





摘要
肺动脉平滑肌细胞(PA-SMCs)的过度增殖和血管周围炎症导致肺动脉高压(PAH)的进展,但目前的治疗方法并不是专门针对它们的。由于瘦素(Ob)及其主要受体ObR-b有助于全身血管细胞增殖和炎症,我们质疑靶向Ob/ObR-b轴是否会是一种有效的抗PAH的抗增殖和抗炎策略。
在特发性PAH (iPAH)中,使用人肺组织和原代细胞培养(早期传代≤5),我们证明肺内皮细胞(P-ECs)过度产生Ob, PA-SMCs过度表达ObR-b。此外,我们获得了Ob促进人PA-SMCs增殖的证据在体外在慢性缺氧性肺动脉高压(PH)模型中,ob处理小鼠右心室收缩压升高。在人类细胞中,我们还发现Ob导致单核细胞活化并增加p - ec中细胞粘附分子的表达水平。我们还发现Ob/ObR-b轴与PH敏感性有关,通过使用obr缺陷大鼠,这些大鼠表现出较不严重的缺氧诱导PH(肺血流动力学,动脉肌肉化,PA-SMC增殖和血管周围炎症)。重要的是,我们证明了在缺氧诱导的PH下使用可溶性Ob中和剂和二氯乙酸两种治疗策略的有效性。
我们在此证明Ob/ObR-b轴可能代表PAH的抗增殖和抗炎靶点。
摘要
靶向Ob/ObR-b轴是PH抗增殖和抗炎策略的重要工具http://ow.ly/I00jh
介绍
肺动脉高压(PAH)肺血管重构的部分原因是肺动脉平滑肌细胞(PA-SMCs)过度增殖和血管周围炎症浸润,这是平均肺动脉压(PAP)升高导致PAH患者功能进行性下降的原因。此外,在浸润肺血管壁的炎症细胞中,单核/巨噬细胞谱系细胞最近被清楚地确定为促进肺动脉高压肺血管重构的关键免疫细胞[1,2]。除此之外,介导PA-SMC增殖和单核细胞/巨噬细胞活化和积累的机制和危险因素尚不完全清楚。到目前为止,逆转PAH肺血管重构的治疗方法尚未被确定并转化为临床实践,这些治疗方法直接涉及PA-SMC增殖和单核/巨噬细胞谱系肺血管周围浸润的机制。已批准的多环芳烃治疗是弱抗增殖和抗炎药,尽管目前可用的医学治疗取得了进步,但特发性多环芳烃(iPAH)患者在诊断后的中位生存期为2.8年,这仍然是不可接受的[3.]。
最近的研究提供了明确的证据,表明瘦素(Ob),一种缺氧诱导因子(HIF)依赖基因,及其主要受体ObR-b有助于全身血管重构,特别是在心血管疾病中,作为全身血管SMCs的增殖和迁移因子,并作为血管壁炎症细胞浸润的有效免疫调节剂[4- - - - - -7]。事实上,Ob/ObR-b轴通过多种机制增强和加速动脉粥样硬化,包括刺激血管SMCs增殖,内膜单核细胞募集,巨噬细胞向泡沫细胞转化,以及促粥样硬化细胞因子的进一步分泌[8]。虽然我们知道iPAH患者的循环Ob水平升高,并可能在疾病的生理病理中发挥作用[7,9- - - - - -11], Ob/ObR-b轴在肺血管增殖和血管周围单核细胞/巨噬细胞谱系细胞积累中的翻译相关性尚不清楚。
因此,我们假设Ob/ObR-b轴可能有助于PAH的发生和/或进展,我们质疑:1)Ob和ObR-b是与人和实验性肺动脉高压(PH)相关的肺血管重构和血管周围单核细胞/巨噬细胞谱系细胞积累的关键因素;2) Ob/ObR-b信号抑制剂可能是治疗PAH的有效策略。综上所述,我们的研究结果首次为基于ob的多环芳烃干预提供了一个框架,并确定了对这种毁灭性疾病可能具有治疗潜力的分子。
方法
本研究经当地伦理委员会(CPP Ile-de-France VII, Paris, France)批准,所有患者签署书面知情同意书。动物研究由巴黎第一大学(Paris, France)动物护理管理小组批准。
主题
在常规随访期间采集iPAH患者和对照组的血样(表1)。纳入标准为:年龄0 ~ 18岁,经右心导管确诊PAH,近3个月临床及血流动力学状态稳定。排除标准是遗传形式的多环芳烃,糖尿病和代谢综合征。诊断和随访时的特征根据法国生物伦理法(国家信息和自由委员会)存储在法国PH网络登记处。
为原位和在体外研究中,肺标本是在肺移植时从iPAH患者身上获得的,或者从没有任何肺血管疾病证据的患者身上获得的,这些患者因局部肺癌而接受了肺叶切除术或全肺切除术,正常组织是在远离肿瘤的地方收集的(Marie Lannelongue Hospital, Le Plessis-Robinson, France) (表2)。
人肺内皮细胞、PA-SMCs和外周血单个核细胞的分离、培养和处理
从肺动脉远端分离出人肺内皮细胞(P-ECs)和PA-SMCs并按先前描述的方法进行培养[12- - - - - -14]。细胞(早期传代≤5)置于无血清培养基中24 h,分别与二甲基氧酰甘氨酸(DMOG, 0.25 mM) (Enzo Life Sciences, Villeurbanne,法国)、二氯乙酸(DCA, 5 mM) (Sigma-Aldrich, Lyon,法国)和重组Ob(10和100 ng·mL)接触−1) (R&D Systems, Lille, France) 24小时。为在体外治疗时,生理剂量(10 ng·mL)−110倍以上(100 ng·mL)−1)被使用,基于文献[15- - - - - -17]。5-溴-2-脱氧尿苷(BrdU)掺入法评估增殖[12]。
取血后,用标准菲科尔梯度离心新鲜分离对照组外周血单个核细胞(pbmc)。细胞在含1%胎牛血清和重组Ob(10或100 ng·mL)的RPMI培养基中孵育−1) 24小时。
动物模型和血流动力学测量
为在活的有机体内研究中,啮齿动物暴露于正常缺氧或缺氧3周(吸入氧含量10%)。雄性C57BL/6j小鼠(5周龄)(法国Janvier实验室,St. Berthevin)分为六组:两组对照组,未经治疗,暴露于常氧或缺氧环境;两组小鼠从第2天至第21天每天腹腔注射重组Ob (3 μg·g)−1初始体重[18]),暴露于常氧或缺氧;两组每日用药i.p。第14 ~ 21天注射可溶性重组蛋白ObR (ObR:Fc) (R&D Systems) (100 μg·小鼠)−1(19]),暴露于常氧或慢性缺氧。将4周龄的Sprague-Dawley雄性大鼠(Charles River Laboratories, Larberesle, France)分为四组:两组对照组(饮水中载具)暴露于常氧或缺氧环境;两组在第14天至第21天给予DCA (1 g·L)−1(在饮用水中)暴露于常氧或慢性缺氧环境中。两组雄性朱克糖尿病脂肪转基因大鼠(ZDF)/ Lepr fa / fa(Charles River Laboratories),大鼠4周龄,对照大鼠(ZDF/ LeprCrl)(查尔斯河实验室)暴露于正常缺氧或慢性缺氧。我们还测试了PH的大鼠单碱模型,该模型不是研究Ob/ObR-b轴的特异性模型(图S3)。
用异氟醚麻醉动物,并测量小鼠的血流动力学参数(右心室收缩压(RVSP)) [20.大鼠(平均PAP和肺血管阻力(PVR)) [12]。然后,取血进行PBMC分析,打开胸腔,立即取出左肺并冷冻。用福尔马林缓冲液将右侧固定在膨胀状态。右室肥厚(RVH)富尔顿指数和肌肉化血管百分比按先前描述测定[12]。
实时定量PCR
使用Trizol (Invitrogen, St. Aubin, France)和RNeasy mini kit (Qiagen, Courtaboeuf, France)从冷冻肺中分离总RNA。总RNA(2µg)按照制造商的说明使用Superscript II (Invitrogen)进行逆转录。ObR、Ki67、Cyclin D1、细胞内粘附分子(ICAM)-1、血管细胞粘附分子(VCAM)-1和e-选择素的基因表达水平使用预先验证的assays -on- on TaqMan引物/探针集(Applied Biosystems, St. Aubin, France)进行定量,并使用比较周期阈值(Ct)方法归一化为18S核糖体RNA (2-ΔΔCt) [13,21,22]。
Western blot, ELISA和免疫染色
细胞/组织匀浆,在含有蛋白酶和磷酸酶抑制剂的PBS中超声处理,用30µg蛋白质检测ObR-b (R&D Systems和Santa Cruz Biotechnology (Le Perray-en-Yvelines, France)分别用于人和啮齿动物)和β-actin (Sigma-Aldrich),如前面所述[21]。根据制造商说明,使用Quantikine (R&D Systems)评估p - ec条件培养基中Ob的浓度。免疫组织化学和免疫细胞荧光染色针对α-平滑肌细胞肌动蛋白(α-SMA)和增殖细胞核抗原(PCNA)的抗体(Dako, Les Ulis, France), Ob,平滑肌(SM)22, Tie2, CD68, F4/80和CD206 (Santa Cruz Biotechnology), ObR-b (R&D Systems)或凝集素蛋白(Sigma-Aldrich)进行了如上所述[21]。使用LSM700共聚焦显微镜,然后使用Zen软件(蔡司,Marly-le-Roi,法国)拍摄图像。
流式细胞术分析
对照和iPAH患者以及啮齿类动物的PBMCs用以下抗体荧光标记:荧光团偶联单克隆抗cd14、抗cd25和抗cd11b (Becton Dickinson, Rungis, France)、抗cd4 (MiltenyiBiotec, Paris, France)和抗obr -b (R&D Systems),如上文所述[7]。流式细胞术门控设置如前所述[7]。流式细胞仪(MACSQuant Miltenyi Biotec)获得流式细胞仪数据,并通过FlowJo软件程序(Tree Star, Inc.)进行分析。阿什兰,OR, USA)。
统计分析
结果以平均值±表示扫描电镜。所有分析均采用p<0.05的显著性水平。采用非参数Mann-Whitney检验或Bonferroni双因素方差分析检验统计显著性事后测试。所有统计程序均使用GraphPad Prism 5.0版本(GraphPad Software Inc.)进行。圣地亚哥,加州,美国)。
结果
iPAH患者肺血管Ob/ObR-b轴的上调
为了确定Ob/ObR-b轴是否在iPAH患者的肺血管中局部上调,我们用Ob或ObR-b抗体对10名iPAH患者和10名对照组的人肺组织进行了免疫染色,同时对α- smc特异性表面标记物SM22和血管内皮进行了凝集素染色。
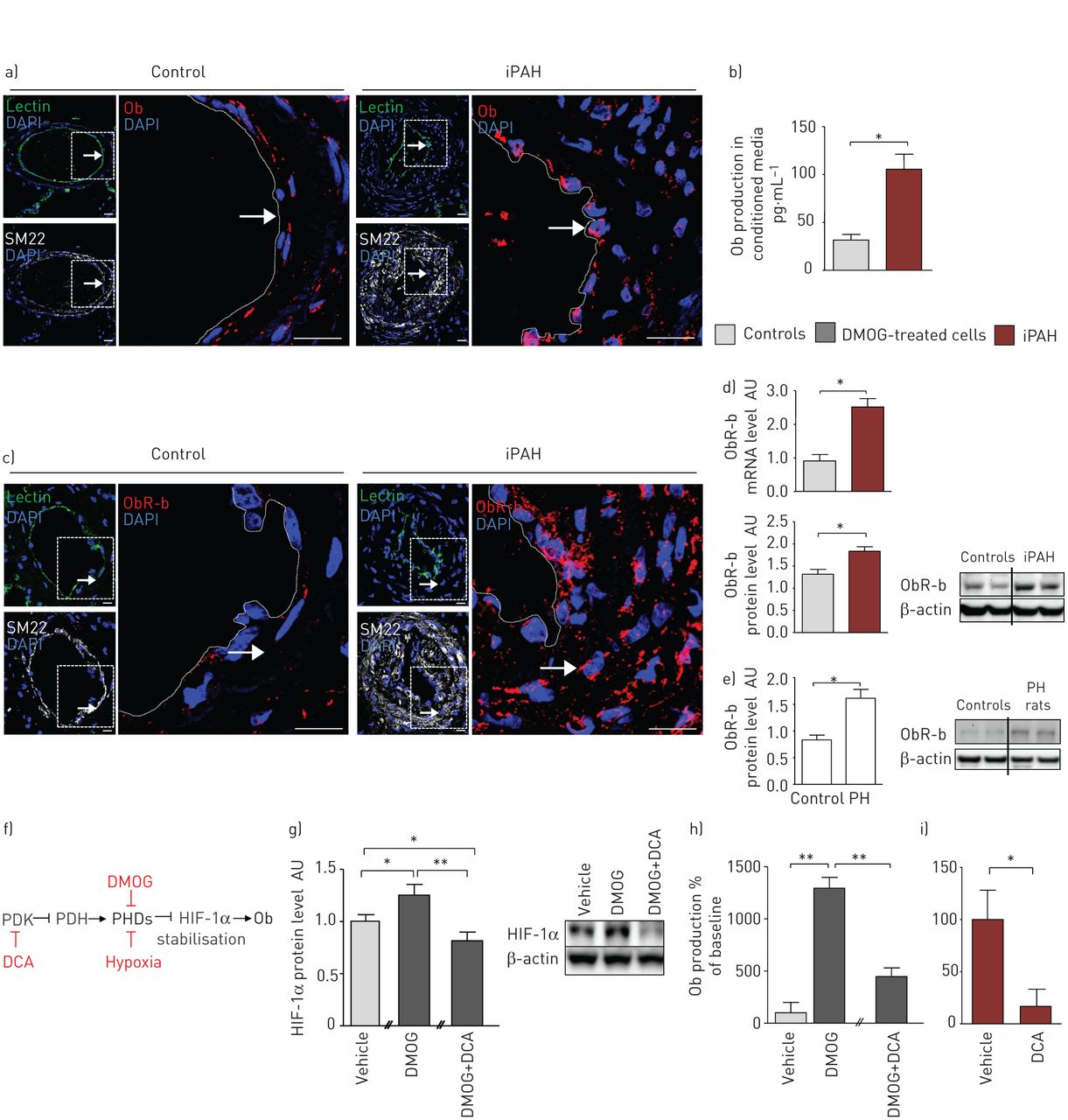
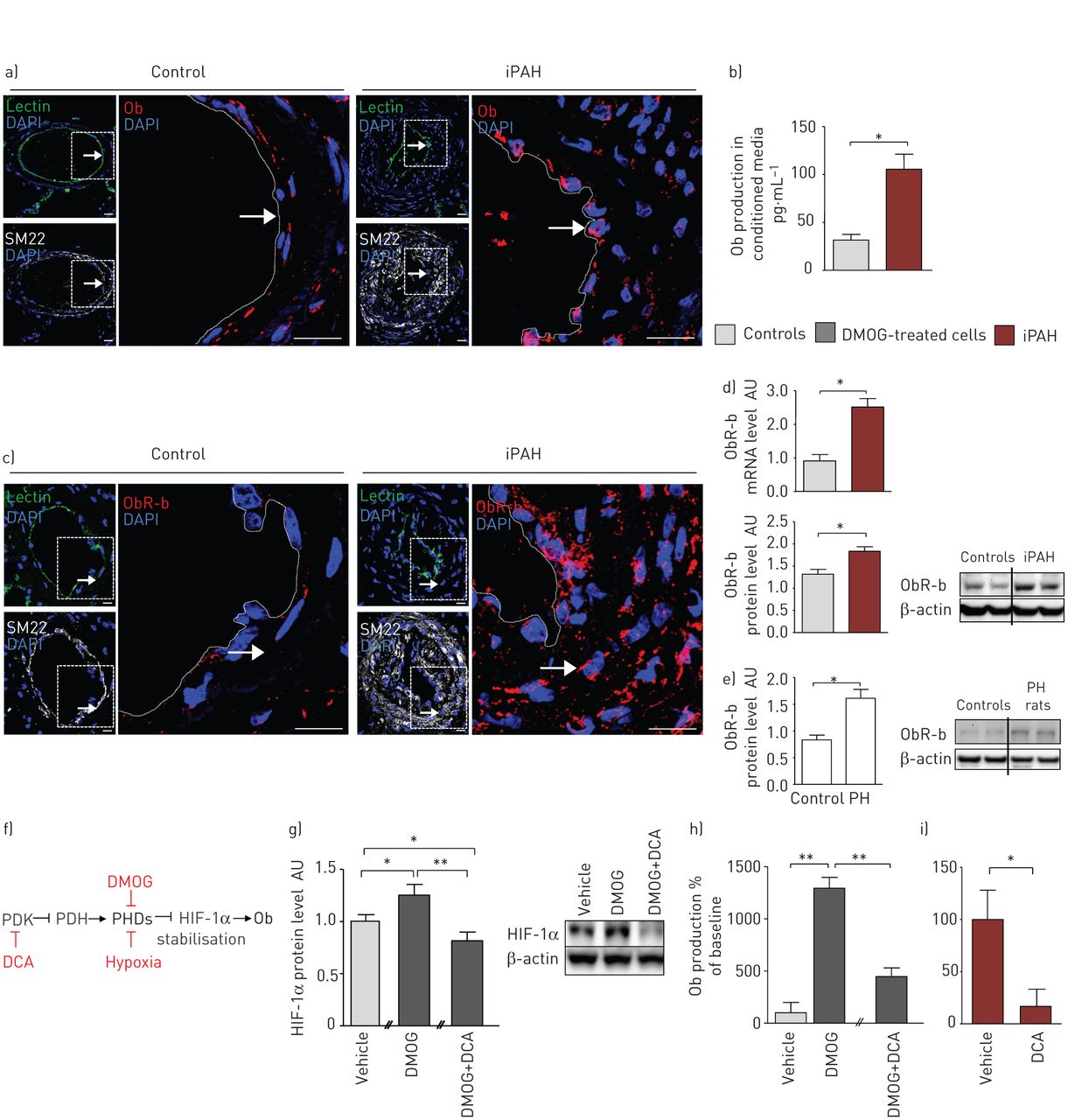
一方面,我们的共聚焦显微镜分析显示,与对照标本相比,iPAH患者远端肺动脉内皮内Ob染色强烈(图1a)有趣的是,这些原位观察结果被重复在体外在人类新分离的p - ec中经ELISA免疫测定,来自iPAH (n=5例患者)的P-EC初级早期传代(≤5)培养物与对照细胞(n=5例受试者)相比,Ob产量显著增加,P-EC条件培养基中的Ob含量增加了4倍(图1b).值得注意的是,我们也能够排除PA-SMCs作为iPAH中Ob的另一个局部来源原位和在体外分析(图S1A)。
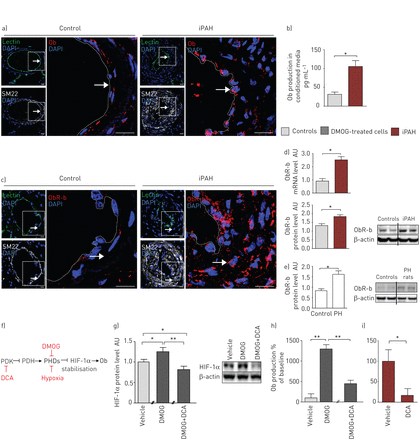
人特发性肺动脉高压(iPAH)中瘦素(Ob)/Ob主受体(ObR-b)轴的上调a和b)人肺内皮细胞(P-ECs)在PAH中的作用。a)的代表形象原位来自对照组和iPAH患者的Ob染色(红色),其中P-ECs凝集素(绿色)阳性,肺动脉平滑肌细胞(PA-SMCs) SM22(白色)阳性。b)在体外对照(n=15)和iPAH患者(n=10)的人P-ECs初代(≤5)传代培养的条件培养基中Ob产量的定量。c-e)人类和啮齿动物的PA-SMCs在PAH中过表达ObR-b。c)对照组和iPAH患者肺中PA-SMCs中ObR-b(红色)染色,SM22(白色)阳性,其中P-ECs中凝集素(绿色)阳性。d)定量对照(n=10)和iPAH (n=10)人PA-SMCs原代早期(≤5)传代培养物中ObR-b mRNA水平,用代表性的Western blot定量对照(n=5)和iPAH (n=5)人PA-SMCs原代早期(≤5)传代培养物中ObR-b/β-actin比值。e)定量大鼠肺中ObR-b/β-actin比值,分别来自对照组(n=5)和慢性缺氧引起的肺动脉高压(PH) (n=5),具有代表性的Western blot。f-i)二氯乙酸(DCA)消除过量的内皮源性Ob的产生。f)二甲基氯酰甘氨酸(DMOG)和DCA在Ob信号通路中的作用示意图。PDK:丙酮酸脱氢酶激酶;PDH:丙酮酸脱氢酶;博士:脯氨酸羟化酶结构域; HIF: hypoxia-inducible factor. g) Quantification of HIF-1α/β-actin ratio and a representative Western blot in primary early (≤5) passage cultures of human P-ECs from controls in untreated (vehicle, n=3), DMOG-treated (n=3) or DMOG+DCA-treated (n=3) conditions. h and i) Quantification of Ob production in conditioned media of primary early (≤5) passage cultures of human P-ECs from h) controls in untreated (n=3), DMOG-treated (n=3) or DMOG+DCA-treated (n=3) conditions and from i) human P-ECs from iPAH patients in untreated (n=3) or DCA-treated conditions (n=3). DAPI: 4',6-diamidino-2-phenylindole; AU: arbitrary unit. *: p<0.05; **: p<0.01. Scale bar=20 µm.
另一方面,通过共聚焦显微镜分析和ObR-b和SM22的双重标记,我们发现了一个很强的原位iPAH患者PA-SMCs内的ObR-b染色,与对照组PA-SMCs内的弱染色相比(图1c)我们也确认了我们的原位的观察,在体外来自iPAH和对照组的PA-SMCs的初级早期传代(≤5)培养研究;我们发现,与对照组相比,iPAH患者的PA-SMCs在mRNA和蛋白质水平上的ObR-b表达增加(图1d).这些发现也被重复了在活的有机体内,与对照组相比,出现PH的大鼠肺部ObR-b蛋白水平增加(图1e).值得注意的是,我们发现ObR-b在P-ECs中表达较弱,iPAH患者的P-ECs与对照组的ObR-b表达无差异(图S1B)。
因为我们的原位和在体外数据表明P-ECs是iPAH中Ob的异常肺源,我们进行了后续研究在体外以测试缺氧条件下Ob是否会诱发。事实上,缺氧是Ob表达的强诱导剂,因为缺氧反应元件的存在Ob募集HIF-1α的基因启动子[23,24]。因此,我们首先用DMOG处理来自对照对象的分离p - ec的新鲜早期传代(≤5)培养物,DMOG是一种细胞渗透性脯氨酸-4-羟化酶抑制剂,可提高缺氧诱导的HIF-1α的稳定性在体外通过抑制脯氨酸羟化酶依赖性HIF-1α降解(图1 f) [25]。与未处理的细胞相比,dmog处理的P-ECs产生了非常高水平的Ob,证实了HIF-1α可以在P-ECs中诱导Ob (图1g).由于新出现的证据表明,线粒体信号的激活可以影响HIF-1α的稳定,我们测试了丙酮酸脱氢酶激酶抑制剂DCA的功效[26,27) (图1f)使用来自对照肺标本的原代人P-ECs使内皮来源的Ob生成正常化。我们检测了HIF-1α蛋白的表达在体外,控制p - ec服从DMOG和DCA。我们发现HIF-1α在dmog处理的细胞中升高,添加DCA后恢复正常(图1g).同样,我们发现dmog诱导的P-EC条件培养基中Ob含量的增加被DCA完全抑制在体外(图1h)。此外,我们发现DCA处理消除了条件培养基中iPAH P-ECs中的Ob含量(图1i).这些结果与先前的数据一致,表明在常氧和缺氧条件下,与对照细胞相比,iPAH肺中HIF-1α的内皮细胞活化异常[28]。此外,有研究表明HIF-1α缺陷小鼠和HIF-2α缺陷小鼠均可免受慢性缺氧PH的影响,这表明HIF-1α和HIF-2α可能在PH中起关键的致病作用[29- - - - - -31]。
总之,我们的数据强调,在iPAH中,Ob是由功能失调的p - ec异常产生的,这可以部分解释为HIF-1α持续稳定。考虑到其受体ObR-b在iPAH中被PA-SMCs过度表达,这一现象更加重要。
Ob/ObR-b轴有助于iPAH中PA-SMCs的过度增殖
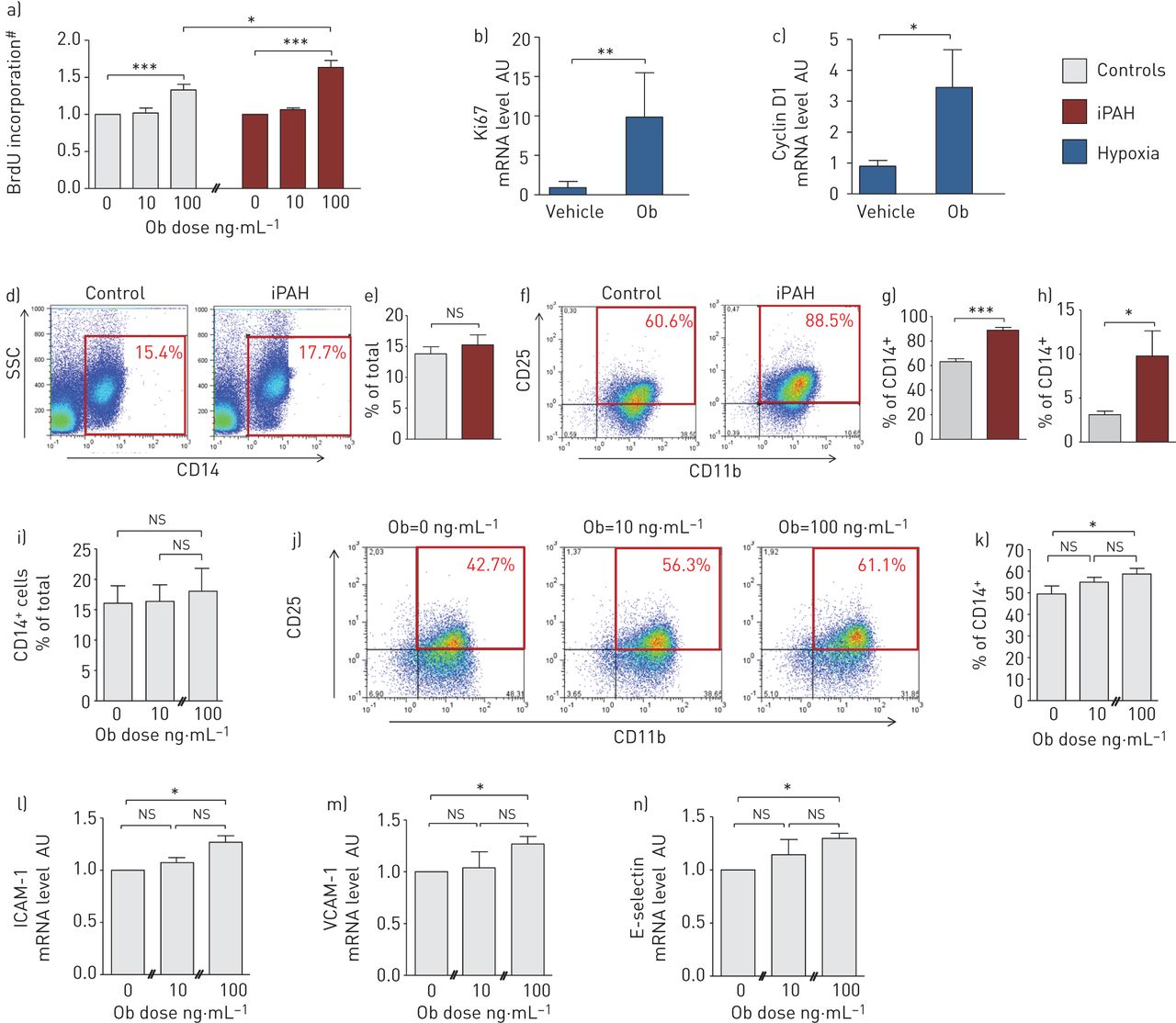
转基因小鼠缺乏Ob (ob/ob)或其受体ObR (db/db)在血管损伤的反应中被保护免受新内膜的形成,而外源性给药Ob以受体特异性的方式促进实验性病变的形成[6,8,32,33]。基于此,我们测试了Ob/ObR-b轴是否在iPAH肺血管重构中发挥类似的作用。我们首先研究了Ob with的增殖能力在体外使用从iPAH患者和对照组的肺组织中分离的原代人PA-SMCs进行研究。通过BrdU掺入,我们证明来自iPAH患者的PA-SMCs与对照PA-SMCs相比,在外源性Ob的反应中增殖更多(图2一个)。
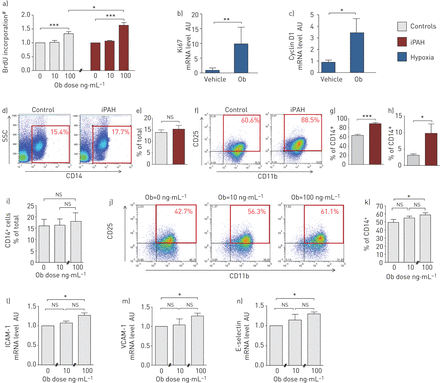
瘦素(Ob)/Ob主受体(ObR-b)轴参与特发性肺动脉高压(iPAH)患者肺血管重构a-c) Ob/ObR-b轴诱导人和啮齿动物肺动脉平滑肌细胞(PA-SMC)增殖。a)定量测定5-溴-2-脱氧尿苷(BrdU)在早期(≤5)代培养的来自对照的PA-SMCs和增加剂量的重组Ob处理的iPAH(每种条件n=3,重复3次)中的掺入情况。b)用重组Ob处理或未处理的慢性缺氧小鼠肺组织中Ki67和c)细胞周期蛋白D1 mRNA水平的定量分析(每组n=5)。d-k) Ob/ObR-b轴诱导iPAH单核细胞活化。d)对照(n=10)和iPAH患者(n=10)外周血单个核细胞(PBMCs)的代表性荧光活化细胞分选仪(FACS)点图,并用抗cd14抗体染色。红色象限表示CD14 (CD14)阳性的pbmc+细胞)。SSC:侧散射光。e) CD14的定量+PMBCs。f)对照(n=10)和iPAH患者(n=10)提取pbmc的代表性FACS点图,并进行抗cd11b和CD25抗体双染色。点图中的红色象限代表CD11b和CD25 (CD11b)双阳性细胞+CD25+细胞)。g) CD11b的定量+CD25+PBMCs。h) CD14数量的定量+对照组(n=10)和iPAH患者(n=10)通过FACS表达ObR-b的细胞。i) CD14数量的定量+对照PBMCs (n=3)用增加剂量的重组Ob(每种条件n=3,重复3次)处理。j)用抗cd11b和CD25抗体双染色增加剂量的重组Ob处理的对照pbmc (n=3)的代表性FACS点图定量。点图中的红色象限代表CD11b和CD25 (CD11b)双阳性细胞+CD25+细胞)。k) CD11b的定量+CD25+增加重组Ob剂量处理PBMCs(每种条件n=3,重复3次)。l-n) Ob/ObR-b轴诱导人肺内皮细胞(P-ECs)关键粘附分子的过表达。l)用增加剂量的重组Ob(每种条件n=3,重复3次)处理对照(n=3)的人P-ECs初代(≤5)传代培养物中细胞间粘附分子(ICAM)-1、血管细胞粘附分子(VCAM)-1和e -选择素mRNA水平的定量。AU:任意单位;ns:不重要的;#:正常化至未治疗状态。*: p < 0.05;* *: p < 0.01;* * *: p < 0.001
为了进一步研究Ob/ObR-b轴在PA-SMC增殖中的作用,我们在慢性缺氧暴露诱导的ph下,每天给小鼠注射重组Ob。有趣的是,缺氧诱导的Ki67 (图2 b)和Cyclin D1 (图2 c),通过qRT-PCR测量,在ob处理的小鼠中甚至更大。我们的在体外和在活的有机体内数据表明Ob/ObR-b轴诱导PA-SMC增殖,从而促进PAH中PA-SMC过度增殖。
Ob/ObR-b轴参与iPAH中单核/巨噬细胞异常活化
最近的证据表明,单核细胞/巨噬细胞谱系作为关键免疫细胞的慢性激活是促进肺动脉高压肺血管重构的根本原因[2]。另一方面,已知Ob影响巨噬细胞的行为[34]。因此,我们试图研究Ob/ObR-b轴在这种致病现象中的作用。我们首先评估了iPAH患者单核/巨噬细胞谱系的激活,使用的是来自iPAH患者和对照组的新提取的pbmc。我们分别用单核细胞表型和活化标记CD14和CD11b/CD25对pbmc进行三重标记,并用流式细胞术对其进行分析。我们发现,与对照组相比,iPAH患者中活化标记CD11b和CD25过表达,而两组受试者显示相同数量的CD14+单核细胞(图2d)。重要的是,我们发现与对照组相比,iPAH患者中活化的单核细胞过度表达ObR-b (图2h),提示Ob/ObR-b轴在iPAH单核细胞活化中的作用。为了进一步验证这一假设,我们将新退出的对照pbmc暴露于外源性Ob在体外。我们发现,24小时后在体外与未处理的细胞相比,ob处理的单核细胞更活跃,呈剂量依赖性(图2i (k)。
我们进行了后续研究,以进一步研究在实验和人PAH中观察到的Ob/ObR-b轴的过度激活在血管周围炎症细胞募集中的作用。我们将从iPAH患者和对照组肺组织中分离的原代人P-ECs与外源性重组Ob接触24小时。有趣的是,与未处理的细胞相比,ob处理的P-ECs在mRNA水平上显示ICAM-1, VCAM-1和e -选择素的表达增加(图2l-n)。
来验证这些的相关性在体外我们建立了缺氧暴露3周,每天给药Ob的小鼠Ob/ObR-b轴慢性激活模型(图3 gydF4y2Baa).有趣的是,我们验证了在常氧条件下,与未处理的动物相比,ob处理的小鼠体重下降,缺氧后体重进一步下降(图3 gydF4y2Bab).通过使用该小鼠模型,我们还能够证明慢性给药Ob会加重慢性缺氧小鼠的PH严重程度。事实上,经ob处理的小鼠表现出更高的肺血流动力学参数,这反映在右心导管测量的RVSP增加(图3 c),与缺氧3周后未治疗的小鼠相比。然而,在RVH中没有观察到变化,肺动脉小肌肉化(图3 d- - - - - -f)和壁厚(图S2A)。有趣的是,长期注射Ob可增强PA-SMC的增殖(图3 g和h)和血管周围巨噬细胞浸润(无花果3我和j和S4)。重要的是,与未治疗的小鼠相比,Ob注射增强了缺氧诱导的单核细胞/巨噬细胞活化(图3 k),通过表面标记物CD206表达来测定。
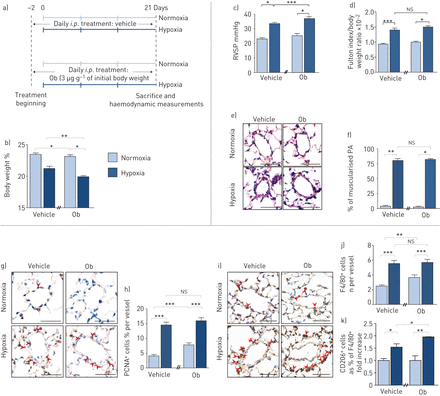
每日瘦素(Ob)注射对慢性缺氧性肺动脉高压(PH)起作用。a和b) Ob/Ob主受体(Obr-b)轴慢性激活的小鼠模型。a)慢性重组Ob腹腔注射实验策略(i.p。)注射给暴露于常氧或缺氧的小鼠(每种情况n=5)。b)体重。c-f)慢性Ob注射增加缺氧诱导的ph右心室收缩压(RVSP)。c) RVSP。d)用体重归一化的Fulton指数测量右心室肥厚。e)血红素和伊红染色的代表性图像。标尺=50 μm。f)小鼠肺动脉肌化率(PA)定量。g-k)慢性Ob注射增强肺动脉平滑肌细胞增殖和血管周围巨噬细胞活化。g)增殖细胞核抗原(PCNA)代表图像,标尺=50µm; i) F4/80标记,标尺=20µm。 h and j) Quantification of cells expressing PCNA and F4/80 markers, respectively, in mice PA. k) Quantification of double-positive cells for CD206 and F4/80 in mice PA.ns:不重要的;*: p < 0.05;* *: p < 0.01;* * *: p < 0.001。
综上所述,我们的数据表明,Ob/ObR-b轴的过度激活有助于PH下肺血管的重塑,不仅促进PA-SMC的增殖,还促进血管周围巨噬细胞的积累。
Ob/ObR-b轴增强啮齿动物慢性缺氧诱导的PH敏感性
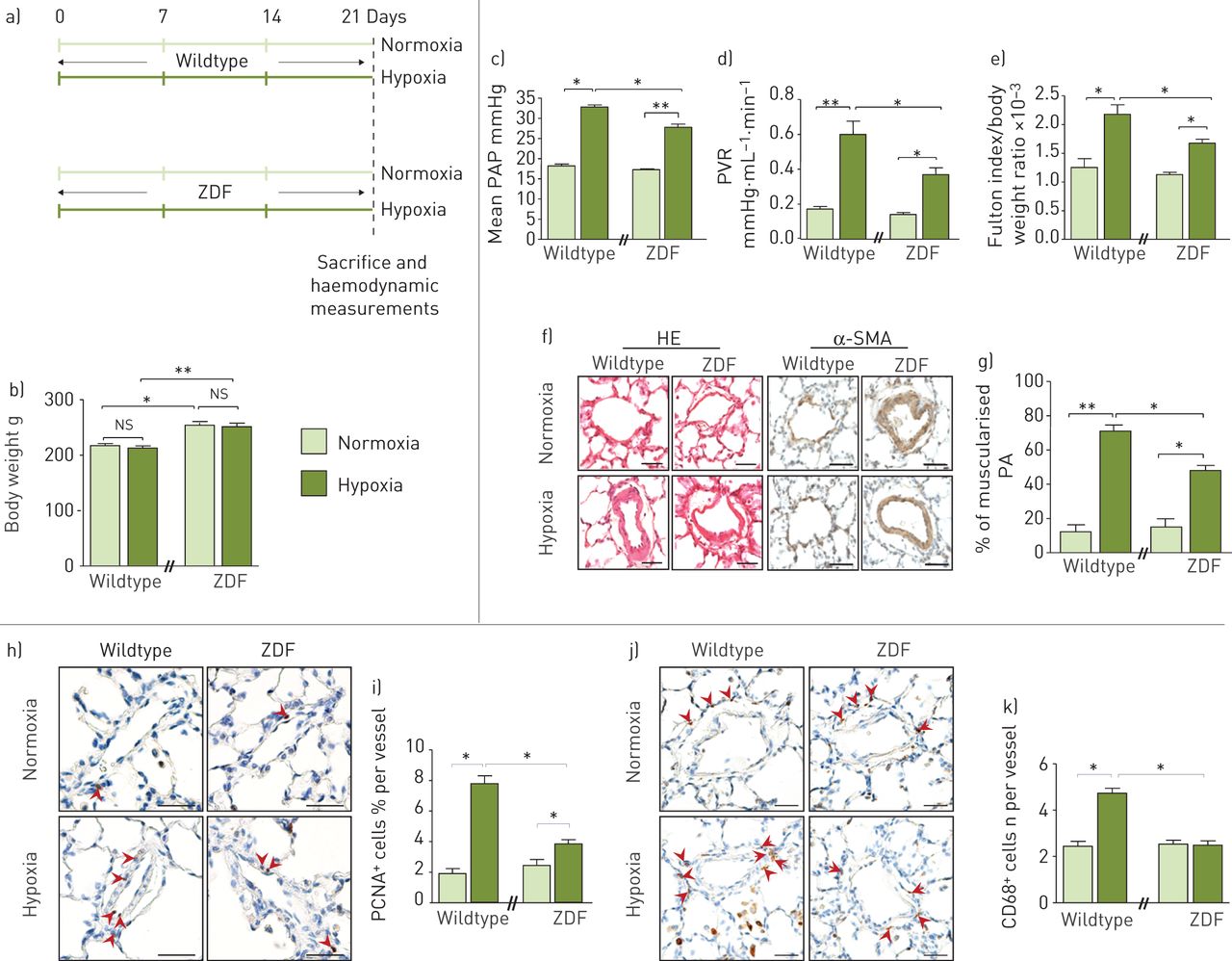
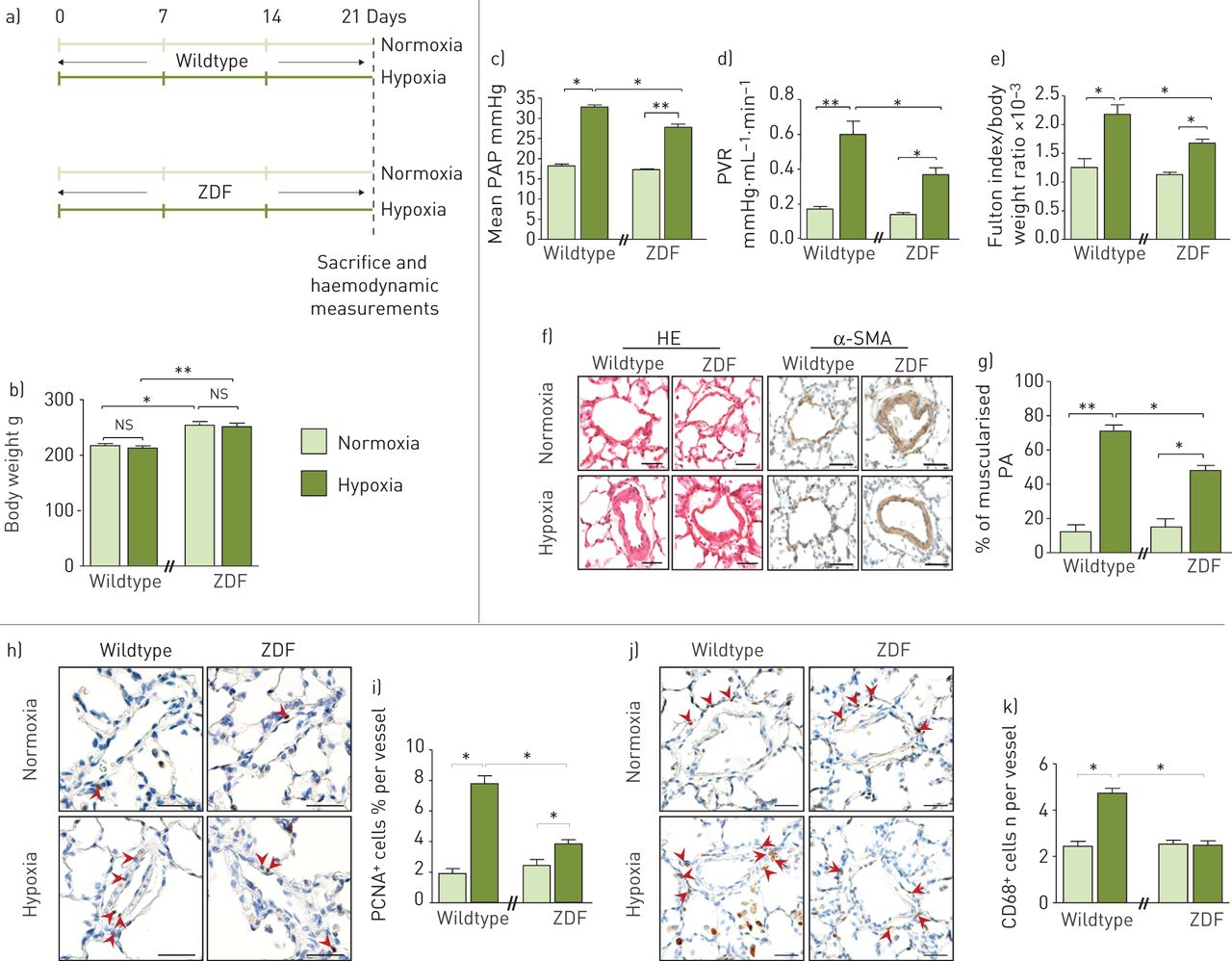
接下来,我们研究了Ob/ObR-b轴是否会增加缺乏ObR (ZDF)的ZDF转基因大鼠的PH敏感性/ Lepr fa / fa),并将他们暴露于慢性缺氧或常氧环境(图4和b)。
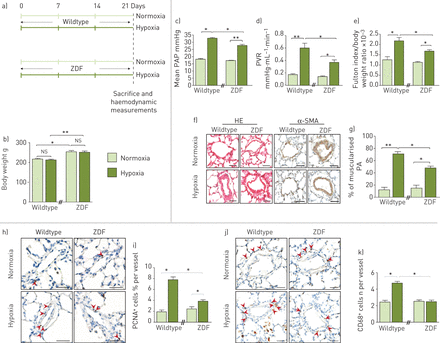
瘦素(Ob)/Ob主受体(ObR-b)轴增强慢性缺氧诱导的肺动脉高压(PH)易感性a和b) obr缺失的Zucker糖尿病脂肪(ZDF)转基因大鼠。a) ZDF转基因大鼠在常氧或缺氧条件下的实验策略(每种条件n=5)。b)体重。c-g) obr缺乏可防止慢性缺氧诱导的ph发育。c)平均肺动脉压(PAP), d)肺血管阻力(PVR), e)体重归一化富尔顿指数测量右心室肥厚。f)血红素和伊红染色(HE)和α-平滑肌细胞肌动蛋白(α-SMA)的代表性图像;g)大鼠肺肌化肺动脉(PA)百分比定量。h-k) obr缺乏降低肺动脉平滑肌细胞增殖和血管周围巨噬细胞积聚。h、j)大鼠PA增殖细胞核抗原(PCNA)和CD68染色的代表性图像。i和k)大鼠PA中分别表达PCNA和CD68标记的细胞定量。ns:不重要的;*: p < 0.05;* *: p < 0.01。比例尺=50µm。
有趣的是,在慢性缺氧暴露3周后,与野生型幼鼠(ZDF)相比,ZDF大鼠出现的疾病不那么严重/ LeprCrl),反映在平均PAP、PVR、RVH、肺动脉肌肉化和壁厚(图S2B)与对照大鼠(图4c g)。此外,ZDF大鼠表现出较低的PCNA数量+细胞(图4h和i)和CD68+细胞(图4J和k),与它们的野生型对照相比。综上所述,这些发现表明Ob/ObR-b轴参与慢性缺氧诱导的PH敏感性。
靶向Ob/ObR轴可防止慢性缺氧诱导的PH进展
为了研究Ob/ObR-b轴抑制作为PAH治疗的潜在治疗靶点的有效性,我们在啮齿动物缺氧诱导的PH中使用两种不同的策略进行了一项临床前研究。
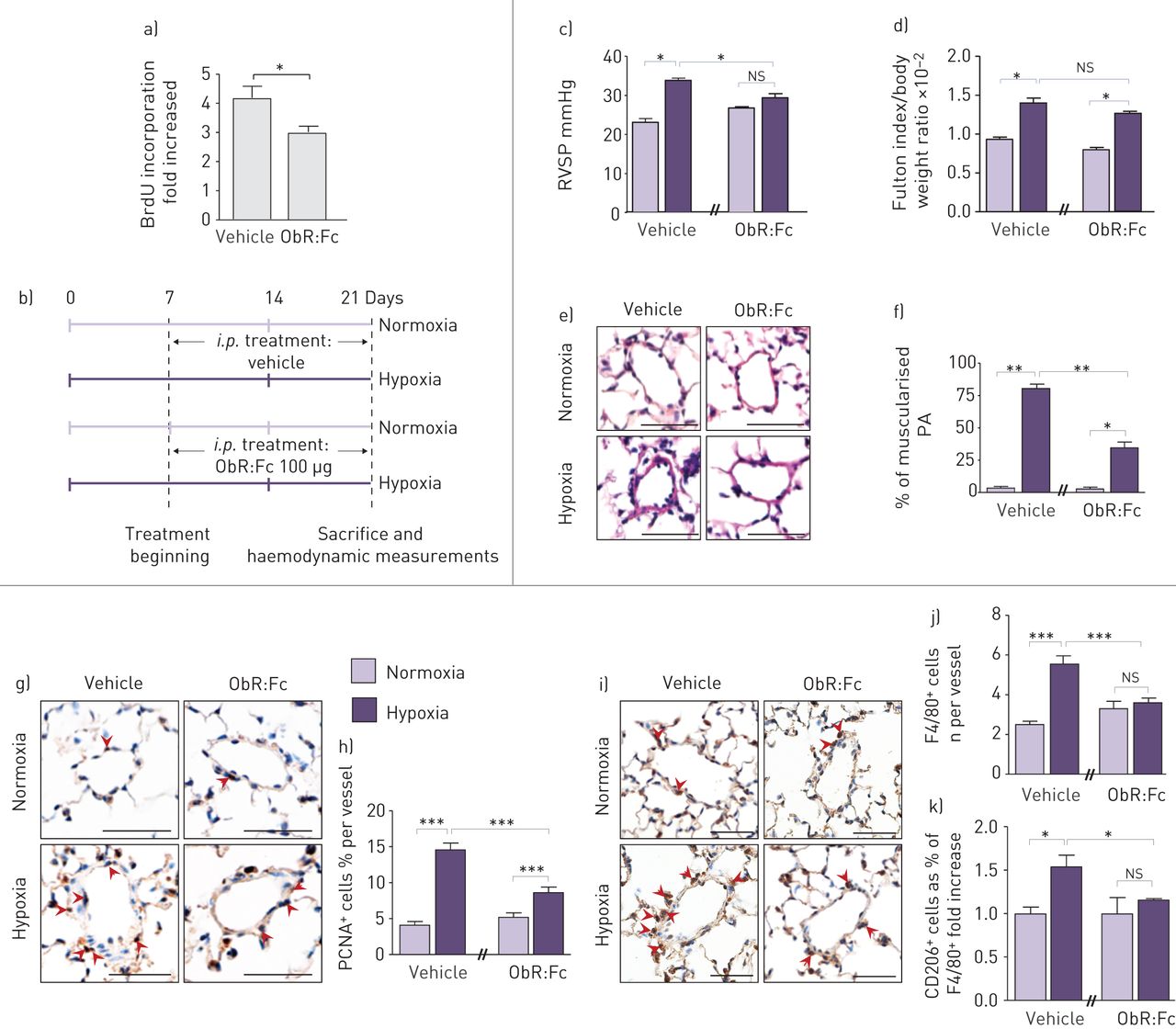
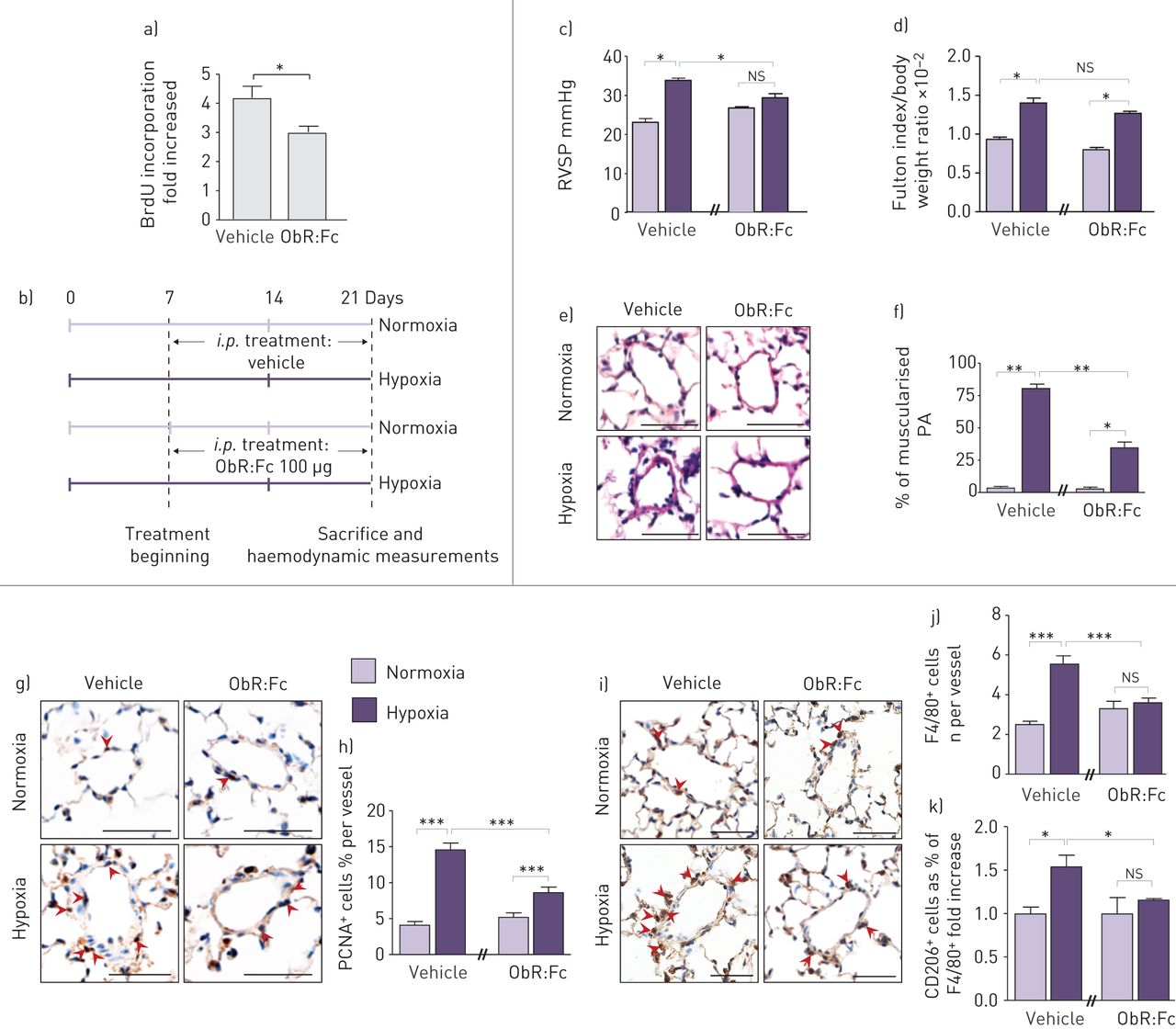
首先,我们的实验策略是测试在体外可溶性重组ObR蛋白(ObR:Fc)在人原代细胞中和Ob/ObR-b轴的效果。我们将对照PA-SMCs暴露于iPAH P-ECs的条件培养基中,无论是否用ObR:Fc处理。通过测量BrdU掺入,我们发现暴露于ObR: fc处理的P-EC条件培养基中的PA-SMCs比暴露于未处理的P-EC条件培养基中的PA-SMCs增殖更少(图5a).基于这些观察结果,我们在慢性缺氧诱导的PH模型小鼠中慢性注射ObR:Fc (图5b)。与未治疗的小鼠相比,我们有趣地发现,在小鼠中慢性注射ObR:Fc后,RVSP值的增加幅度较低,远端血管部分或完全肌肉化的百分比(图5c-f)和壁厚(图S2C)。与这些数据一致,我们发现ObR:Fc治疗显著降低了PCNA的数量+细胞在慢性缺氧诱导的PH值中,与载药处理(图5g和h)。此外,通过原位F4/80染色(无花果。5我和j与未治疗的小鼠相比,S5)。重要的是,通过CD206表达测量,ObR:Fc治疗降低了缺氧诱导的巨噬细胞活化(图5k)。
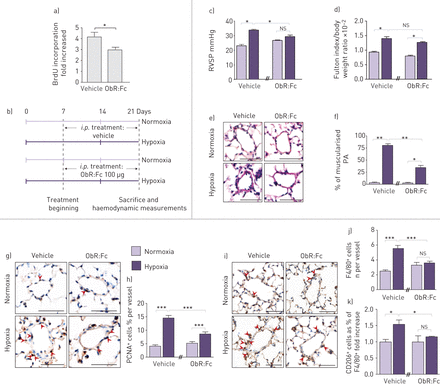
用Ob中和剂(ObR:Fc)靶向瘦素(Ob)/Ob主受体(ObR-b)轴可预防肺动脉高压(PH)进展。a)定量测定5-溴-2-脱氧尿苷(BrdU)掺入原发性早期(≤5)代培养的人肺动脉平滑肌细胞(PA-SMCs),这些细胞来自未经治疗(对照)或用ObR:Fc治疗的特发性肺动脉高压(iPAH)患者的肺内皮细胞(P-ECs)的条件培养基。b)慢性ObR:Fc腹腔注射的实验策略(i.p。)注射给暴露于常氧或缺氧的小鼠(每种情况n=5)。c-f) ObR:Fc治疗可防止慢性缺氧诱导的PH恶化。c)右心室收缩压(RVSP), d)用体重归一化的Fulton指数测量的右心室肥厚,e)血红素和伊红染色的代表性图像,f)小鼠肌肉化肺动脉(PA)百分比的量化。g-k) ObR:Fc治疗增加PA-SMC增殖和血管周围巨噬细胞活化。g)增殖细胞核抗原(PCNA)和i) F4/80染色代表图。h和j)小鼠PA中分别表达PCNA和F4/80标记的细胞的定量。k)小鼠PA中CD206和F4/80双阳性细胞的定量。ns:不重要的;*: p < 0.05;* *: p < 0.01;* * *: p < 0.001。比例尺=50µm。
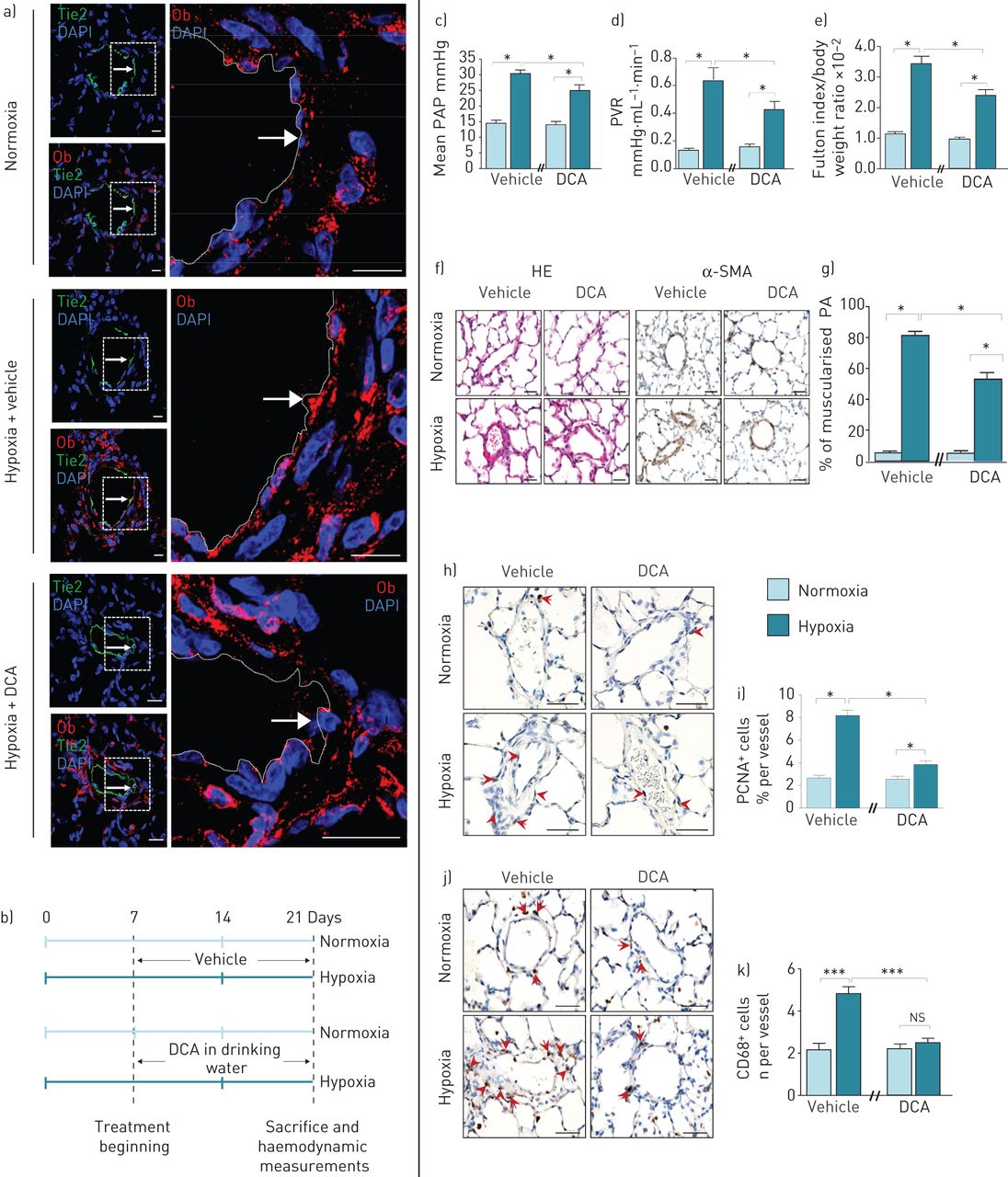
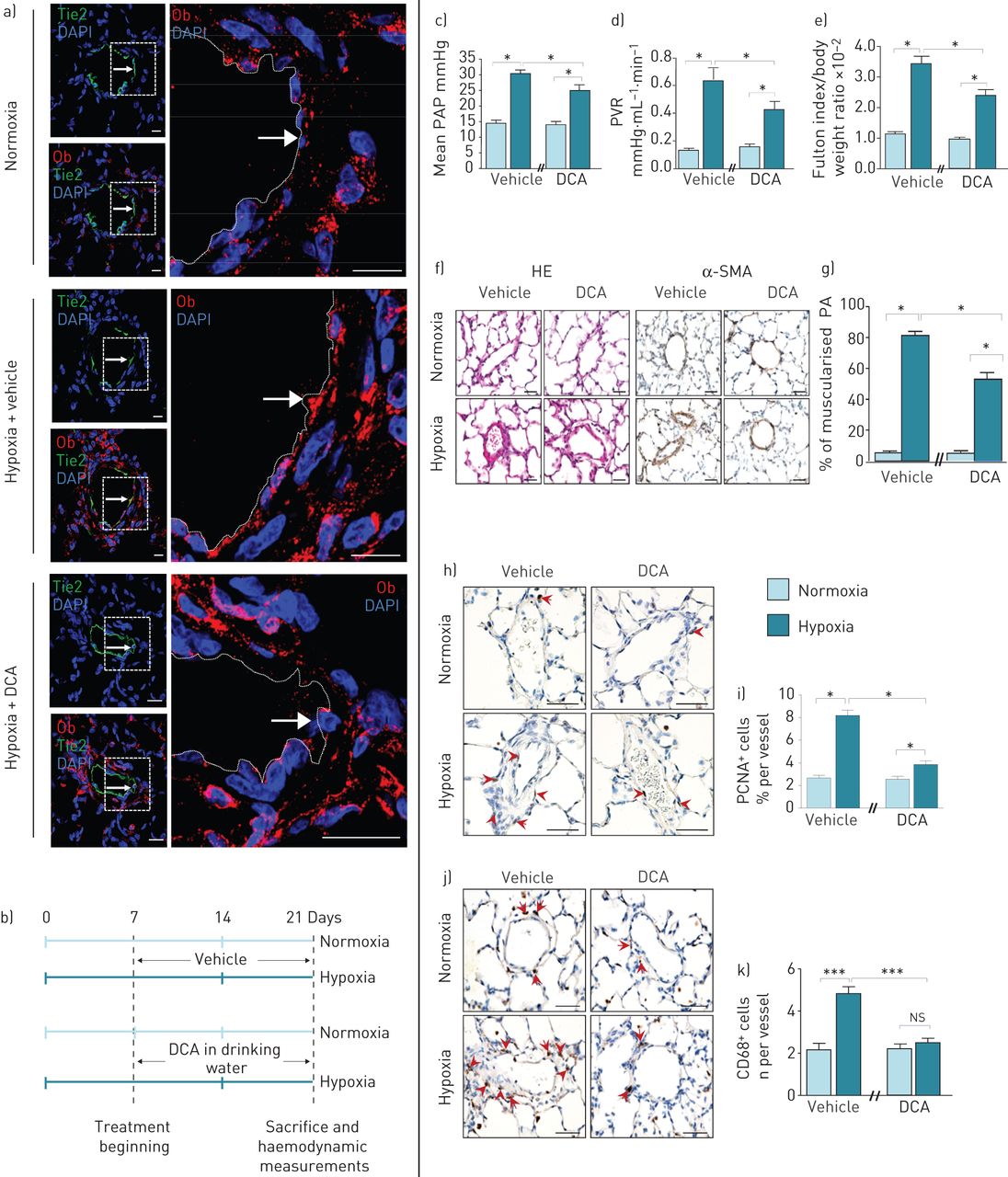
我们的第二个治疗策略是使用DCA间接调节Ob/ObR-b轴。我们首先研究了缺氧是否会触发p - ec衍生的Ob合成在活的有机体内以及DCA是否能抑制这种缺氧诱导的作用。与我们的在体外观察(图1g),我们在活的有机体内数据表明,与正常缺氧的大鼠相比,暴露于慢性缺氧的大鼠在P-ECs中显示出更高的Ob含量,最重要的是,DCA治疗消除了缺氧诱导的内皮源性Ob生成(图6a).因此,我们测试了DCA治疗的疗效在活的有机体内(图6b)并发现dca治疗的大鼠表现出较轻的疾病,通过较低的平均PAP, PVR和RVH (图6C-e)以及较低的肌肉化肺动脉远端百分比(图6f和g)和壁厚(图S2D)。此外,通过PCNA染色检测,DCA处理还降低了PA-SMC的增殖(图6h和i),通过CD68表达(图6J和k),与未治疗的大鼠相比。
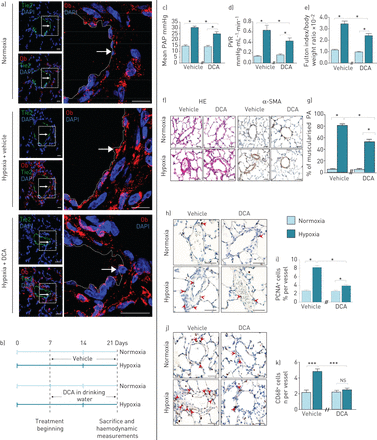
慢性二氯乙酸(DCA)治疗可调节低氧性肺动脉高压(PH)中瘦素(Ob)/Ob主受体(ObR-b)轴。a和b)慢性DCA治疗可消除缺氧情况下内皮源性Ob的产生。a)的代表形象原位用浓度为1 g·L的DCA处理或未处理的大鼠肺,在常氧或缺氧条件下的Ob染色(红色)1,其中肺内皮细胞(P-ECs) Tie2阳性(绿色)。b)常氧或缺氧大鼠饮水中慢性DCA处理的实验策略(每种情况n=5)。c-g) DCA治疗可防止慢性缺氧诱导的PH恶化。c)平均肺动脉压(PAP), d)肺血管阻力(PVR), e)体重归一化富尔顿指数测量右心室肥厚。f)血红素和伊红染色(HE)和α-平滑肌细胞肌动蛋白(α-SMA)的代表性图像。g)定量大鼠肺肌化肺动脉(PA)百分比。h-k) DCA治疗可降低肺动脉平滑肌细胞增殖和血管周围巨噬细胞积聚。染色的代表图像h)增殖细胞核抗原(PCNA)和j) CD68+。i和k)大鼠PA中分别表达PCNA和CD68标记的细胞定量。6-diamidino-2-phenylindole DAPI: 4;ns:不重要的;*: p < 0.05;* * *: p < 0.001。比例尺=50µm。
综上所述,我们的数据清楚地表明,使用Ob中和剂ObR:Fc和DCA,针对Ob/ObR-b轴治疗慢性缺氧诱导PH的两种治疗策略的有效性。
讨论
我们在这里首次证明了肺血管壁Ob/ObR-b轴的异常过度激活及其对慢性缺氧诱导ph的易感性和进展的贡献原位,在体外和在活的有机体内实验表明,与对照组相比,在iPAH中:1)人类和啮齿动物的P-ECs过量产生Ob,这可以部分解释为hif持续稳定;2)人类和啮齿动物PA-SMCs对外源性Ob的反应是过度表达ObR-b并过度增殖;3) Ob/ObR-b轴通过激活循环单核细胞,促进血管周围巨噬细胞增加,诱导ICAM-1、VCAM-1和e -选择素在p - ec中的过表达来驱动血管周围单核细胞/巨噬细胞谱系细胞的积累。与我们在人类中的研究结果一致,我们证明慢性注射重组Ob可以加重慢性缺氧诱导PH的小鼠模型中的PH严重程度。我们还证明Ob/ObR-b轴增加PH敏感性,使用obr缺陷大鼠,慢性缺氧暴露后表现出较轻的疾病,通过肺血流动力学,心输出量,RVH,肺动脉肌肉化,SMC增殖和血管周围单核/巨噬细胞谱系细胞积累。重要的是,我们还证明了针对PH中Ob/ObR-b轴的两种治疗策略的有效性:可溶性Ob中和剂和DCA。我们的研究证实了Ob/ObR-b轴在PH发病机制中的作用。
肺血管结构重构是PAH患者平均PAP增加的原因。虽然PA-SMCs的过度增殖和血管周围单核细胞/巨噬细胞的积累/激活是这一过程的主要因素,但介导这些作用的机制尚不完全清楚[35]。在此,我们表明功能失调的p - ec代表了Ob的异常局部来源,而Ob反过来又作用于不同的细胞类型通过自分泌和旁分泌作用:分别是p - ec, PA-SMCs和单核/巨噬细胞谱系细胞。事实上,通过获得人类数据原位和在体外,以及与在活的有机体内实验,我们在这里证明Ob/ObR-b轴能够诱导PA-SMC增殖,血管周围单核细胞/巨噬细胞谱系细胞积累,并促进P-EC促炎表型,这是PAH肺血管重构的主要因素[36,37]。与这些发现一致,我们发现慢性Ob注射会加重小鼠的PH严重程度。Ob对血管重构影响的缺失可能与内皮功能障碍导致的肺血管反应性改变有关[9]。
尽管全身和肺血管周围炎症的增加在PAH发病机制中的确切作用尚不清楚,但越来越多的证据表明,血管周围炎症的程度与疾病的严重程度有关,特别是与肺血管壁的重塑和厚度有关[38,39]。使用新生儿慢性缺氧诱导的PH模型,F掉et al。[40]发现肺外膜重构是由于表达α-SMA的单核/巨噬细胞谱系细胞的强大募集,而α-SMA可以产生胶原。最近,Vergadiet al。[2]提供的证据表明,不仅肺泡巨噬细胞与慢性缺氧诱导的PH发展有关在活的有机体内但缺氧肺泡巨噬细胞也能诱导PA-SMC增殖在体外。相反,已知PA-SMCs在哮喘中释放多种促炎因子,包括细胞因子、趋化因子和生长因子[41]和急性肺部炎症[42]。综上所述,所有这些发现表明PA-SMC增殖和血管周围单核细胞/巨噬细胞谱系细胞积累可能不是肺血管重构的两个不同组成部分,而是两个密切相关的过程。然而,关于这些现象的触发因素的关键问题仍未得到解答。在此,我们证明了靶向Ob/ObR-b轴的过度激活代表了PAH的潜在创新治疗方法,不仅对平滑肌增生起作用,而且对血管周围单核细胞/巨噬细胞谱系细胞积累起作用。除了这些发现,还需要进一步的研究来更好地确定单核/巨噬细胞系细胞诱导PA-SMC增殖的确切机制,以及PA-SMC是否有助于血管周围单核/巨噬细胞系细胞的募集、粘附和浸润。
大量临床前研究支持DCA对实验性PH的有益作用,包括慢性缺氧和单苦杏仁碱诱导的PH [28,43- - - - - -46]。已知DCA以细胞糖酵解/葡萄糖氧化比为目标,并将细胞代谢从厌氧糖酵解转变为氧化磷酸化。通过丙酮酸脱氢酶激酶抑制,导致HIF-1α降解[27]。虽然HIF-1α已被发现是缺氧依赖性反应的主要调节因子,但越来越多的证据表明HIF-1α也与炎症密切相关[25,47]。特别是,研究表明HIF-1α与主要促炎转录因子之一核因子-κB之间存在调节相互作用[48,49]。在条件HIF-1α缺陷小鼠中,也有研究表明HIF-1α通过诱导巨噬细胞存活和/或分化在先天反应的调节中发挥积极作用[50,51]。在我们的研究中,我们通过作用于PA-SMC增殖和血管周围单核/巨噬细胞谱系细胞积累,证明了DCA治疗对实验PH的临床相关有益作用。除了抑制HIF稳定外,DCA还具有广泛的作用,包括产生活性氧和改变细胞凋亡/增殖比,但没有明显的毒性。因此,DCA的有益作用是多因素的。然而,Ob/ObR-b清楚地出现在PAH代谢转移和肺血管重构之间的界面。
综上所述,我们的研究结果揭示了PAH发病的新机制,即肺血管壁Ob/ObR-b轴异常过度激活,促进了PH易感性和进展。此外,我们证明抑制Ob/ObR-b轴是逆转PAH肺血管重构的相关治疗策略,其目标是PA-SMC增殖和血管周围单核细胞/巨噬细胞谱系细胞积累。
致谢
作者感谢Yuichi Tamura(法国巴黎第一大学,Kremlin-Bicêtre), Roland Jovan (AP-HP, Service de Pneumologie,东华大学胸腔创新,Hôpital Bicêtre, Kremlin-Bicêtre,法国)和Jennifer Bordenave(法国巴黎第一大学,Kremlin-Bicêtre, INSERM UMR_S 999, Marie Lannelongue外科中心,LabEx LERMIT, Le Plessis-Robinson和学院,Le Plessis-Robinson)。感谢他们在研究中的帮助。
脚注
本文的补充资料来自www.qdcxjkg.com
支持声明:这项研究得到了法国国家卫生与医学研究所(INSERM)、法国国家研究机构的资助:资助ANR_12_JSV1_0004_01)、葛兰素史克公司(PAH资助2013)和辉瑞公司(IIR资助WI182054)的资助。本文的资金信息已存入FundRef
利益冲突:可以在以下网址找到与本文在线版本一起披露的信息www.qdcxjkg.com
- 收到了2014年10月17日。
- 接受2015年1月19日。
- 版权所有©ERS 2015











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}