





肺科医生很清楚药物引起的急性和慢性肺毒性;吸入或全身给药可影响气道张力,引起咳嗽、气隙病导致的呼吸困难、弥漫性肺泡损伤、肺毛细血管炎或间质性肺纤维化等症状和组织表现。经充分研究的导致肺毒性的药物包括甲氨蝶呤[1]、博莱霉素[2]和胺碘酮[3.],所有这些都可引起间质性肺病,铂化合物可产生肺超敏反应[4],或丙基硫脲嘧啶,可引起抗中性粒细胞细胞质抗体阳性的血管炎和非特异性间质性肺炎[5,6].多年来,一份药物性肺毒性的目录已被整理出来[7]以及最近在血液系统疾病患者中发现的药物引起的呼吸道疾病[8]以及与全身治疗非小细胞肺癌相关的毒性[9已经被审查过了。
相比之下,PubMed对文献的反复研究没有发现一份药物性肺气肿的临床报告。这种情况引出了一个问题,药物是否不会引起导致人类肺泡毛细血管细胞改变和肺气肿性肺实质破坏的毒性,或者药物引起的肺气肿是否根本没有报道。
在这里,我们试图以适当的谨慎来探讨这个话题,首先声明,在理论基础上,根据来自动物研究的数据,应该有药物导致易感患者的肺气肿。众所周知,并非所有患肺气肿的病人都是吸烟者;据估计,约15%的肺气肿患者不吸烟[10].然而,可以解释这些患者肺气肿的发病机制在罕见的联系后缺乏,如厌食症[11]、镉接触[12或者演奏管乐器的可能性已经被排除在外。在接下来的社论中,我们为我们的药物性肺气肿假设提供了一个基本原理,没有推测在某些情况下,非吸烟者的肺气肿可能与药物或饮食有关。
用于解释肺气肿性肺组织破坏的病理生物学的传统概念集中在炎症细胞释放的介质、蛋白酶和活性氧,所有这些都针对肺泡结构的构建块;主要是上皮细胞和内皮细胞。此外,一些研究人员认为,肺泡结构细胞本身,如内皮细胞,在不需要与炎症细胞相互作用的情况下被激活和损伤。无论如何,肺泡气体交换单元的损伤性攻击也可以在受损的肺结构维护计划中看到[13,14].成人肺结构维持概念整合了遗传和表观遗传控制机制,以稳态平衡原则运作,并为我们理解炎症机制如何参与肺气肿发生提供了一个框架。肺结构的维护理念也提供了空间本身非炎症机制,包括药物毒性。需要明确的是,临床相关药物性肺气肿的假设目前是从动物研究中推断出来的;一个反向翻译的例子。
第一个动物数据来自酪氨酸激酶/血管内皮生长因子(VEGF)受体抑制剂Sugen 5416的测试结果,该抑制剂是通过对抑制VEGF依赖性内皮细胞生长的化合物的高通量筛选而获得的[15].这种高亲脂性小分子化合物在一次皮下注射后4周内引起成年大鼠的气道显著扩大[16].在进行动物实验时,共识是多种肺细胞类型产生VEGF,而VEGF受体主要存在于内皮细胞上,VEGF受体阻断导致肺内皮细胞凋亡。这些实验以及严重肺气肿患者肺中肺血管内皮细胞凋亡的鉴定[17],为肺气肿中凋亡细胞丢失的假设和讨论铺平了道路[13,14].后来发现VEGF作用于大量肺组织细胞,包括肺泡II型细胞和血管平滑肌细胞[17].使用VEGF受体靶向药物治疗导致肺气肿发展的发现得到了其他研究的支持,这些研究表明抗VEGF抗体治疗[17]或在基因工程小鼠中肺靶向VEGF失活[18产生肺气肿。VEGF受体/酪氨酸激酶抑制剂Su5416 (semaxinib)作为抗血管生成癌症药物进入临床阶段[19,20.],但是否有任何接受西莫西尼治疗的患者出现肺气肿尚不清楚,因为不幸的是,接受抗血管生成治疗的癌症患者的生存期很短,肺部药物毒性通常只被认为是“附带损害”。c-Jun n端激酶/应激激活蛋白激酶似乎调节fgf诱导的血管生成[21]也可能参与vegf诱导的血管生成的控制[22].米izunoet al。[23]最近的研究表明,对黏附激酶的抑制会导致空域的急剧扩大,综合这些报道,药物诱导的抑制许多激酶诱导的磷酸化反应可能会干扰血管生成和成人肺细胞的生长(图1).
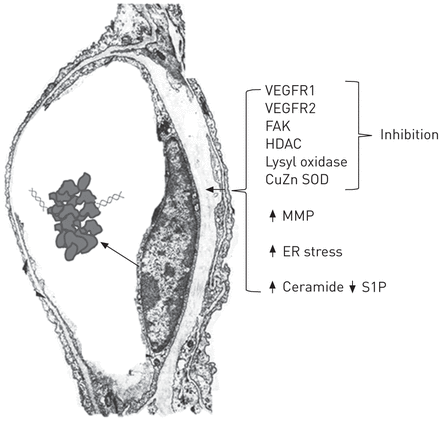
电镜下可见单个肺毛细血管,细胞核大,细胞体薄而伸展。潜在的细胞损伤药物靶点是受体激酶和酶。受体激酶可能被抑制,而酶可能被激活(如。金属蛋白酶)或被抑制(如。赖氨酸氧化酶和超氧化物歧化酶)。血管内皮生长因子(VEGF)信号通过其膜和核受体完好时,鞘氨醇-1磷酸(S1P)/神经酰胺平衡负细胞存活,作为成人肺结构维护计划的一部分。毛细血管腔内的聚集反映了细胞凋亡过程中产生的细胞核片段和游离DNA。VEGFR: VEGF受体;FAK:黏附激酶;组蛋白去乙酰化酶;SOD:超氧化物歧化酶;MMP:基质金属蛋白酶;ER:内质网。
我来et al。[24]研究了组蛋白去乙酰化酶(HDAC)依赖的炎症基因表达,随后检测了慢性阻塞性肺疾病(COPD)患者肺组织中HDACs的表达。尽管作者讨论了炎症背景下肺组织标本中HDACs的低表达和活性,但我们想知道HDACs是否也参与了肺结构维持计划的工作,以及HDAC抑制是否通过降低基因表达导致肺气肿。事实上,非特异性HDAC抑制剂曲古菌素A,当每天给药时,在几周内引起大鼠肺气肿[25].气道扩大与细胞凋亡、缺氧诱导因子(HIF)-1α、VEGF和赖氨酸氧化酶(LOX)表达抑制有关,而肺组织p53和miR34a表达增加[25].人肺微血管内皮细胞HDAC2基因沉默抑制HIF-1α和VEGF的表达[25].因此,抑制HDAC可以抑制炎症,但也会损害肺细胞的生长。
除了蛋白激酶和表观遗传控制器参与转录控制许多细胞生物学的基本过程,如衰老和自噬,鞘脂已被调用在肺气肿的病理生物学中发挥作用[26,27];例如,吸烟肺气肿患者的肺组织脂质提取物中神经酰胺水平升高[26].鞘氨脂-1磷酸(S1P)和神经酰胺的平衡作用,也被称为鞘脂变阻器,可能作为细胞凋亡的控制器参与肺结构的维持[28].目前的理解是S1P促进血管生成和抗凋亡,而神经酰胺触发凋亡[28].合成的视黄醇N-(4-羟基苯基)视黄胺(4-HPR, fenretinide)目前正在研究作为一种抗癌剂[29];其促凋亡活性被认为是由于神经酰胺的产生[30.].鉴于已知的神经酰胺合成被fenretinide激活,我们证明了用fenretinide治疗成年大鼠会导致肺空间扩大,这可以通过腹腔内给药S1P的同时治疗来防止。fentretinide诱导的肺气肿与caspase 3数量的增加有关+HIF-1α、VEGF和HDAC2蛋白表达降低,提示新创神经酰胺的合成影响了极其重要的转录因子HIF-1α的表达[31,32],正如之前用HDAC抑制剂治疗动物后观察到的那样(见上文)。
多年来,人们已经认识到类固醇可引起淋巴细胞和嗜酸性粒细胞的凋亡,并可激活基质金属蛋白酶(MMP)。也有充分的文献证明糖皮质激素会损害产后肺发育[33,34].有了这个信息C锄头et al。[35]给予成年大鼠每日剂量甲基强的松龙(2 mg·kg−1)并在治疗4周后发现空域扩大。类固醇治疗增加了MMP-9的肺组织活性,而泛mmp -抑制剂可防止类固醇治疗引起的肺气肿扩大[35].地塞米松诱导培养的人晶状体上皮细胞凋亡[36]和软骨细胞[37],而曲安奈德会导致骨骼肌细胞凋亡[38].因此,虽然人们普遍认为类固醇具有细胞毒性并促进骨质疏松症的发展,但目前还没有发表关于长时间系统给药类固醇治疗患者的高剂量类固醇对肺实质的影响的信息。
最后,丙烯醛;这种高活性的醛不是一种药物,丙烯醛在这里讨论其潜在的协同毒性。丙烯醛是吸烟者吸入的数千种化学物质之一。根据香烟品牌的不同,一支香烟的烟雾会吸入200-400 μg这种挥发性醛[39].然而,丙烯醛也是在炎症过程中内源性产生的通过髓过氧化物酶介导的苏氨酸降解及胺氧化酶介导的精胺和亚精胺降解[40,41].丙烯醛也是环磷酰胺的代谢产物,具有潜在的临床意义[42].具有侵略性的乙醛在血液中循环,并随尿液排出。最近,研究表明,系统给药丙烯醛可在成年大鼠中产生内质网应激和肺气肿[43],可能是因为诱导内皮细胞凋亡[44,45].环磷酰胺治疗是否会导致肺气肿性肺组织破坏尚不清楚(表1).
饮食习惯或营养缺乏是另一个潜在的问题。我们知道微量金属铜对许多酶的正常功能至关重要。CuZn超氧化物歧化酶和LOX(胶原蛋白和弹性蛋白纤维交联的关键酶)的功能已被充分了解[23].铜是血管生成所必需的[46],突变的铜转运体基因Atp7a是导致门克斯病患儿先天性肺气肿的原因[47].有一种这种疾病的动物模型,即斑鼠,它携带了这种特定基因的突变,并自发地发展成肺气肿[48].饮食中的铜缺乏相当普遍[49,50]和对COPD患者呼出的空气冷凝物的铜测量表明,这些患者在一定程度上缺乏铜[51].米izunoet al。[23]给成年老鼠喂食缺铜的食物,并用铜++螯合剂;他们发现这些动物出现了严重的肺气肿,偶尔还出现了胸膜下水泡。在这些肺中,HIF-1α活性和VEGF表达降低,许多细胞被标记为cleaved caspase 3阳性,这是细胞凋亡的标志。因此,有趣的是,在这些实验研究铜++消耗和螯合再现了在携带突变的小鼠中观察到的结果++转运基因[48,52].
综上所述,我们认为成人肺结构是不稳定的,可以受到环境的攻击通过吸入,但也通过的循环。实验表明,营养缺乏和药物诱导细胞凋亡均可引起肺空间扩大,但无明显炎症反应。VEGF受体抑制剂,HDAC抑制剂和抵消S1P/神经酰胺变阻器和环磷酰胺的药物(通过丙烯醛)是可能引起易感患者肺气肿的药物的例子。易感人群可能是吸烟者、戒烟者和营养不良患者。药物或饮食不足本身可能对正常肺结构的影响有限,但对吸烟者和免疫功能低下的患者或α患者,其影响可能具有协同作用和相关性1-抗胰蛋白酶缺乏[53].可以理解的是,这样的概念与研究者的观点相冲突,研究者不认为结构细胞凋亡是“肺气肿的主要引发因素,并且肺气肿凋亡模型中空气空间的扩大反映的是急性肺损伤和表面张力的变化”[54].最后,我们并不打算将我们所认为的COPD/肺气肿或未被认可的肺气肿合并肺纤维化的一组疾病的复杂性降到最低[55到目前为止,这种疾病只在吸烟者身上观察到。
脚注
利益冲突:没有声明。
- 收到了2013年5月3日。
- 接受2013年7月29日。
- ©2013人队











{kind=link}
{kind=link}