病毒可诱导肺泡上皮细胞坏死,坏死是感染的病理特征之一,是感染动物高发病率和高死亡率的原因[17].RIPK3缺失导致细胞因子表达降低,减轻甲型H7N9流感病毒诱导的小鼠急性肺损伤[18].最近有报道称RSV可引发肺泡上皮细胞和THP-1细胞坏死[19,20.].然而,RSV是否诱导AM坏死,其作用对宿主是保护作用还是有害作用尚有待证实。
在这里,我们报道了活跃的RSV感染以RIPK1-, RIPK3-和mlkl依赖的方式触发原系小鼠巨噬细胞和人单核细胞的坏死是必要和充分的。此外,坏死途径的激活不利于病毒的清除。Ripk3−−/小鼠免于rsv引起的体重减轻,肺部病毒载量降低。令人惊讶的是,AM已被证明在RSV感染中起有害作用,AM耗尽取消了RSV的上调Ripk3而且MlklRSV感染诱导的肺组织mRNA水平。此外,自分泌肿瘤坏死因子(TNF)介导rsv触发的巨噬细胞坏死Tnfr1−−/小鼠的蛋白表达明显下降Ripk3而且MlklmRNA和肺中死亡AMs数量的急剧减少。这些数据表明,tnf介导的AM坏死在RSV感染的发病机制中起着重要作用。
方法
病毒的准备
RSV A2毒株(由Fernando Polack捐赠,Fundación Infant, Argentina)的病毒生产是在VERO细胞(ATCC CCL-81)中获得的,在Opti-MEM培养基(Gibco by Life Technologies, Grand Island, NY, USA)中培养,含2%胎牛血清(FBS) (Gibco by Life Technologies),温度37℃,温度5% CO2.为了评估病毒滴度,VERO细胞在不含血清的培养基中感染RSV,然后进行羧甲基纤维素斑块试验。裂解板滴定使用抗rsv F蛋白抗体(Millipore, Billerica, MA, USA)进行,病毒滴度表达为空斑形成单位(PFU)。病毒分份储存在−80°C。
RSV临床分离株制剂
两名患者的鼻咽吸入液被DMEM低葡萄糖(Gibco by Life Technologies) (5 mL)稀释,含1000µg·mL−1并接种到VERO细胞培养中。37℃孵育1 h,完全吸附病毒后,去除病毒接种物,加入10 mL添加2%胎牛血清和1000µg·mL的Opti-MEM−1在烧瓶中加入庆大霉素。37℃,5% CO孵育2直到细胞病变效果显现出来。收集细胞,离心,细胞颗粒进行快速冻融循环,以获得新的病毒颗粒。将蔗糖冷冻保护剂添加到病毒分份中,并在- 80°C保存,直到滴定或进一步分析。使用抗rsv抗体进行裂解平板滴定,病毒滴度表达为PFU。病毒命名基于隔离城市/毒株编号/年(RSV POA10/2018和RSV POA43/2018)。对临床分离株进行实时PCR检测,鉴定结果如下:RSV POA10/2018为RSV B亚型,RSV POA43/2018为RSV A亚型。
小鼠巨噬细胞的分离与刺激
肺泡巨噬细胞
通过支气管肺泡灌洗(BAL)收集AMs,用1ml加EDTA (5 mM)的冷冻PBS冲洗肺部三次。灌洗重复两次,并收集了几只小鼠的AMs。细胞在3×10上播种5·好−1在AIM-V介质(Gibco by Life Technologies)中过夜。然后通过洗涤去除非贴壁细胞,用不同的RSV感染(MOIs)(0.1-10)在37℃、5% CO下刺激贴壁细胞2、4或6小时2.然后,巨噬细胞用固定活力染料eFluor 780 (eBioscience公司,Thermo Fisher Scientific, Waltham, MA, USA)进行标记,通过轻轻刮取,流式细胞术分析死亡细胞的百分比,如下所述。
腹膜巨噬细胞
腹腔内灌注3 mL 3%硫代巯基乙酸盐,用冷冻PBS冲洗腹腔,3天后腹腔巨噬细胞获得。细胞在3×10上播种5·好1在AIM-V中过夜。然后通过洗涤去除非贴壁细胞,用不同的RSV MOIs(0.1-10)在37℃、5% CO下刺激贴壁细胞2,4或6 h2.然后,用eFluor 780固定活力染料标记巨噬细胞,收获后用流式细胞术分析死亡细胞的百分比。
人单核细胞的分离和刺激
从签署了知情同意书的健康志愿捐献者(平均年龄29岁,男女均有)中分离出外周血单个核细胞(pmcs)。采集20 mL全血,进行histoapque -1077 (Sigma-Aldrich, Saint Louis, MO, USA)梯度离心30分钟。pbmc在3×10上播种5·好−1在RPMI 1640培养基(Gibco by Life Technologies)中过夜,含有10%的FBS。然后通过清洗去除非贴壁细胞,在37°C下,MOI为1的条件下,用RSV感染贴壁细胞6小时。然后,用eFluor 780固定活性染料标记细胞,收获后用流式细胞术分析死亡细胞的百分比。
细胞系
人单核细胞样细胞系(THP-1;ATCC TIB-202)和促单核细胞细胞系(U937;ATCC CRL-1593.2)在添加10%胎牛血清的RPMI 1640培养基中培养。为了向巨噬细胞样细胞分化,U937细胞以2×10的密度接种5·厘米−2在RPMI 1640中加入10%含12-肉豆蔻酸佛波酯13-乙酸酯(PMA)的FBS (50 ng·mL)−1) 24小时[21].人腺癌肺泡上皮细胞系(A549;ATCC CCL-185)在DMEM低葡萄糖+ 10%胎牛血清中培养。对所有细胞系进行支原体污染检测。在37℃下,在MOI为1的条件下感染RSV 6 h,感染THP-1细胞和分化为巨噬样细胞的U937细胞。然后用eFluor 780固定活性染料标记细胞,流式细胞术分析死亡细胞的百分比。
肿瘤坏死因子测定
小鼠巨噬细胞上清液、人单核细胞上清液和RSV鼻洗液中TNF的浓度+和RSV−采用ELISA (Sigma-Aldrich),按照制造商说明进行检测。
乳酸脱氢酶释放测定
根据制造商说明,使用CytoTox 96非放射性细胞毒性检测试剂盒(Promega, Madison, WI, USA)检测巨噬细胞或单核细胞上清液中的乳酸脱氢酶(LDH)释放。读数在490 nm波长下进行,使用EZ Read 400微板阅读器(Biochrom, Holliston, MA, USA),并表示为% LDH释放。
Ripk3,Mlkl而且处于受控实时PCR检测
老鼠肺或AMs (3×105·好−1)悬浮在Brazol试剂(LGC biotecologia, Cotia, Brazil)中(1 mL),总RNA提取方案根据制造商的说明进行。RNA使用高容量cDNA逆转录试剂盒(Applied Biosystems by ThermoFisher Scientific, Waltham, MA, USA)反转录成cDNA,荧光定量使用Qubit (ThermoFisher Scientific)。的表达式Ripk3,Mlkl而且处于受控使用特异性引物和探针进行TaqMan测定(分别为Mm00444947_m1, Mm01244222_m1和Mm04207460_m1)β肌动蛋白基因(Mm02619580_g1)作为内源性对照。基因表达的定量是使用StepOne (ThermoFisher Scientific的Applied Biosystems)进行的。每个反应共使用15 ng cDNA。目的基因表达用2−ΔCt方法(对控制表达式折叠更改)。的循环阈值(Ct)值减去Δ值β肌动蛋白的Ct值Ripk3,Mlkl而且处于受控对于每个样本。
Western blotting检测MLKL
AMs(3×105·好−1)在指定时间内感染RSV (MOI 1)。然后收集细胞并在含有蛋白酶抑制剂鸡尾酒的RIPA缓冲液中裂解。将细胞裂解物煮沸,并在还原条件下在sds -聚丙烯酰胺凝胶(10%)中进行电泳。将定量的蛋白质转移到甲醇激活的PVDF膜上,在4°C下放置2小时。然后,用脱脂牛奶- tbs - t溶液在室温下阻塞膜1小时,并与抗mlkl抗体(1:1000;cat# AP14272, Abgent, San Diego, CA, USA),其次是辣根过氧化物酶(HRP)抗兔二抗(1:5000;猫#31460,ThermoFisher Scientific)。作为内源性对照,抗gapdh抗体(1:5000;cat# MA5-15738, ThermoFisher Scientific),其次是HRP抗小鼠二抗(1:5000;猫# 31430,ThermoFisher Scientific)。 MLKL expression was detected using the ECL system (Amersham ECL Prime Western Blotting Detection reagent; GE Healthcare Life Sciences, Chicago, IL USA) and the membrane was imaged using the ChemiDoc Imaging System (BioRad, Hercules, CA, USA). Densitometry analysis was performed using ImageJ 1.43 software (US National Institutes of Health, Bethesda, MD, USA). MLKL bands were normalised to GAPDH bands.
动物与RSV感染
为体外实验中,8 - 10周龄的雌性Balb/c小鼠由Biológicos Experimentais (CeMBE/PUCRS, Porto Alegre, Brazil)的育种设施提供。动物实验由PUCRS动物使用伦理委员会(CEUA/PUCRS)根据第8049号议定书审查并批准。为在活的有机体内实验中,8 - 10周龄女性TNF-α受体p55 (p55tnf - knockout or TNF受体1 (Tnfr1)−−/)和C57/BL6野生型(WT)小鼠由Criação de Camundongos Especiais da ade de Medicina de Ribeirão Preto (FMRP/USP, Ribeirão Preto, Brazil)育种设施提供,而8 - 10周龄的雌性RIPK3−−/C57/BL6 WT小鼠由Vishva Dixit (Genentech, Inc, San Francisco, CA, USA)提供[22]并安置在FMRP/USP的繁殖设施中。所有的老鼠都被关在有过滤盖的笼子里,免费获得无菌水和食物。动物程序由生物研究所动物使用伦理委员会审查和批准(协议号4740-1/2017和4740-1(A)/2018),并根据《赫尔辛基公约宣言》执行。
对于RSV感染,用5%异氟醚麻醉小鼠,并鼻内感染RSV A2菌株(107每只动物PFU)。所有动物每天都称重。感染后5天进行数据分析。收集BAL液,灌注福尔马林后取左肺进行组织病理和免疫组织化学分析。
BAL液体收集
腹腔注射氯胺酮(0.4 mg·g)麻醉小鼠−1)/xylazine (0.2 mg·g−1)溶液和气管插管。用1ml冷冻的RPMI 1640培养基清洗肺两次。离心BAL液,收集上清液进行细胞因子分析,悬浮微球进行总细胞计数和差异细胞计数及流式细胞术。为了进行细胞计数,细胞旋切片用苏木精和伊红染色(H&E),计数过程由有经验的研究者以盲法进行。
组织病理学和免疫组织化学分析
为了进行组织病理学分析,左肺用石蜡块包埋,切成4µm切片,用H&E染色。支气管周围和血管周围炎症按B阿伦兹et al。[23]为不存在(0)、最小(1)、轻微(2)、中度(3)、标记(4)或严重(5)。采用盲法进行玻片分析。对于免疫组化,肺切片进行抗原回收,随后用抗rsv F蛋白抗体标记。
我耗尽
通过鼻内给药氯膦酸脂质体(Sigma-Aldrich)(每只动物500µg)或PBS脂质体作为对照来消耗AMs。小鼠感染RSV (107每只动物PFU) 24小时后。如上所述,每天对动物称重,感染后5天进行数据分析。BAL标本经流式细胞仪分析证实巨噬细胞耗竭。
流式细胞术
目的分析RSV感染后细胞死亡的百分比在体外,细胞用eFluor 780固定活力染料(1:1000)在避光的冰上标记30分钟,然后用轻轻刮取。然后用PBS清洗细胞,加入细胞计数缓冲液。使用BD FACS Verse (BD Biosciences, San Jose, CA, USA)流式细胞仪采集样品,并使用FlowJo软件(version 10, Tree Star Inc., San Jose, CA, USA)进行分析。
从感染小鼠的BAL液中分离的细胞用小鼠Fc Block孵育20分钟,然后用eFluor 780(1:1000)在冰上标记30分钟。然后用抗cd11c表面抗体(1:200;克隆N418, BioLegend, San Diego, CA, USA), anti-F4/80 (1:200;克隆BM8, bilegend)和抗cd170 (Siglec-F) (1:200;克隆1RNM44N, eBioscience by ThermoFisher Scientific)在冰上放置30分钟。使用BD FACS Verse流式细胞仪采集样品,并使用FlowJo软件进行分析。
实时PCR定量病毒载量
按照制造商的说明,使用GoScript逆转录酶试剂盒(Promega)提取肺总RNA,并合成cDNA。采用指定的特异性引物和探针进行RSV F蛋白基因的实时PCR扩增:正向引物:5 ' -AACAGATGTAAGCAGCTCCGTTATC-3 ';反向引物:5 ' - gatttttattggatgctgtacatt3 ';探针:5 ' FAM/TGCCATAGCATGACACAATGGCTCCT-TAMRA/-3 '。引物序列被合成并克隆到pUC57质粒(GenScript, Piscataway, NJ, USA)中进行10倍稀释,并生成用于计算病毒载量的标准曲线。从病毒拷贝中获得的值(基于质粒对照的浓度)相对于预称重的肺部分的重量(拷贝·g肺)计算−1).
人类的样本
鼻咽灌洗样本取自2016年至2018年出生第一年因毛细支气管炎住院的婴儿。这些样本来自有72小时以下呼吸道感染临床症状的患者,如咳嗽、喘息和/或呼吸窘迫。所有患者在住院第一天进行数据收集和鼻咽冲洗以识别呼吸道病毒。通过免疫荧光和RT-PCR检测患者样本中RSV的存在。采集后,鼻咽洗液用1ml生理盐水稀释,均质离心。收集上清液并在- 80°C冷冻,直到进一步分析。
道德声明
对于婴儿样本采集,所有参与者的父母或法定监护人在样本采集前签署知情同意书。该研究由PUCRS研究伦理委员会(CEP/PUCRS)审查并批准,协议号为1.158.826和2.471.678。所有程序都遵循《赫尔辛基宣言》规定的标准。
统计分析
数据以均数±表示扫描电镜.所有在体外实验一式三次,重复至少两次。在活的有机体内实验共进行2次(每次实验每组n≥3只)。使用GraphPad Prism 6统计软件包(GraphPad software, La Jolla, CA, USA)对所得结果进行分析。多组间比较采用单因素方差分析(one-way ANOVA)和a事后Tukey或Bonferroni检验。适当时采用非配对t检验或Mann-Whitney检验。显著性水平设定为p≤0.05。
结果
RSV可诱导小鼠原代巨噬细胞、人单核细胞和巨噬样细胞系坏死
为了评估RSV对AM坏死的影响,细胞被RSV (MOI 1)感染6小时,并通过渗透死细胞受损膜的活性染料评估细胞死亡。我们的结果显示RSV引发AM坏死(图1一个),而且RSV对巨噬细胞死亡的影响依赖于病毒复制,因为紫外线(UV)灭活会损害RSV诱导的死亡(图1一个).为了证实AMs可被RSV感染,我们在感染6小时后定量了病毒载量。事实上,RSV能够感染小鼠AMs (图1 b).有趣的是,RSV也能杀死腹膜巨噬细胞(图1 c),同样,紫外线灭活的RSV没有引发细胞死亡(图1 c),表明活跃的RSV感染是促进巨噬细胞死亡所必需的。rsv诱导坏死的进一步特征显示AM和腹腔巨噬细胞坏死均以浓度依赖的方式发生(补充图S1a, d).在感染后定量测定乳酸脱氢酶的释放时,也观察到类似的结果(补充图S1b, e).急性脑脊髓炎可在感染后2小时内检测到细胞死亡(补充图S1c)和感染后4小时腹腔巨噬细胞(补充图S1f).此外,我们发现RSV能够诱导人单核细胞坏死(补充图S2a, d),在人巨噬细胞样THP-1细胞(补充图S2b, e)和人U937单核细胞经PMA分化为巨噬细胞样细胞(补充图S2c, f).我们证实RSV在所有这些细胞类型中感染和复制(补充图S2g),并以人肺上皮细胞系A549作为RSV复制的阳性对照(补充图S2g).最后,我们测试了RSV临床分离株对人单核细胞坏死的影响。两种不同的RSV临床分离株能够促进人单核细胞坏死(图1 d).总之,这些数据表明,肺泡和腹膜小鼠巨噬细胞、人单核细胞和人巨噬细胞样细胞系易发生RSV感染诱导的坏死。此外,我们的研究结果表明,从患者身上分离出的临床RSV菌株也会引发人单核细胞坏死。
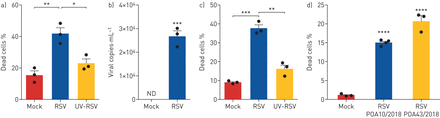
呼吸道合胞病毒(RSV)可诱导小鼠原代巨噬细胞、人单核细胞和巨噬样细胞系坏死。a) Balb/c小鼠源性肺泡巨噬细胞(AMs) (3×105·好−1)用RSV(感染多重性(MOI) 1)或用紫外线灭活(UV) RSV (MOI 1)在37℃、5% CO下感染6 h2.用eFluor 780固定活性染料标记细胞,收获后用流式细胞术分析死亡细胞的百分比。b) AMs (3×105·好−1)感染了RSV (MOI 1)或在37°C和5% CO下保持未感染6小时2.收集RNA,实时PCR检测RSV病毒载量。c) Balb/c小鼠来源的腹膜巨噬细胞(3×105·好−1)感染RSV (MOI 1)或UV-RSV (MOI 1) 6 h。收集细胞,用eFluor 780标记,流式细胞术分析死亡细胞的百分比。d)人类单核细胞(3×105·好−1)用两种不同社区分离的RSV (MOI 1)感染6 h。用eFluor 780固定活性染料标记细胞,收获后用流式细胞术分析死亡细胞的百分比。数据代表三次独立实验,每次重复三次,用均数±表示扫描电镜.数据分析采用单因素方差分析(a, c, d)或未配对t检验(b)。ND:未检出。*: p < 0.05;* *: p < 0.01;* * *: p < 0.001;* * * *: p < 0.0001。
RIPK1, RIPK3和MLKL在rsv诱导的巨噬细胞坏死中是必需的
大多数关于坏死的研究通常在缺乏caspase信号的情况下,用炎症剂刺激免疫细胞和非免疫细胞(使用苄氧羰基- val - ala - asp -氟甲基酮(zVAD-FMK)) [24].我们的研究结果表明,在RSV感染之前用zVAD-FMK治疗巨噬细胞,与RSV单独治疗相比,并不能减少细胞死亡(图2一个),提示RSV感染不诱导细胞凋亡,也不需要zVAD-FMK抑制caspase诱导巨噬细胞坏死。为了确定RIPK1在rsv驱动的巨噬细胞死亡中的作用,研究人员用坏死抑制素-1 (nec1)预处理细胞,nec1靶向RIPK1的激酶活性[25].用NEC-1预处理RSV引起的巨噬细胞死亡显著受损(图2 b).RIPK3在rsv引发的巨噬细胞死亡中的作用是通过感染巨噬细胞来检测的Ripk3−−/对照组WT小鼠注射RSV 6 h。值得注意的是,Ripk3−−/此时,巨噬细胞完全抵抗rsv诱导的细胞死亡(图2 c).同样,GW42X对RIPK3的药理抑制作用(Synkinase, Parkville, VIC, Australia) [26]保护人类单核细胞免受RSV感染导致的死亡(图2 d)和necrosulfonamide (NSA) (Millipore, Billerica, MA, USA)对MLKL的抑制作用[14]阻断rsv引发的人单核细胞坏死(图2 e).接下来,我们试图研究抑制坏死途径是否会影响RSV复制。事实上,RIPK1、RIPK3和MLKL的药物阻断显著降低了人单核细胞中的RSV病毒载量(图2 f).同样的,Ripk3−−/与感染RSV的WT巨噬细胞相比,巨噬细胞表现出明显较低的病毒载量(图2 g).有趣的是,zad - fmk抑制caspase并不影响人单核细胞的病毒载量(图2 f),表明caspases在RSV感染过程中不参与病毒复制。然后我们决定验证RSV是否会诱导巨噬细胞中RIPK3和MLKL的表达。RSV感染导致巨噬细胞显著Ripk3而且Mlkl感染后6 h mRNA表达(图2 h, i)以及感染后2小时和4小时MLKL蛋白水平的升高(补充图S3).综上所述,这些结果表明RSV通过激活RIPK1、RIPK3和MLKL充分促进小鼠巨噬细胞和人单核细胞坏死,坏死途径不利于病毒清除。
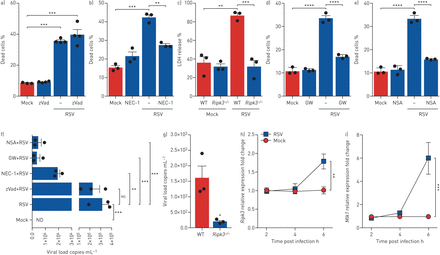
RIPK1, RIPK3和MLKL在呼吸道合胞病毒(RSV)诱导的巨噬细胞坏死中是必需的。a, b) Balb/c小鼠来源的巨噬细胞(3×105·好−1)用zVad - fmk(20µM) (zVad) (a)或用necrostatin-1 (nec1)(30µM) (b)在RSV感染(多重感染(MOI) 1)前1小时治疗6小时。巨噬细胞用eFluor 780固定活性染料标记,收获后用流式细胞术分析死亡细胞的百分比。c)肺泡巨噬细胞(AMs) (3×105·好−1)Ripk3−−/感染同窝对照野生型(WT)小鼠体外RSV (MOI 1)作用6小时。通过巨噬细胞上清液中乳酸脱氢酶(LDH)的释放来评估细胞死亡。d, e)人类单核细胞(3×105·好−1)在RSV感染(MOI 1)前6小时,用GW42X (GW)(2µM) (d)或用尸磺酰胺(NSA)(2µM) (e)处理1小时。用eFluor 780固定活性染料标记细胞,收获后用流式细胞术分析死亡细胞的百分比。f)在RSV感染(MOI 1) 6小时之前,用zVAD-FMK(20µM)、NEC-1(30µM)、GW42X(2µM)或NSA(2µM)处理巨噬细胞1小时。收集RNA,实时PCR检测RSV病毒载量。g)巨噬细胞(3×105·好−1)Ripk3−−/对照组WT小鼠感染体外RSV (MOI 1)作用6小时。收集RNA,实时PCR检测RSV病毒载量。h, i) AMs感染RSV (MOI 1) 2、4或6 h。收集RNARipk3mRNA (h)和MlklmRNA (i)的表达采用实时荧光定量PCR,使用2−ΔCt方法。数据代表至少两个独立的实验,每次重复三次,并以均数±表示扫描电镜.数据采用Tukey's单因素方差分析事后检验(a-f, h, i)或未配对t检验(g)。ns:不显著;ND:未检测到。*: p < 0.05;* *: p < 0.01;* * *: p < 0.001;* * * *: p < 0.0001。
RIPK3在RSV感染期间以部分依赖于细胞死亡的方式加剧肺部病理
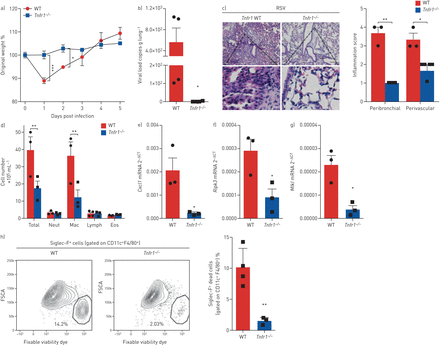
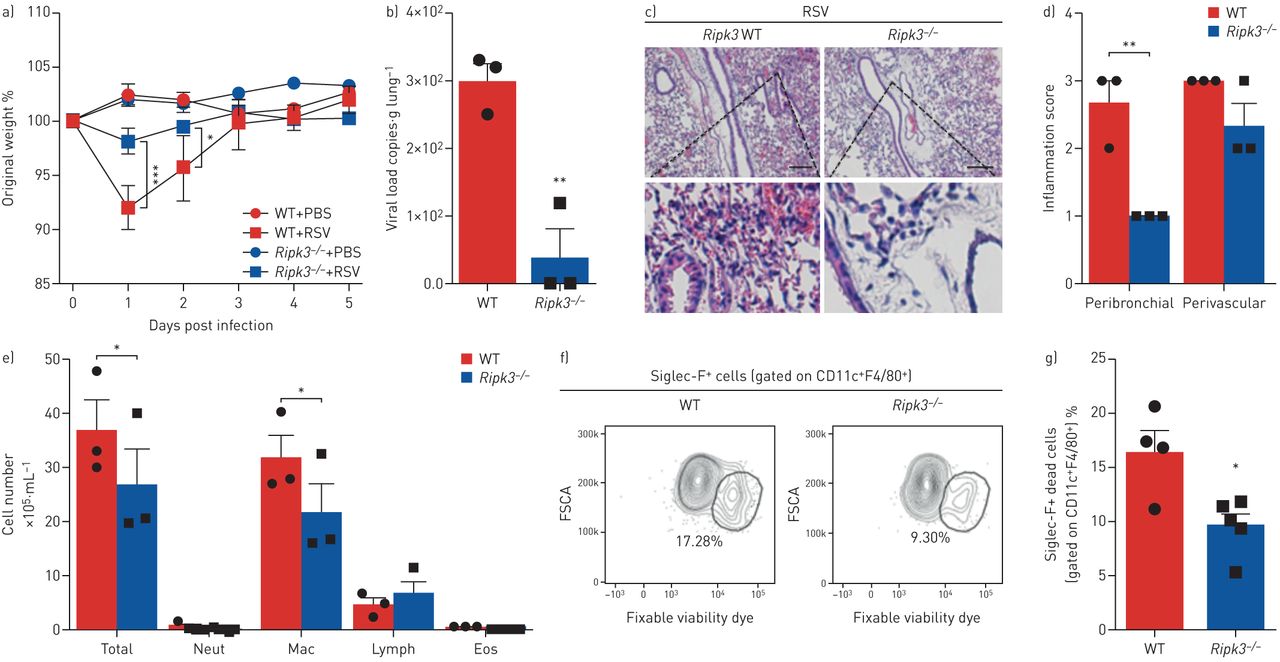
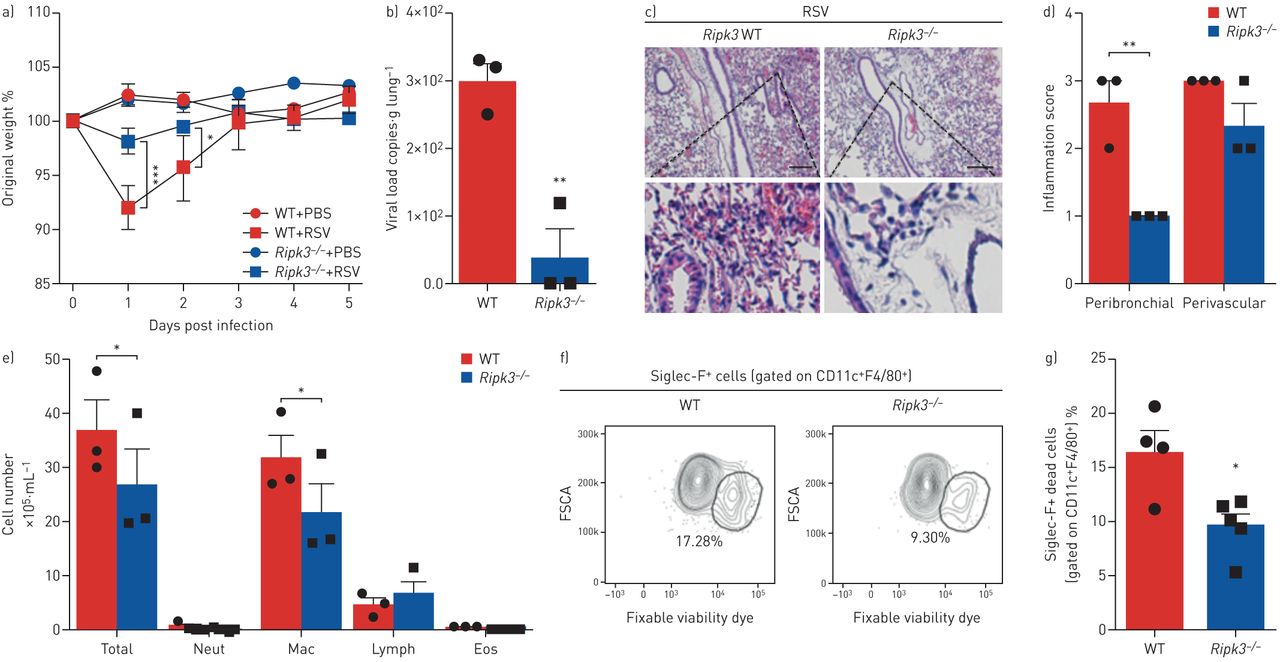
此前已有研究表明,RIPK3基因缺失可保护ciap2缺陷小鼠免受甲型H1N1流感病毒感染引起的发病和死亡[17].此外,Ripk3−−/与WT小鼠相比,感染H7N9流感病毒的小鼠存活率更高,体重减轻和炎症细胞浸润更少[18].基于文献和我们的发现,RSV通过RIPK3激活诱导巨噬细胞坏死,我们试图研究RIPK3在RSV感染过程中的作用在活的有机体内.Ripk3−−/与WT小鼠相比,rsv小鼠免于体重减轻(图3一),这与肺部病毒载量的显著降低有关(图3 b).Ripk3−−/与WT小鼠相比,小鼠的肺支气管周围炎症评分也显著降低(图3 c, d)。此外,ripk3缺陷小鼠BAL液中巨噬细胞和总细胞数量显著减少(图3 e).然后,我们通过检查BAL样本中坏死的AM群体,探讨RIPK3是否会在RSV感染期间调节AM坏死Ripk3−−/和WT小鼠。Ripk3−−/小鼠表现出坏死AMs (CD11c+F4/80+Siglec-F+可固定活性染料+)在RSV感染后的BAL液中(图3 f, g和补充图S4).这些结果表明RIPK3在RSV感染期间加剧了肺部病理在活的有机体内这部分与AM死亡结果相关。
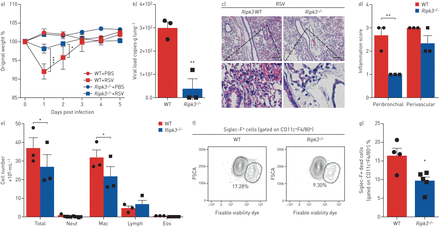
RIPK3在呼吸道合胞病毒(RSV)感染期间加重肺部病理,部分依赖于细胞死亡。Ripk3−−/RSV感染小鼠和同窝对照野生型小鼠(107空斑形成单位·动物−1).感染后5天进行数据分析。a)感染后体重下降相对于初始体重的百分比(第0天)(n=5)。b) real-time PCR检测肺组织中RSV病毒载量(n=3)。c)苏木精、伊红染色肺组织切片代表性图像。比例尺:200µm。d)各自的炎症评分(n=3)。e)支气管肺泡灌洗液(BAL)中总细胞数和分化细胞计数(n=3)。f)坏死的肺泡巨噬细胞百分比(CD11c .+F4/80+Siglec-F+可固定活性染料+)和g)其代表的荧光激活细胞分选谱(n= 4-5)。数据代表两个独立的实验,用均数±表示扫描电镜.数据采用Bonferroni's单因素方差分析事后(a)或Mann-Whitney检验(b-e)。中性粒细胞:中性粒细胞;麦克:巨噬细胞;淋巴:淋巴细胞;Eos:嗜酸性粒细胞;FSCA:正向散射区。*: p < 0.05;* *: p < 0.01;* * *: p < 0.001。
AMs在RSV感染过程中起着不利作用在活的有机体内
AMs在呼吸道病毒感染中所起的作用一直是一个有争议的问题。AMs似乎对RSV感染的早期抗病毒反应至关重要[27,28].然而,AMs的消耗与疾病的显著改善有关,包括减毒体重减轻,在新生感染小鼠RSV再感染期间不改变病毒载量[29].为了确定AMs在RSV感染过程中的作用,我们在小鼠感染前24小时通过鼻内给药氯膦酸脂质体来消耗这些细胞。利用抗cd11c、抗f4 /80和抗siglec - f抗体识别小鼠AM,通过流式细胞术分析BAL细胞,首次证实AM耗竭。与PBS脂质体处理的小鼠相比,氯膦酸脂质体处理24小时的小鼠BAL液中AM数量显著减少3倍(补充图S5a, b).在RSV感染后,am耗尽的小鼠表现出明显较少的体重减轻(图4一),与未耗尽的小鼠相比,肺部病毒载量急剧下降(图4 b).RSV感染am缺失小鼠肺组织免疫组化分析未检测到RSV F蛋白,与病毒载量下降相对应(图4 c).此外,AM耗竭导致感染小鼠的肺组织学炎症评分显著降低(图4 b, e).正如预期的那样,am耗尽小鼠在BAL液中显示出较低数量的巨噬细胞,但令人惊讶的是,大量的中性粒细胞(图4 f).作为细胞损伤的标志,我们测量了小鼠BAL样品中的LDH活性。与未感染的小鼠相比,我们能够检测到rsv感染小鼠BAL液中LDH的水平升高;然而,氯膦酸脂质体治疗不影响小鼠LDH的释放(图4 g).有趣的是,AM的消耗消除了rsv诱导的上调Ripk3而且Mlkl肺组织中的mRNA (图4 h, i).综上所述,这些数据表明AMs在RSV感染过程中起着有害的作用,AMs是感染后肺组织中RIPK3和MLKL表达的主要原因。
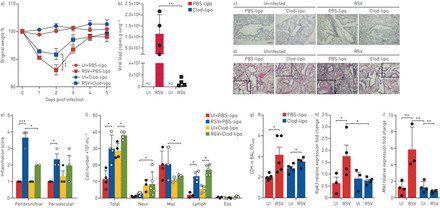
肺泡巨噬细胞在呼吸道合胞病毒(RSV)感染中起着有害的作用在活的有机体内.通过鼻内给药氯膦酸脂质体(Clod-lipo)(500µg·动物),Balb/c小鼠肺泡巨噬细胞减少−1)或PBS脂质体(PBS-lipo)。小鼠感染RSV (107空斑形成单位·动物−1) 24小时后。感染后5天进行数据分析。a)感染后体重下降相对于初始体重的百分比(第0天)(n=5)。b) real-time PCR检测肺组织RSV病毒载量(n= 4-5)。c)使用抗rsv融合蛋白抗体进行肺组织免疫组化染色的代表性图像。比例尺:200µm。d)苏木精、伊红染色肺组织切片代表性图像。比例尺:200µm。e)与d所示组织学图像相对应的支气管周围和血管周围炎症评分(n=3)。f)支气管肺泡灌洗液中总细胞数和细胞计数(n= 4-5)。 g) Lactate dehydrogenase (LDH) activity measured at an optical density of 490 nm (OD490) (n= 4-5)。h,我)Ripk3mRNA (h)和MlklmRNA (i)在肺组织中的表达,实时荧光定量PCR,使用2−ΔCt方法,并表示为折叠变化(n= 3-4)。数据代表两个独立的实验,用均数±表示扫描电镜.数据采用Tukey's单因素方差分析事后测试。UI:未感染;ND:未检测到;中性粒细胞:中性粒细胞;麦克:巨噬细胞;淋巴:淋巴细胞;Eos:嗜酸性粒细胞。*: p < 0.05;* *: p < 0.01;* * *: p < 0.001。
自分泌TNF介导rsv诱导的巨噬细胞坏死在体外
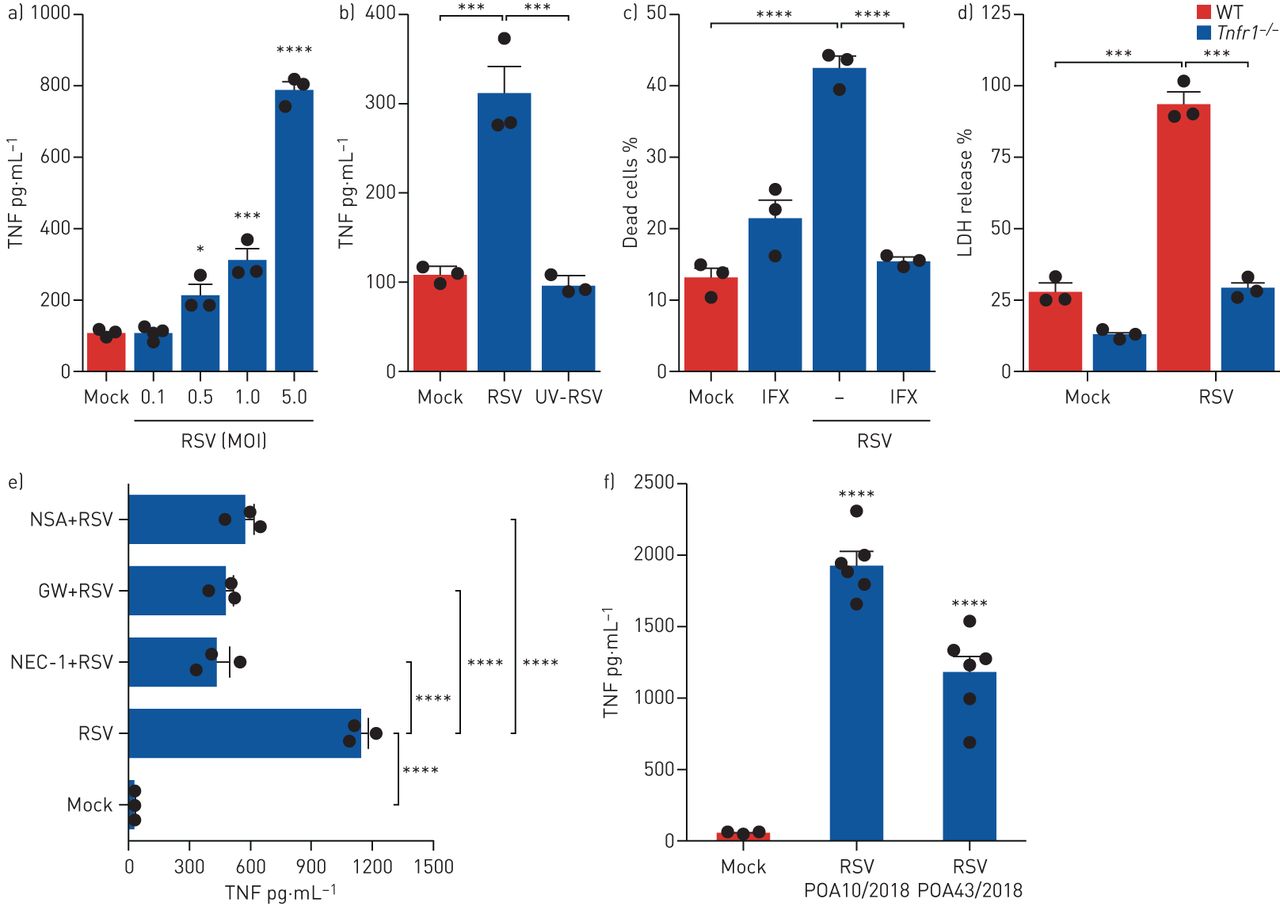
我们最近证明了巨噬细胞对体外RSV感染通过释放TNF [30.].因为TNF作为坏死触发因子已被广泛研究[31],我们试图阐明TNF在rsv诱导的巨噬细胞坏死中的作用。RSV以浓度依赖的方式诱导巨噬细胞分泌TNF (图5一个),但只有复制病毒能够诱导TNF释放(图5 b).为了探索TNF是否在RSV引发的巨噬细胞坏死中发挥作用,我们用治疗性TNF特异性中和抗体(英夫利昔单抗)处理细胞,这是一种与小鼠TNF交叉反应的嵌合抗体(小鼠/人)[32].在RSV感染前立即用英夫利昔单抗治疗巨噬细胞。英夫利昔单抗中和TNF可消除rsv诱导的巨噬细胞坏死在体外(图5 c).有趣的是,用brefeldin A(一种著名的蛋白质转运抑制剂)阻断TNF分泌[33],巨噬细胞上清中TNF浓度降低至对照水平(模拟)(补充图S6a)和大幅减少RSV引起的巨噬细胞死亡(补充图S6b).然后我们感染巨噬细胞Tnfr1−−/对照组WT小鼠为RSV体外.值得注意的是,Tnfr1−−/巨噬细胞完全免受rsv诱导的死亡(图5 d).接下来,我们试图进一步阐明坏死途径在RSV感染诱导的TNF释放中的作用。药物抑制RIPK1、RIPK3和MLKL显著降低RSV促进人单核细胞TNF分泌(图5 e),提示坏死途径对于rsv引发的TNF释放是必要的。最后,我们观察到用两种不同的RSV临床分离株感染人单核细胞能够诱导TNF释放(图5 f).总的来说,这些结果表明rsv触发的巨噬细胞坏死是由自分泌TNF介导的,坏死途径参与了TNF的释放。
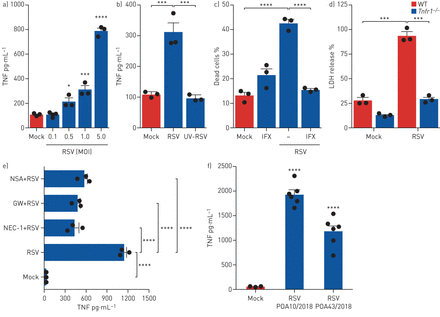
自分泌肿瘤坏死因子(TNF)介导呼吸道合胞病毒(RSV)诱导的巨噬细胞坏死在体外.a, b) Balb/c小鼠来源的巨噬细胞(3×105·好−1)感染RSV感染多重性(MOIs)增加(0.1-5)6 h (a)或活性或紫外线灭活RSV (UV-RSV) (MOI 1) 6 h (b)。收集上清液并用ELISA法测定TNF浓度。c) Balb/c小鼠来源的巨噬细胞(3×105·好−1)在英夫利昔单抗(IFX)存在下(10µg·mL)感染RSV (MOI 1)−1) 6小时。用eFluor 780固定活性染料标记细胞,收获后用流式细胞仪分析死亡细胞的百分比。d)肺泡巨噬细胞(3×105·好−1)Tnfr1−−/感染同窝对照野生型(WT)小鼠体外RSV (MOI 1)作用6小时。通过巨噬细胞上清液中乳酸脱氢酶(LDH)的释放来评估细胞死亡。e)人类单核细胞(3×105·好−1)在RSV感染(MOI 1) 6小时之前,用necrostatin-1 (NEC-1)(30µM)、GW42X (GW)(2µM)或necrosulfonamide (NSA)(2µM)处理1小时。收集细胞上清液,ELISA法测定TNF浓度。f)人类单核细胞(3×105·好−1)用两种不同社区分离的RSV (MOI 1)感染6 h。收集细胞上清液,ELISA法测定TNF浓度。数据代表至少两个独立的实验,每次重复三次,并以均数±表示扫描电镜.数据采用Tukey's单因素方差分析事后测试。*: p < 0.05;* * *: p < 0.001;* * * *: p < 0.0001。
缺乏TNFR1信号可以改善RSV感染诱导的病理,减少AM坏死
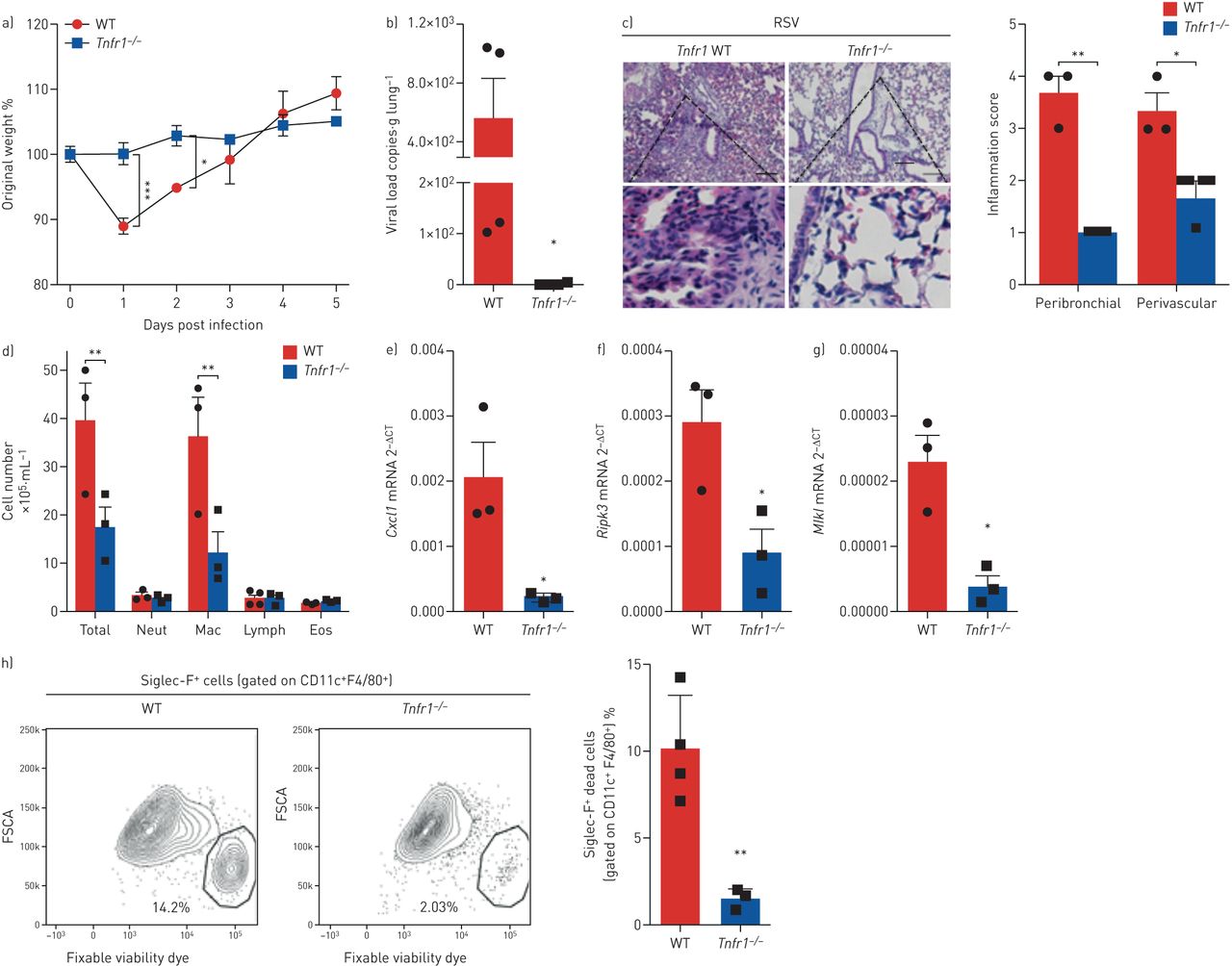
基于我们的发现,自分泌TNF介导rsv诱导的巨噬细胞坏死体外,我们有兴趣阐明TNF在RSV感染和RSV诱导的AM坏死中的作用在活的有机体内.因此,Tnfr1−−/与同窝WT小鼠感染RSV。我们的研究结果表明Tnfr1−−/小鼠免于rsv引起的体重减轻(图6),并显示肺组织的病毒载量显著降低(图6 b)与WT小鼠相比。此外,Tnfr1−−/与WT小鼠相比,小鼠的组织学支气管周围和血管周围炎症评分降低(图6 c).白细胞总数,以及巨噬细胞的数量,在BAL液Tnfr1−−/小鼠RSV感染后明显低于WT小鼠(图6 d).有趣的是,的表达处于受控肺组织中的mRNATnfr1−−/小鼠严重受损(图6 e),证实炎症在这些小鼠感染RSV期间被抑制。类似地,的表达式Ripk3而且Mlkl转录本显著减少Tnfr1−−/老鼠(图6 f, g),提示TNF介导RSV感染后肺组织中RIPK3和MLKL的表达。最后,为了确定缺乏TNFR1信号是否会影响RSV感染期间AM的坏死,我们分析了BAL样本中坏死的AM种群Tnfr1−−/和WT小鼠。值得注意的是,Tnfr1−−/小鼠的坏死性AMs (CD11c)频率下降了6倍+F4/80+Siglec-F+可固定活性染料+)在RSV感染后的BAL液中(图6 h而且补充图S7).总之,这些数据强烈表明TNFR1信号通路介导由RSV感染引发的肺病理和AM坏死。
肿瘤坏死因子受体1 (TNFR1)信号缺失改善呼吸道合胞病毒(RSV)感染诱导的病理,减少肺泡巨噬细胞坏死。Tnfr1−−/RSV感染小鼠和同窝对照野生型小鼠(107空斑形成单位·动物−1).感染后5天进行数据分析。a)感染后体重下降相对于初始体重的百分比(第0天)(n=5)。b) real-time PCR检测肺组织中RSV病毒载量(n=4)。c)苏木精和伊红染色的肺组织切片代表性图像及其各自的炎症评分(n=3)。比例尺:200µm。d)支气管肺泡灌洗液中总细胞数和分化细胞计数(n=3)。eg)处于受控信使rna (e),Ripk3mRNA (f)和MlklmRNA (g)在肺组织中的表达用实时PCR定量2−ΔCt方法(n = 3)。h)坏死的肺泡巨噬细胞百分比(CD11c .+F4/80+Siglec-F+可固定活性染料+)及其具有代表性的荧光激活细胞分选谱(n=4)。数据代表两个独立的实验,用均数±表示扫描电镜.数据采用非配对t检验进行分析。中性粒细胞:中性粒细胞;麦克:巨噬细胞;淋巴:淋巴细胞;Eos:嗜酸性粒细胞;FSCA:正向散射区。*: p < 0.05;* *: p < 0.01;* * *: p < 0.001。
RSV细支气管炎患儿洗鼻液中TNF水平升高与疾病严重程度相关
我们通过测量患有RSV细支气管炎的婴儿洗鼻样品中的TNF水平来评估TNF在急性RSV感染中的作用。RSV患者的年龄(p=0.6784)、性别(p=0.1507)和出生体重(p=0.4484)差异无统计学意义+与RSV相比−集团(补充表S1).RSV的鼻样本中TNF的浓度明显更高(p=0.0014)+婴儿(209.41±23.55 pg·mL)−1, n=39)−婴儿(113.38±14.16 pg·mL)−1, n=5) (图7).接下来,我们分析了TNF水平与需要补充氧疗的关系。TNF水平与需氧量之间呈正相关趋势,尽管不显著(p=0.0649) (图7 b).相反,洗鼻液中TNF水平与住院时间呈显著正相关(p=0.0010) (图7 c).有趣的是,住院时间越长(≥7天)的婴儿在洗鼻液中TNF浓度越高(341.6±60.24 pg·mL)−1(119.7±17.93 pg·mL)高于住院时间短(<7天)的患者−1) (p=0.0002) (图7 d).这些数据表明,婴儿RSV感染时,洗鼻液中TNF水平升高与疾病严重程度呈正相关。
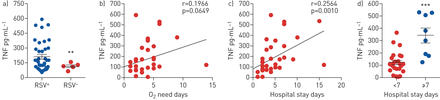
呼吸道合胞病毒(RSV)细支气管炎婴儿鼻腔洗液中肿瘤坏死因子(TNF)水平升高与疾病严重程度相关。从因急性细支气管炎入院的12个月以下婴儿中收集鼻洗液样本。实时荧光定量PCR检测RSV的存在。a)用ELISA法测定鼻洗液上清中的TNF水平,并在RSV阳性的婴儿中进行比较(RSV+)和婴儿RSV阴性(RSV−).b) TNF水平与O时间呈Spearman线性相关2RSV需求+患有急性细支气管炎的婴儿。c) TNF水平与RSV住院时间Spearman线性相关+患有急性细支气管炎的婴儿。d) RSV中TNF水平的比较+住院时间<7天的婴儿与住院时间≥7天的婴儿。数据以均数±表示扫描电镜采用未配对t检验(a, d)进行分析。**:p=0.0014;* * *: p = 0.0002。
讨论
呼吸道病毒已被证明可引发不同类型细胞的坏死[16,34- - - - - -36].据此,近期研究报道RSV可诱导肺泡上皮细胞和THP-1细胞坏死[19,20.].然而,RSV是否能够引发原代巨噬细胞的坏死,而巨噬细胞是对抗这种病毒感染的免疫反应的重要细胞,尚未得到研究。在本研究中,我们证明RSV感染以浓度和时间依赖的方式诱导小鼠肺泡和腹膜巨噬细胞坏死。此外,考虑到紫外线灭活RSV不能引发巨噬细胞死亡,RSV的复制活性对于诱导坏死至关重要。我们通过显示人类单核细胞、THP-1细胞和U937细胞分化为巨噬细胞样细胞都易受RSV感染引发的坏死扩展了我们的研究结果。重要的是,为了努力使用具有临床代表性的RSV菌株,我们用两种不同的RSV群落分离物感染人类单核细胞,并证明这些分离物能够促进坏死。
当caspase-8活性降低时,就会发生坏死,而众所周知,病毒编码的caspase-8抑制剂可能会将细胞死亡程序转换为坏死,就像痘苗病毒的情况一样。由于caspase-8抑制剂B13R的表达,痘苗病毒感染的细胞容易发生tnf诱导的RIPK1-和ripk3依赖性坏死[15,37,38].迄今为止,文献中还没有rsv编码的caspase-8抑制剂的描述。因此,为了抑制caspase-8活性,我们在RSV感染前用泛caspase抑制剂zVAD-FMK处理巨噬细胞。然而,在我们的研究中,zVAD-FMK不能进一步增加RSV诱导的巨噬细胞坏死,这表明RSV感染足以诱导坏死。我们的数据得到了先前研究结果的支持,这些研究表明RSV自然触发肺泡上皮细胞坏死[20.].
RIPK1和RIPK3通过RIP同型相互作用基序(RHIM) -RHIM结构域的相互作用是坏死信号传导的核心,并导致坏死体的形成[39],导致RIPK1和RIPK3的相互磷酸化和激活[40].RIPK3的激活导致其底物MLKL的磷酸化,MLKL依次寡聚并易位到质膜,在那里形成膜破坏孔,导致细胞裂解[41- - - - - -44].为了描述rsv引起的AM坏死过程中RIPK1的需求,我们用NEC-1处理细胞,它可以阻断RIPK1的激酶活性[25].用NEC-1处理巨噬细胞可显著降低RSV诱导的细胞死亡。此外,鉴于此,我们已经证明RIPK3对于rsv引发的坏死至关重要Ripk3−−/巨噬细胞完全抵抗病毒促进的细胞死亡,RIPK3的药理抑制实际上消除了RSV感染引起的人单核细胞坏死。同样,NSA对MLKL的抑制严重损害rsv刺激的人单核细胞坏死。重要的是,我们发现RSV诱发了这两种疾病Ripk3而且Mlkl感染后6小时mRNA表达。我们还观察到,对坏死途径的药理学抑制或基因缺失显著降低了AMs和人类单核细胞中的病毒载量,这表明坏死促进了RSV在这些细胞中的持久性。与我们的研究结果一致,RIPK3已被证明促进柯萨奇病毒在肠上皮细胞中的复制[45].因此,AMs和人类单核细胞明显依赖RIPK1、RIPK3和MLKL来进行细胞死亡,这些分子作为RSV复制的正向调节因子。
基于RIPK3的重要性在体外rsv诱导的巨噬细胞坏死,我们认为RIPK3对rsv诱导的疾病至关重要在活的有机体内.事实上,RIPK3的遗传缺陷保护了小鼠免受rsv诱导的体重减轻,并降低了肺部的病毒载量。此外,Ripk3−−/小鼠显示巨噬细胞向肺的招募受损,这可能是RIPK3缺失时病毒复制减少的继发原因。因此,我们为RIPK3在RSV感染过程中的有害作用提供了证据。同样,RIPK3缺陷通过减少体重减轻和肺部炎症细胞的浸润来保护小鼠免受H7N9流感病毒的感染[18].此前已有研究表明,RIPK3同时激活mlkl驱动的坏死和fas相关蛋白与死亡结构域(FADD)介导的凋亡,以防止甲型H1N1流感病毒感染的传播[16].然而,据报道RIPK3在不引发细胞死亡的情况下促进炎症和调节病毒复制[46].在我们的研究中,RIPK3在一定程度上通过促进炎症和AM坏死来加重肺损伤。因此,RIPK3在病毒感染期间的生理相关性可能由病毒亚型和宿主免疫反应中可能的干扰因素决定。作为坏疽体形成的重要组成部分,RIPK1在小鼠病毒性细支气管炎中也是有害的,因为RIPK1的药理学阻断和遗传缺陷都保护小鼠免于体重减轻、中性粒细胞炎症和alarmin释放[20.].因此,RIPK1和RIPK3是RSV感染过程中的病理参与者。
AMs对呼吸道病毒诱发疾病的影响是有争议的。虽然AMs似乎对早期对抗RSV感染的抗病毒反应至关重要[27,28,47,48]及预防致命流感肺炎[49],在小鼠感染肺冠状病毒和人偏肺病毒后,AMs的消耗与疾病的显著改善有关,包括减轻体重和减少气道阻塞[27,50].此外,AM耗损延长了小鼠急性肺炎病毒感染的生存期,并在不改变新生感染小鼠RSV再感染期间的病毒载量的情况下减少了体重减轻[29,51].我们的数据清楚地表明,AMs增强RSV在肺部的复制,并显著促进临床疾病和肺部病理。在am耗尽小鼠中,肺部病毒载量降低,体重减轻和支气管周围炎症评分受损,始终强调了这些发现。因此,我们有理由认为AMs可能是RSV感染肺部的第一个免疫细胞靶点,在疾病早期扮演病毒工厂的角色。重要的是,AM耗尽消除了rsv诱导的上调Ripk3而且MlklmRNA,提示AMs占感染后肺组织中RIPK3和MLKL的大部分表达,导致肺坏死。
由于RSV诱导的巨噬细胞坏死需要病毒复制,我们假设在RSV复制周期中坏死机制被间接激活,如。通过病毒诱导的促坏死细胞因子(如TNF)的自分泌活动。与这一假设一致,我们最近发现巨噬细胞对RSV感染的反应是通过分泌TNF [30.].我们扩展了这些发现,并表明RSV以浓度依赖的方式触发TNF的释放,并且TNF的释放是RSV复制的结果,因为紫外线灭活的RSV不能诱导巨噬细胞分泌TNF。然后我们研究TNF是否介导rsv促进的巨噬细胞坏死。用英夫利昔单抗(一种治疗性抗TNF抗体)中和TNF,可消除rsv诱导的巨噬细胞坏死。有趣的是,当TNF分泌被brefeldin A阻断时,RSV对巨噬细胞死亡的影响消失了。此外,tnfr1缺陷的巨噬细胞完全抵抗rsv引发的细胞死亡。因此,我们的研究结果证实,TNF在AMs中RSV复制期间分泌,进而以自分泌方式促进巨噬细胞坏死。此外,最近有研究表明,即使在质膜完整性丧失后,RIPK3的激活也会导致细胞因子的产生[52].因此,我们决定阐明RIPK1、RIPK3和MLKL是否会促进rsv诱导坏死的TNF合成。坏死蛋白的药理学阻断显著损害rsv诱导的TNF分泌。这些数据表明,RSV除了通过其在巨噬细胞中的复制活性诱导TNF生成外,还通过激活坏死机制导致TNF合成,即使巨噬细胞已导致细胞死亡。坏死激活分泌TNF可导致正反馈循环,恶化RSV感染引起的肺部病理。与我们的研究结果一致,流感病毒也引发ripk3介导的坏死[16,35],已被报道促进了这种现象[52].
鉴于我们对TNF参与的观察在体外我们试图阐明TNF在RSV感染和RSV诱导的AM坏死中的作用在活的有机体内.在我们的研究中,TNFR1信号的缺乏对rsv诱导的体重减轻和肺部炎症具有保护作用。此外,Tnfr1−−/小鼠肺部的病毒载量和巨噬细胞浸润明显减少。相应地,缺乏TNF和白细胞介素1 (IL-1)受体的小鼠表现出肺部炎症减轻和H5N1病毒致命挑战引起的死亡率显著延迟[53].此外,TNF和巨噬细胞对rsv诱导的小鼠哮喘加重至关重要[54].我们发现的表达式Ripk3而且Mlkl肺部有明显损伤吗Tnfr1−−/RSV感染小鼠后,这可能反映了TNFR1信号缺失时坏死AMs百分比的显著降低。我们提供了令人信服的证据,TNF通过增加病毒载量和肺部病理,以及促进RIPK3和mlkl依赖的AM坏死,在RSV感染期间与临床疾病的严重程度密切相关。为了支持我们的发现,先前的研究表明,在不同的细胞类型和小鼠模型中,tnf诱导的坏死需要激活RIPK1、RIPK3和MLKL [41,55,56].此外,在tnf诱导的全身炎症反应综合征中,ripk1 - ripk3介导的坏死细胞损伤导致死亡[57].
先前的研究表明,在感染RSV的婴儿洗鼻液中,TNF与其他促炎细胞因子一起升高[58- - - - - -60].我们的研究结果证实了先前的发现,rsv感染的婴儿在鼻洗液中比感染其他呼吸道病毒的婴儿表现出更高水平的TNF。呼吸道内TNF信号通路控制呼吸道的不同病理生理功能,包括炎症细胞招募和病原体激活[61].高水平的TNF可能增加疾病严重程度的一个可能机制是通过上调小毛细血管上的粘附分子,从而诱导炎症细胞的募集,如。中性粒细胞、巨噬细胞和嗜酸性粒细胞进入气道[62].最近也有报道称TNF参与小鼠RSV感染期间的支气管收缩[63].我们根据住院时间和补充氧疗的持续时间来衡量婴儿的疾病严重程度。我们无法发现TNF水平与氧疗需求之间的正相关;然而,我们确实发现鼻洗液中TNF水平与住院时间之间存在显著的正相关。我们发现住院≥7天的婴儿在鼻洗液中TNF浓度高于住院<7天的婴儿。总的来说,这些数据表明,婴儿RSV感染时,洗鼻液中TNF水平升高与疾病严重程度呈正相关。此外,先前的研究表明RSV细支气管炎与TNF单倍型相关[64],表明基因介导的TNF上调可能支持气道炎症的加剧,从而促进RSV感染的更严重过程。
总之,我们的数据表明,自分泌TNF介导由RSV感染引发的RIPK1-、RIPK3-和mlkl依赖的AM坏死,坏死途径不利于病毒清除。此外,tnf介导的AM坏死在RSV感染的发病机制中发挥重要作用(图8).我们提供的证据表明,鼻TNF水平升高与婴儿RSV细支气管炎的疾病严重程度相关。我们认为,靶向TNF和/或坏死机制可能是一种有价值的额外治疗方法,以减少幼儿和婴儿RSV感染引起的呼吸道发病率。
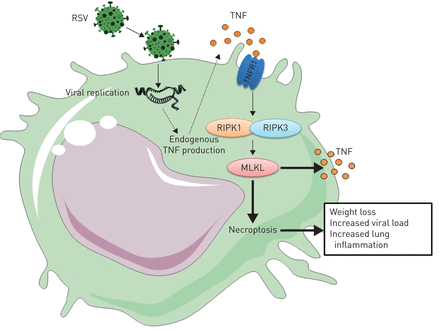
呼吸道合胞病毒(RSV)诱导巨噬细胞坏死的机制。在RSV感染巨噬细胞期间,由于病毒复制和坏死机制的激活,肿瘤坏死因子(TNF)产生和分泌。以自分泌的方式,TNF与TNF受体1 (TNFR1)结合,导致受体相互作用蛋白激酶1 (RIPK1)和RIPK3磷酸化。作为RIPK3激活的结果,混合谱系激酶结构域(MLKL)被磷酸化和激活,导致巨噬细胞坏死。在小鼠中,rsv诱导的tnf介导的肺泡巨噬细胞坏死导致体重减轻,肺部病毒载量增加,肺部炎症加重。
补充材料
可共享的PDF
确认
我们感谢Géssica Antunes, Deise do Nascimento de Freitas, Rodrigo Dornelles, Janaína Pasetti Nunes(都来自PUCRS)和Rosemeire Florença de Paula (UNICAMP)提供的优秀技术援助。我们也感谢São Lucas医院的风湿病服务(Porto Alegre,里约热内卢Grande do Sul, Brazil)慷慨捐赠英夫利昔单抗(Remicade;Centocor, Horsham, PA, USA)。
脚注
这篇文章有补充资料可从www.qdcxjkg.com
作者贡献:B.N.波尔图构思了这项研究。b·n·波尔图和m·a·r·维诺洛设计并监督了实验。B.N.波尔图和L.D.桑托斯在M.A.R.维诺洛、R. Weinlich和A.P.杜阿尔特·德·索萨的帮助下撰写了手稿。L.D. Santos, K.H. Antunes, S.P. Muraro, G.F. de Souza, A.G. da Silva, J.S. Felipe, L.C. Zanetti, R.S. Czepielewski和K. Magnus进行了实验并分析了数据。F. Maito进行肺组织病理和免疫组织化学分析。M. Scotta, R. Mattiello和A.P. Duarte de Souza提供了患者的鼻腔清洁剂,并对数据分析做出了贡献。所有作者都对手稿进行了审阅和评论。
利益冲突:L.D. Santos在研究进行期间报告了CAPES的资助。
利益冲突:K.H.安图内斯没有什么可透露的。
利益冲突:S.P.穆拉罗没有什么可透露的。
利益冲突:G.F. de Souza没有什么可透露的。
利益冲突:a·g·达·席尔瓦没有什么可透露的。
利益冲突:J.S.费利佩没什么可透露的。
利益冲突:L.C.萨内蒂没什么可透露的。
利益冲突:R.S. Czepielewski没有什么可透露的。
利益冲突:K. Magnus没有什么可透露的。
利益冲突:斯考塔先生没什么可透露的。
利益冲突:R. Mattiello没有什么可透露的。
利益冲突:F. Maito没有什么可透露的。
利益冲突:A.P.杜阿尔特·德·索萨没有什么可透露的。
利益冲突:R. Weinlich没有什么可透露的。
利益冲突:M.A.R.维诺洛没有什么可透露的。
利益冲突:B.N.波尔图报告了Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)在研究期间的资助。
支持声明:本研究由Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)资助号456282/2014-9(给B.N. Porto)和Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) -财务代码001。本文的资助信息已存入交叉参考基金注册.
- 收到了2020年10月7日。
- 接受2020年11月27日。
- 版权所有©ERS 2021
本版本根据知识共享署名非商业许可4.0的条款发布。











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}