





摘要
我们假设来自慢性阻塞性肺病(COPD)的受试者的原发性支气管上皮细胞(COPD)对不同的反应铜绿假单胞菌卷烟烟雾提取物(CSE)之后的脂多糖(LPS)比没有气流阻塞和非吸烟者获得的PBEC。
在气液界面培养16例COPD患者、10例无气流阻塞吸烟者和9例非吸烟者的PBECs。培养物在刺激前先用CSE培养P.铜绿假单胞菌有限合伙人。ELISA法检测白细胞介素(IL)-6和IL-8,荧光活化细胞分选仪检测toll样受体(TLR)-4的表达。采用Western blotting和ELISA检测NF -κB的活化,Western blotting检测MAPK和caspase-3的活性。Annexin-V染色和末端转移酶介导的dUTP刻痕末端标记法检测细胞凋亡。
COPD培养中IL-8和IL-6的结构性释放最大。但是,CSE预处理之后P.铜绿假单胞菌LPS刺激减少COPD PBECs中IL-8的释放,但增加了没有气流阻塞的吸烟者和不吸烟者的细胞中IL-8的释放。COPD培养中TLR-4表达、MAPK和NF-κB活化在CSE治疗后降低,但在无气流阻塞的吸烟者和非吸烟者组中没有降低,这与细胞凋亡增加有关。
CSE可减弱COPD患者细胞中对LPS的炎症反应,但对不吸烟和无气流阻塞吸烟的人无效。
慢性阻塞性肺病(COPD)是全球卫生问题,主要死因[1].由感染继发的COPD加重是发病率、死亡率和医疗费用负担的一个重要原因。吸烟、感染、炎症与慢性阻塞性肺病之间的关系尚不清楚。
白细胞介素(IL)-6和IL-8对烟雾烟雾提取物(CSE)刺激的原发性支气管上皮细胞(PBEC)的反应是特别的兴趣2].IL-8是一种重要的中性粒细胞趋化剂,与炎症性肺病有关[3.,常用于细胞培养研究的炎症测量[4].吸烟可诱发肺组织中的IL-6 [5和与慢性阻塞性肺病严重程度相关的水平[6].在全身,IL-6诱导骨骼肌耗损[7]可能提高了恶化的风险[8],两者都是COPD中的主要合并症。
核因子(NF)-κB是COPD患者气道炎症基因调控的重要转录因子[9].先前的研究人员报道,暴露于CSE后,PBECs中NF-κB的激活减少[10],而其他人则报道在吸烟者和慢性阻塞性肺病患者的支气管活检中表达增加[9].活化的NF-κB在某些细胞中具有促凋亡作用,但在其他细胞中具有保护凋亡作用[11].据报道,CSE旨在诱导PBEC中的细胞凋亡[12]和原代鼻上皮细胞[13,但其他调查人员没有[10].此外,最近使用PBECs的研究表明,CSE诱导坏死而非凋亡[14].
CSE提供了一种工具来探索香烟烟雾对PBEC培养的影响,并有助于我们理解关键的细胞内信号通路。关于CSE在上皮细胞中诱导促炎症反应的能力,目前尚无共识。制备CSE所采用的方法存在很大差异,且没有商定的金标准。然而,大量证据支持CSE主要具有促炎症作用[2,15,16]而不是免疫抑制作用[17,18在支气管上皮细胞上。虽然有证据表明,鼻上皮细胞可以作为支气管上皮细胞在某些端点的令人满意的替代品,但我们最近表明,支气管上皮细胞对鼻上皮细胞对CSE治疗的反应不同[19].
在本研究中,我们调查了COPD受试者、无气流阻塞吸烟者和非吸烟对照组受试者对CSE治疗的PBECs反应。为了解决这个问题,我们在气液界面(ALI)处培养细胞,它最接近于在活的有机体内环境,并刺激细胞铜绿假单胞菌脂多糖(LPS)作为感染的替代物,有或没有CSE预处理。我们的目的是确定CSE对炎症反应的影响P.铜绿假单胞菌COPD上皮细胞中LPS和凋亡水平与无气流阻塞吸烟者和非吸烟者的比较
方法
研究对象
16名根据英国胸科学会指南诊断为COPD的受试者(其中13名是当前吸烟者,3名是已戒烟者),10名没有气流阻塞的吸烟者和9名非吸烟者被纳入研究。科目人口统计资料详列于表1.所有研究对象都提供了进行纤维支气管镜检查的书面知情同意,在直接目视指导下,通过保护刷从第三代支气管获得4至6个支气管刷。刷毛置于支气管上皮生长培养基中,运至实验室培养。排除标准包括其他重要的肺部病变,包括肺癌。在研究之前,没有患者口服糖皮质激素至少8周,也没有患者的支气管扩张剂可逆性大于10%。COPD患者近期无病情加重,目前的治疗方法见表1.10名吸烟者没有气流阻塞,需要支气管镜检查的临床需要,如持续咯血,但在过程中没有明显的异常。这9名非吸烟者是自愿参加的。这项研究得到了北爱尔兰研究伦理委员会办公室的批准(REC: 09/NIR03/42)。
细胞培养
PBEC最初在支气管上皮生长培养基(BEGM; PROMOCELL,HEIDELBERG,德国)中涂上纯化的牛胶原蛋白(PRECECOL;先进的BIOMATRIX,SAN DIEGO,CA,USA)T10烧瓶,然后用青霉素链霉素抗生素(Invitrogen,大岛,纽约,美国)和牧民(Invivogen,圣地亚哥,加州,美国)。通过针对细胞角蛋白表达的免疫细胞化学染色来确认细胞以随机染色的培养物进行上皮,用于细胞角蛋白表达(数据未显示)。所有实验均通过播种细胞在Ali培养物中以1.5×10的播种密度播种在胶原涂层的Transwell(Corning Inc.,Corning,NY,USA)上进行5细胞每孔生长,直到培养物融合并发展紧密连接。在这一阶段,去除顶端培养基,只在基底侧交替喂养细胞,持续28天。
CSE
通过对Richter等等。[15].一种万宝路红色香烟(尼古丁0.8毫克;10毫克焦油;10毫克一氧化碳)用改进的注射器驱动装置燃烧。通过每15秒抽吸35毫升烟,使烟在5分钟内通过25毫升介质冒泡。悬浮液通过0.2 μm孔径的过滤器过滤,去除大颗粒和细菌。该溶液被视为“100% CSE”,每次实验都是新鲜生成的,然后用培养基连续稀释,最终得到5%的工作浓度。
免疫脉冲
用兔抗e -cadherin一抗对选定的培养物进行染色,以证明紧密连接的存在。分别将培养物暴露于1:200稀释的兔抗muc5ac一抗和小鼠抗乙酰化α-微管蛋白抗体,分别显示杯状细胞和纤毛的存在。使用LAS AF(徕卡,Wetzlar,德国)采集软件捕捉和可视化图像。详细信息请参阅在线补充材料。
FACS.
使用荧光激活细胞分选仪(FACS),用藻红蛋白偶联的抗TLR-4单克隆抗体对渗透细胞进行染色,测定toll样受体(TLR)-4 (eBioscience, San Diego, CA, USA)。用特异性抗体获得的结果与用同型匹配对照抗体获得的结果进行了比较。使用Epics XL流式细胞仪(Beckman Coulter, UK Ltd, High Wycombe, UK)对10000例事件进行分析。
使用膜蛋白V和碘化丙啶(PI)染色(eBioscience,UK)分析细胞凋亡。对于使用CSE的实验,将细胞用5%CSE进行处理24小时,并测量细胞凋亡和/或坏死的量。对膜蛋白阳性的事件,但Pi阴性被认为是早期的凋亡,对膜蛋白v和pi晚期凋亡的事件阳性,以及Pi单独坏死的事件阳性。详细信息请参阅在线补充材料。
TUNEL分析
CSE处理PBECs 24小时后,使用Click-It TUNEL分析细胞凋亡(Invitrogen公司,英国)。细胞经过固定和渗透后,在室温下暴露于反应混合物中过夜。然后用反应缓冲添加剂混合物处理电池30分钟,然后用安装介质和DAPI将电池安装到玻璃盖上。在×100的最终放大倍数下随机选取10个高倍电场进行计数,结果表示为凋亡细胞总数除以每个电场的细胞总数。详细信息请参阅在线补充材料。
西方的屁股
用于确定P38,ERK和JNK1 MAPK的磷酸化,4小时刺激后确定P38,ERK和JNK1 MAPK的磷酸化,切割的Caspase 3,IκB-α和磷酸NF-κBP.铜绿假单胞菌LPS,含或不含5% CSE预处理24小时。详细信息可在在线补充材料中提供。
ELISA
根据制造商的说明,使用商业化的IL-6和IL-8 ELISA试剂盒(R&D Systems Europe, Abingdon, UK),在结果部分中概述的适当刺激后,从基底外侧和根尖培养基等份中测量细胞因子浓度。根据制造商说明书使用显色内毒素定量试剂盒检测细菌内毒素(Thermoscientific, Cambridge, UK)。
TransAM NF -κB化验
根据制造商说明,使用Active Motif公司(La Hulpe, Belgium)的核提取试剂盒制备核提取物。详情请参阅在线补充资料。采用Active Motif Trans-AM NF-κB ELISA试剂盒(Active Motif)检测核提取物中p65的水平。简单地说,将核提取物2 μg稀释至20 μL,加入到含有NF-κB共识结合位点的寡核苷酸包被的孔中。用于检测NF-κB的主要抗体识别的是p65上的一个表位,该表位只有在NF-κB被激活并与目标DNA结合时才能访问。加入与HRP和底物结合的二抗后,在450nm处读取吸光度(参考波长650 nm)。为了监测特异性,进行了竞争性结合分析。在加入核提取物之前,将野生型或突变的共识寡核苷酸添加到含有固定寡核苷酸的孔中。
统计数据
使用SPSS 17.0 (SPSS Inc., Chicago, IL, USA)进行统计分析。数据以四分位区间的中位数表示。组间多组比较采用非参数Kruskal-Wallis检验,两组比较采用Mann-Whitney检验。p值小于0.05被认为是显著的。
结果
细胞培养
细胞在Transwells中成功培养,形成紧密连接,使用相衬光显微镜检查时呈现鹅卵石外观。培养约7天后,粘液分泌明显。在培养14天后,纤毛细胞数量增加,在培养28天后,纤毛细胞数量明显增加(补充图S1)。
CSE
与一系列5% CSE制剂相比,5% CSE浓度在450 nm处的光密度没有显著变化(光密度~ 0.25)。使用一根香烟在25ml培养基中制备CSE产生了足够一致的制剂。以前的研究人员也报告了类似的发现[17]. 在我们的5%CSE制剂中未检测到内毒素。
可溶性介质释放
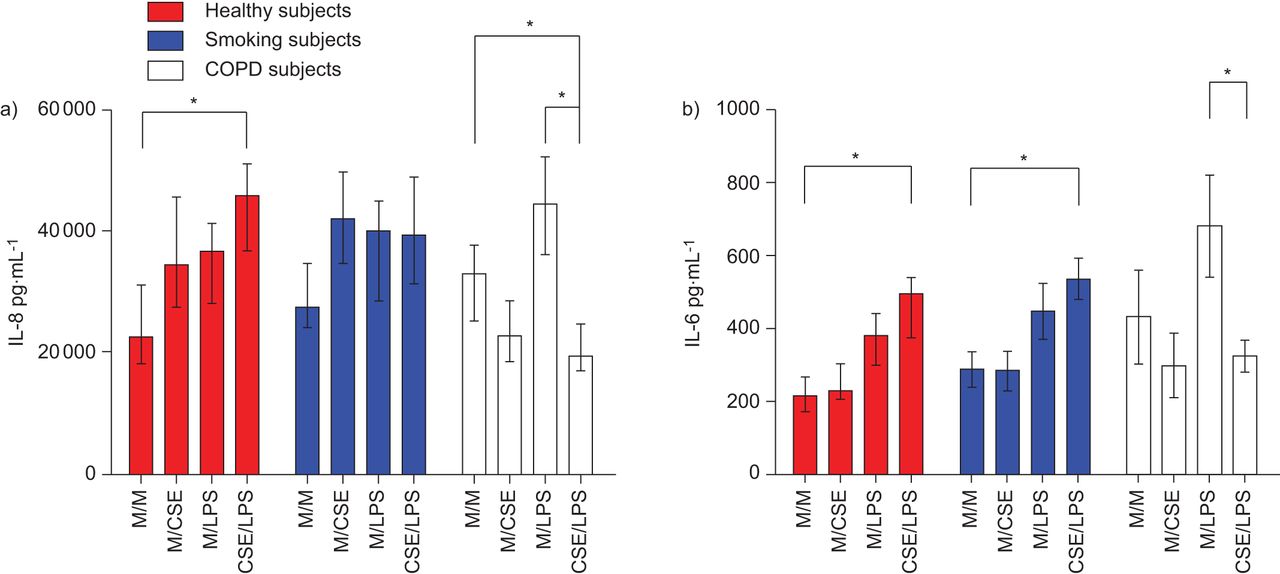
IL-8和IL-6的结构性释放和刺激释放被用来确定上皮细胞的激活。蛋白浓度与通过BCA法随机选择Transwells测定的蛋白浓度没有显著差异(数据未显示),因此可溶介质释放在pg·mL中表达−1而不是正常的蛋白质浓度。的浓度P.铜绿假单胞菌LPS(Sigma-Aldrich,吉尔明厄姆,英国)高于50μg·ml−1在50μg·mL的刺激下,IL-8和IL-6的释放均明显减少−1P.铜绿假单胞菌对照组和无气流阻塞的吸烟者培养物的LPS在CSE预处理24小时后升高。在COPD培养物中,5%的CSE预处理减轻了刺激作用p。铜绿假单胞菌脂多糖(无花果。1).
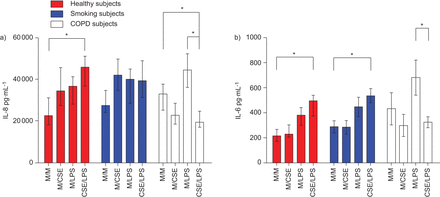
a)Interen介素(IL)-8和b)从慢性阻塞性肺病(COPD)受试者的原发性支气管上皮细胞(PBEC)空气液界面(ALI)培养物中的胰岛介素释放来自慢性阻塞性肺部疾病(COPD),吸烟受试者和健康对照受试者治疗后铜绿假单胞菌脂多糖(LPS) 50 μg·mL−1分别用或不用5%烟抽提物(CSE)预处理24 hP.铜绿假单胞菌有限合伙人(50μg·毫升−1)24小时,有或没有预处理,有5%CSE(或载体)24小时(每组n = 7)。通过ELISA收集并评估EL-8 / IL-6的顶端上清液。数据显示为中位数和四分位数区间。M / M:媒体(24小时)和媒体(24小时);M / CSE:媒体(24小时)和5%CSE(24小时);M / LPS:介质(24小时)和LPS50μg·mL−1(24小时);CSE/LPS: 5% CSE (24 h), LPS 50 μg·ml−1(24 h). *: p<0.05。
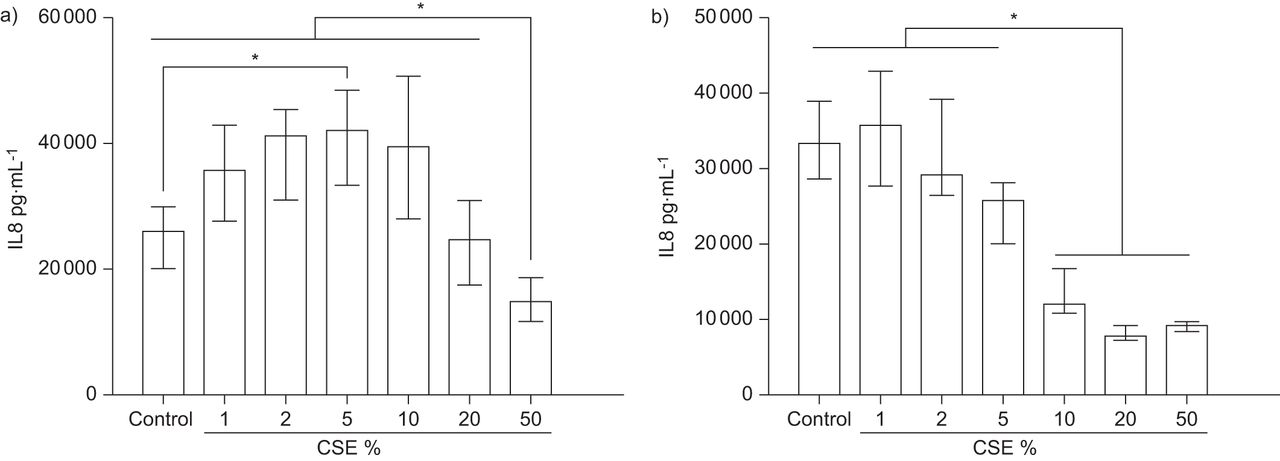
在单独的实验中,从非吸烟受试者获得的细胞与不同浓度的CSE(1-50%)孵卵24小时。CSE刺激培养物释放IL-8至5%浓度,随后在较高浓度时由于细胞毒性而下降(图2).
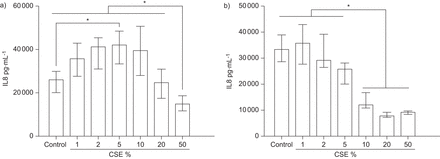
对照原代支气管上皮细胞(PBEC)气液界面(ALI)培养物经烟抽提物(CSE)长期治疗后的白介素(IL)-8剂量反应。对a)非吸烟者和b)慢性阻塞性肺疾病患者的分化良好的PBEC ALI培养物分别用1%、2%、5%、10%、20%或50%的CSE(或载体)治疗24小时。收集根尖段上清,采用ELISA法检测IL-8(每组n=5)。数据显示为中位数和四分位数区间。*:P <0.05。
CSE可诱导原代支气管上皮细胞凋亡
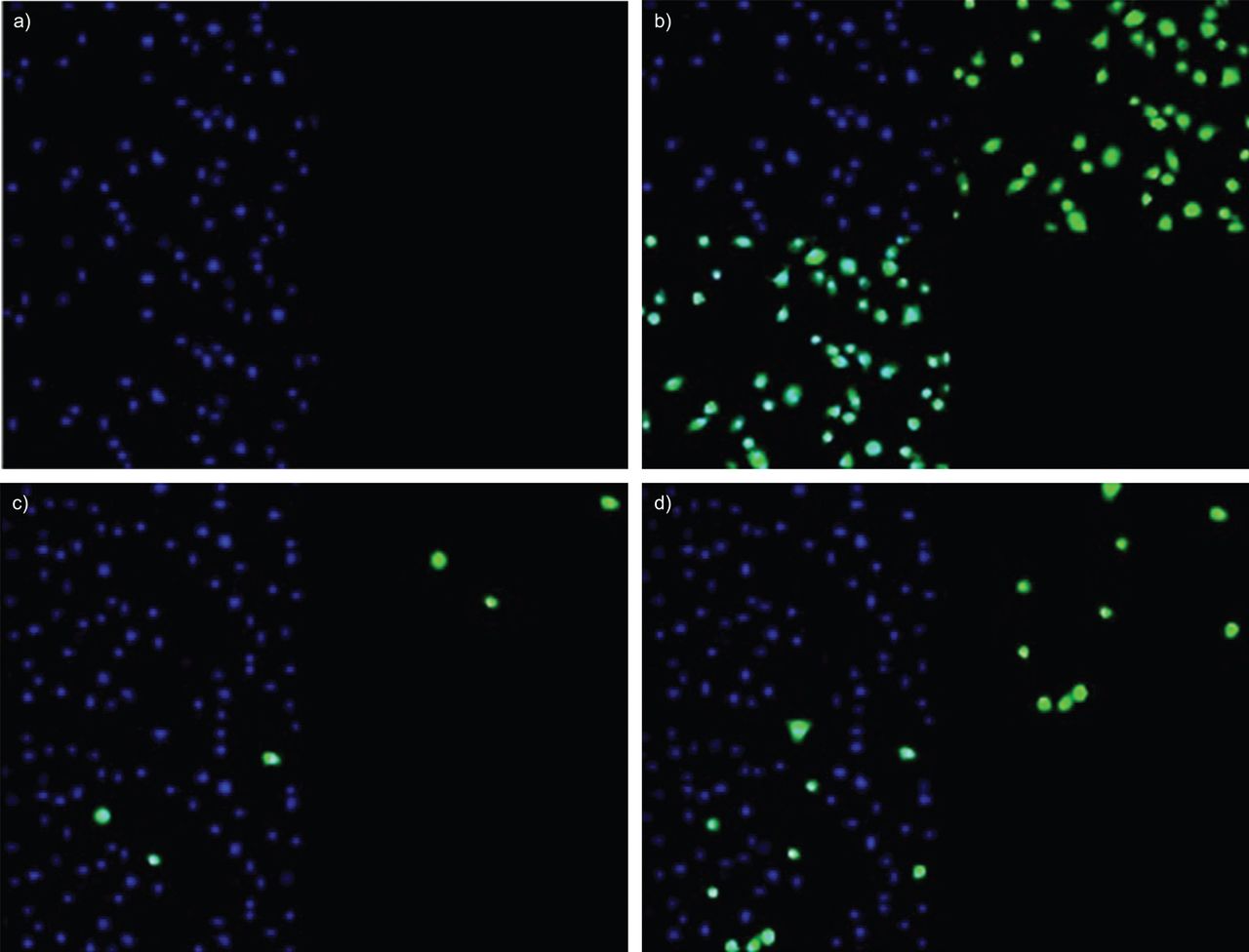
TUNEL法证实细胞凋亡。以浏览具代表性的图像(图3),细胞暴露于CSE 24 h后,该技术测定的凋亡细胞数对健康者为4%,对相应的COPD患者为19%。
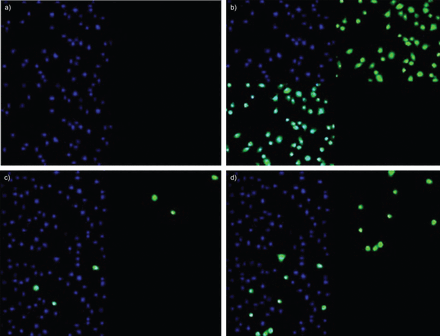
TUNEL法测定5%香烟烟雾提取物(CSE)对健康受试者和气液界面培养吸烟者原代支气管上皮细胞凋亡的影响。非吸烟受试者或无气流阻塞的吸烟者的支气管上皮细胞的代表性图像在盖玻片上生长,并分别用a) PBS或b) DNase I溶液处理阴性和阳性对照。从c)非吸烟受试者和d)慢性阻塞性肺疾病受试者获得的培养物用5% CSE治疗24小时,并根据制造商说明使用Click-iT反应计数凋亡细胞数量。
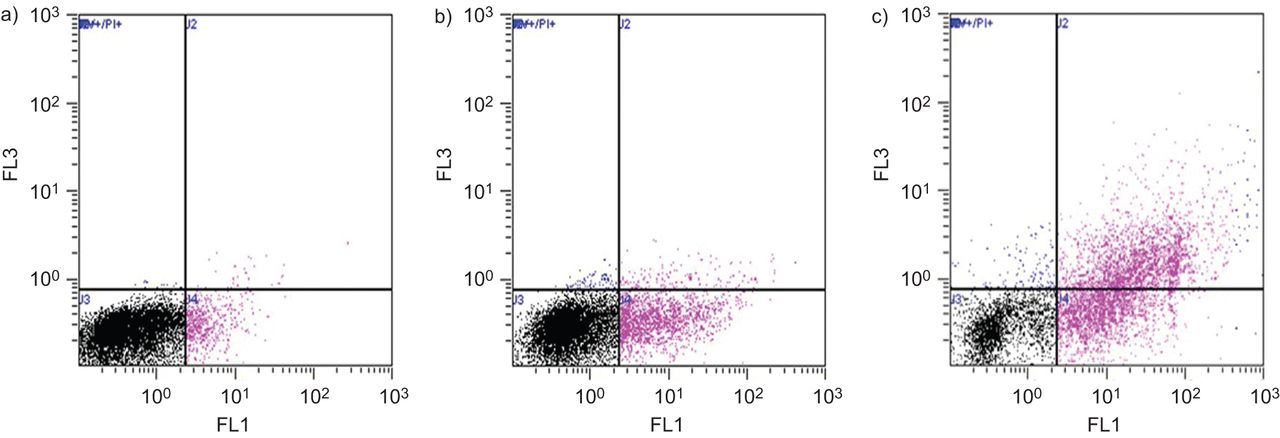
使用FACS技术,也在COPD细胞中用5%CSE进行治疗后处理24小时:51%的细胞保持活性(膜蛋白V和PI阴性);19%发生早期凋亡;26%发生晚期凋亡;4%是坏死(图4).在单独的实验中,三个研究组的细胞(在CSE处理24小时后再次进行)被Annexin V染色,以确定各组间凋亡水平的差异。CSE在COPD培养物中诱导的细胞凋亡百分率最高,在非吸烟者中诱导的细胞凋亡百分率最低。有代表性的点图如图所示图5.

Annexin V/碘化丙啶(PI)分析5%烟提取物(CSE)对慢性阻塞性肺疾病原代支气管上皮细胞气液界面的治疗作用。在每个图中,横轴表示膜联蛋白V的染色强度,纵轴表示PI的染色强度(分别在FL1和FL3图中确定,均为对数标度)。a)未处理细胞,同位素对照染色,b) staurosporin 2 μM处理细胞8 h, c) 0.1% Triton处理细胞8 h, d) 5% CSE处理细胞24 h。5% CSE处理细胞:51%存活;19%发生早期凋亡;26%发生晚期凋亡;4%为坏死。
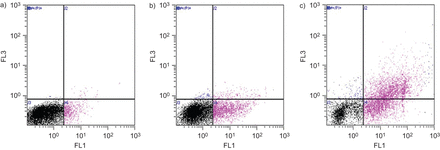
膜蛋白V分析原发性支气管上皮细胞空气液体界面慢性阻塞性肺疾病培养,展示了24小时孵育与5%香烟烟雾提取物对细胞活力的影响。代表性点绘图展示染色阳性阳性阳性的细胞百分比对应于a)不显示器,b)吸烟者,其没有气流阻塞和c)慢性阻塞性肺病受试者。指出了在每个象限中染色的细胞的百分比表2.
CSE在PBEC培养物中切割全长Caspase-3
使用抗体抗裂殖片段的Caspase-3(17kDa),我们通过蛋白质印迹进行治疗P.铜绿假单胞菌LPS和CSE增加了活性Caspase-3的量,而P.铜绿假单胞菌仅LPS一家没有。此外,COPD培养物在培养后有最多的半胱氨酸蛋白酶-3断裂P.铜绿假单胞菌LPS和CSE刺激。通过密度测定法和代表性的印迹法测定的Cleaved caspase与actin的比值如图图6.

的影响铜绿假单胞菌Western blotting检测脂多糖(LPS)和5%香烟烟雾提取物(CSE)对原代支气管上皮细胞气液界面(ALI)中caspase-3激活的影响。a)通道1-3表示用50 μg·mL培养基处理非吸烟(NS)受试者的ALI培养物−1P.铜绿假单胞菌LPS(4h)、5%CSE(24h)和50μg·mL−1P.铜绿假单胞菌LPS(4小时)分别。LANES 4-6表示从吸烟者获得的ALI培养物中的等效刺激,没有气流阻塞(SAWAO)和来自慢性阻塞性肺病的受试者的7-9种ALI培养物。上印迹表示由切割(17kDa)和下β-肌动蛋白产生的Caspase-3的大片段。b)接触暴露于5%CSE后的肌动蛋白比(每组n = 5)。数据显示为中位数和四分位数区间。*:P <0.05。
在单独的实验中,慢性阻塞性肺病培养物用浓度增加的P.铜绿假单胞菌有限合伙人(0-50μg·毫升−1)和CSE(5-50%)和caspase-3的检测使用抗全长caspase (35 kDa)和其剪切片段(17 kDa)的抗体。P.铜绿假单胞菌LPS单独不能切割全长caspase,但使用5%的CSE时,可以明显切割caspase,且CSE浓度越高,切割效果越明显(补充图S2)。
CSE降低COPD细胞培养中TLR-4的表达
检查CSE是否调节TLR-4的表达,PBEC培养物被治疗P.铜绿假单胞菌有限合伙人(50μg·毫升−1),有或没有预处理,24小时5%CSE。尽管CSE处理后,来自对照受试者的培养物中TLR-4的平均荧光强度的绝对值没有变化,但CSE治疗降低了COPD培养物中TLR-4的平均荧光强度。代表性直方图显示在图7.

原代支气管上皮细胞(PBEC)气液界面(ALI)细胞内toll样受体(TLR)-4含量的代表性直方图铜绿假单胞菌含或不含5%香烟烟雾提取物(CSE)预处理的脂多糖(LPS)。a)和b)患有慢性阻塞性肺疾病(COPD)的受试者以及c)和d)没有气流阻塞的吸烟者的高分化PBEC-ALI培养物与5%CSE孵育24小时,然后用P.铜绿假单胞菌有限合伙人(50μg·毫升−1)4小时。e)和f)将来自COPD受试者的PBEC Ali培养物分离,用或没有5%CSE进行24小时治疗。然后将细胞固定,透析化并用针对TLR-4或等效同种型匹配对照的植物粥共轭抗体染色。示出了平均荧光强度(MFI)(每个组n = 4)。数据显示为中位数和四分位数区间。*:P <0.05。
CSE降低COPD细胞培养中的MAPK激活和NF-κB
Western blotting显示,5% CSE治疗24小时后,COPD培养物中p38、JNK和ERK MAPK的磷酸化水平降低。即使没有刺激,所有的MAPK都被激活,之后没有进一步显著增加P.铜绿假单胞菌LPS刺激(所有印迹中的通道1-4;补充图S3-S5)。然而,磷酸化P38,ERK和JNK的水平降低了24小时5%的CSE预处理和P.铜绿假单胞菌LPS刺激COPD培养物(所有印迹中lanes 5-8;补充图S3-S5)。在对照培养中,MAPK的激活减少并不明显。与JNK或ERK相比,p38的激活减弱并不明显,因此也使用FACS进行了测量,证实了5% CSE治疗后,phospho-p38的平均荧光强度降低(补充图S3)。
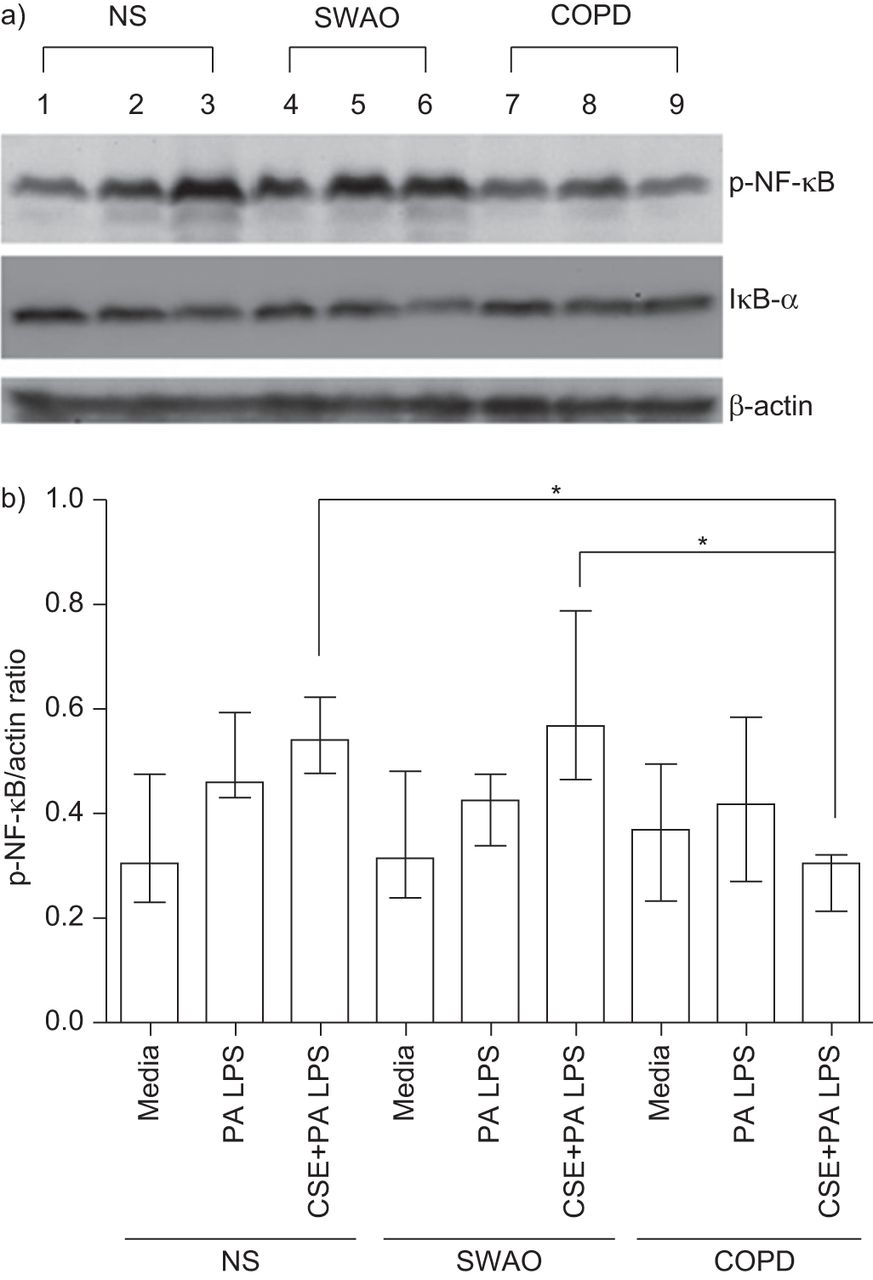
50 μg·mL刺激后,全细胞裂解液中磷酸化nf -κB蛋白水平显著升高−1P.铜绿假单胞菌三组均为LPS。在用5%CSE预处理后,不吸烟者和无气流阻塞的吸烟者组的这一情况进一步加剧。相反,在COPD培养物中,用5%的CSE预处理降低了死亡率P.铜绿假单胞菌LPS诱导NF-κB活化。通过密度法和代表性的印迹法确定的磷酸化- nf -κB与肌动蛋白的比值如图图8.随着磷酸NF-κB增加,IκB-α的水平降低。使用ELISA的方法重复来自核提取物的P65 NF-κB的实验和确定水平,结果表明了类似的结果(无花果。9).
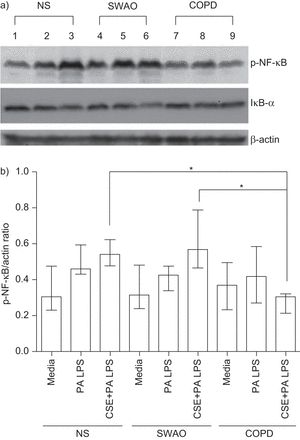
Western blotting检测5%烟提取物(CSE)和不含脂多糖(LPS)对原代支气管上皮细胞(PBEC)气液界面(ALI)核因子(NF)-κB活化的影响。a) 50 μg·mL治疗后,对照组(NS)、无气流阻塞吸烟者(SWAO)和慢性阻塞性肺疾病(COPD) PBEC ALI培养物中p65 NF-κB和IκB-α蛋白表达(β-actin负荷对照)的Western blot检测−1铜绿假单胞菌LPS处理4 h, 5% CSE处理24 h, 50 μg·mL处理−1P.铜绿假单胞菌LPS治疗4小时。1-3通道表示仅用50 μg·mL培养基治疗非吸烟者PBEC−1P.铜绿假单胞菌LPS (4h)和50 μg·mL−1P.铜绿假单胞菌LPS (4h)与5% CSE预处理(24h)。第4 - 6道代表无气流阻塞吸烟者ALI培养的等效治疗,第7-9道代表COPD受试者ALI培养的等效治疗。b) Phospho-NF-κB / actin比值(各组n=5)。数据显示为中位数和四分位数区间。*:P <0.05。
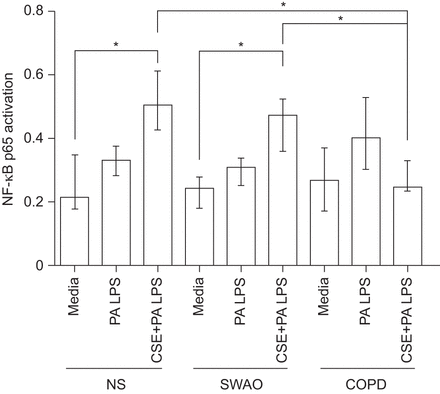
利用TransAM NF-κB试剂盒检测脂多糖(LPS)添加或不添加5%香烟烟雾提取物(CSE)对原代支气管上皮细胞(PBEC)气液界面(ALI)培养物核因子(NF)-κB活化的影响。对照组(NS)、无气流阻塞吸烟者(SWAO)和慢性阻塞性肺疾病(COPD)患者经50 μg·mL治疗后PBEC ALI培养物的核提取物−1铜绿假单胞菌LPS处理4 h, 5% CSE处理24 h, 50 μg·mL处理−1P.铜绿假单胞菌使用TransAM NF-κB p65试剂盒检测LPS处理4 h后NF-κB p65的激活情况。所示结果均来自双孔测定。
讨论
在这一系列的实验中,我们证明了从对照组和无气流阻塞的吸烟者培养物中获得的IL-6和IL-8的刺激释放被5%的CSE预处理放大,但这在COPD组中不明显。我们的结果还表明,5%的CSE在COPD培养中具有细胞免疫抑制作用。这些发现表明,慢性阻塞性肺疾病更易受到CSE的免疫抑制作用,这可能部分解释了这一特殊群体对呼吸道感染的易感性增加。
在我们的实验中,5%的CSE降低了TLR-4的表达。最近使用人类支气管上皮细胞系报告了类似的发现,CSE治疗导致TLR-4表达下调,这与IL-8释放的相应增加有关。受体的内化被认为是其机制,通透性细胞中TLR-4的表达平行增加[4]. 然而,报道的CSE对TLR-4表达的影响并不完全一致。尽管经CSE刺激后,A549细胞中TLR-4 mRNA和蛋白表达呈剂量依赖性下调[20.[次年的出版物表明CSE在BEA-2B细胞中增加TLR-4表达[21.].CSE还可以通过下调RIG-1受体的表面表达来影响其他重要受体,[22.].因此,尽管我们的数据表明CSE下调COPD PBEC中的TLR-4表达,但我们不能绝对信心这是其免疫抑制作用的唯一机制。可以想到,CSE也会影响其他受体,这可能影响细胞刺激与TLR-4无关。
NF-κB的激活调节基因表达,促进细胞存活,并在小鼠B细胞和许多细胞系中保护细胞免于凋亡[23.,24.].此外,通过透明质酸在气道上皮细胞中通过TLR依赖激活激活NF-κB的激活是对细胞凋亡的保护性[25.].有趣的是,由于CSE治疗后COPD上皮细胞培养中TLR-4的绝对数量减少,这与所有MAPKs激活的减少有关,这是这些特定培养物对CSE治疗的凋亡的一个潜在机制。然而,其他数据不支持这一概念,因为从健康受试者获得的PBECs用CSE治疗48小时可导致NF-κB激活抑制,而没有任何凋亡、坏死或caspase-3激活的证据[10].
除了可溶性调解员释放中的差异,我们在我们的研究组中展示了磷酸化NF-κB的差异。虽然治疗从健康对照受试者和没有呼吸道病的吸烟者获得的PBECP.铜绿假单胞菌LPS增加了NF-κB的磷酸化水平,但在COPD ALI培养中并非如此。此外,COPD ALI培养物在5% CSE治疗后,所有MAPK的激活降低。后者与TLR-4和转录因子磷酸化NF-κB的表达减少同时出现。
使用CSE在体外研究被批评为长期令人不满意的模型,吸烟者暴露于[26.].使用的CSE浓度范围为100% CSE,持续15分钟[1724小时至1%CSE(在那些使用单卷烟的研究中准备最初的“100%”股票CSE)[27.].其他研究人员已经准备了多种香烟的CSE,从两支到五支不等。13,28.].许多这样的研究评估可溶性介质释放和单个时间点,但两项研究强调需要谨慎CSE可以延迟LPS-induced释放引发和granulocyte-macrophage原发性上皮细胞集落刺激因子,但是在之后的时间点不完全废除(17,18].因此,如果分离使用较早的时间点,结果会误导地认为CSE仅仅具有免疫抑制作用。当CSE浓度大于5%时,存在不可接受的细胞毒性(补充图S2),因此我们选择使用该CSE制剂来研究从ALI培养中释放的可溶性介质。
本研究中使用的大多数COPD受试者是目前的吸烟者。我们的数据表明,相对于无气流阻塞的吸烟者和对照组受试者,CSE暴露后细胞凋亡的增加和caspase-3激活的增加与吸烟无关本身.似乎COPD本身的发展进一步增加了支气管上皮对CSE的细胞毒性作用的易感性。另外,这些个体的上皮细胞可能本质上更容易凋亡。
Caspase活化并未一致显示在CSE诱导的细胞凋亡中是重要的[29.]并且实际上,细胞死亡可以通过独立于Caspase活性的机制来发生[30.].CSE改变促凋亡和抗凋亡因子是可行的,这些因子在这一日益复杂的细胞死亡途径中发挥着重要作用。CSE改变了PBEC ALI培养中磷酸化NF-κB的水平,我们的数据表明,在这种情况下,NF-κB活化的升高可能对细胞凋亡具有保护作用。
尽管可以认为,细胞培养过程可能通过ALI培养中涉及的多个增殖周期改变上皮表型,但最近的研究表明,这是不太可能的,至少对原代鼻上皮细胞而言[31.].此外,还有多种方法用于获得和建立气道上皮细胞培养物,每个方法都具有自己的优点和局限性。此外,具有COPD的个体的气道可能不是无菌。我们讨论了在线补充材料中的这些重要方面。
综上所述,尽管COPD患者的气道上皮细胞表现出一种促炎表型,但与对照组相比,这些细胞对CSE的炎症反应减弱。此外,这些细胞更容易发生凋亡。目前还不清楚这种异常是由于疾病过程中细胞发生了变化,还是这些特定的细胞从根本上更容易受到细胞死亡的伤害。不管哪种解释是正确的,这种特殊的异常至少表明,旨在减少细胞凋亡数量的治疗可能是有益的。细胞凋亡抑制剂的开发是一个现实的目标。在治疗肝病的临床试验中,已经有广谱凋亡抑制剂,它们是同类药物中的第一种[32.].此外,该数据强调了戒烟对COPD受试者的重要性,以尽量减少上皮细胞的损失,并维持支气管上皮的完整性。我们的研究结果支持吸烟会降低先天性肺防御的观点,并有能力增加呼吸道感染的易感性。
脚注
这篇文章有补充资料可从www.www.qdcxjkg.com
利益陈述书
没有人申报。
- 收到了2012年4月17日。
- 接受2012年7月22日。
- ©2013人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}