





摘要
强效促炎介质的释放对于在感染期间建立有效的宿主反应至关重要。然而,过度的炎症可能导致有害的组织损伤。这在严重肺炎球菌性肺炎中尤为突出,在这种情况下,杀死肺炎球菌所需的强烈炎症反应与器官功能丧失之间的微妙平衡决定了疾病的结局。
我们评估了有效的抗炎细胞因子白细胞介素(IL)-10在肺炎球菌感染中的调节作用通过Western blot, ELISA和染色质免疫沉淀分析。
链球菌引起的肺炎诱导IL-10在小鼠肺和人肺上皮细胞中的表达。肺炎球菌感染可强烈诱导krueppel样因子(KLF)4的表达在活的有机体内和在体外。IL-10和KLF4的诱导都是通过细菌DNA、toll样受体(TLR)9、MyD88和Src激酶等途径介导的。KLF4被招募到il10启动子和小干扰rna介导的KLF4表达下调可阻断肺炎球菌感染期间IL-10的表达。
综上所述,KLF4以细菌DNA - TLR9 - src依赖的方式诱导并调节IL-10的表达,将TLR9检测细菌DNA与炎症反应的控制联系起来。
一个快速,强大和有效的先天免疫反应保护哺乳动物免受入侵病原体。与病原体来源的毒素结合,从人体细胞释放的促炎和毒性物质损害组织功能。因此,先天免疫反应必须足以杀死病原体,但也需要严格控制,以确保最小的组织损伤和宿主存活。这种微妙的平衡在肺炎中尤为突出,肺炎是全球第三大死亡原因[1]。一方面,在肺炎期间,宿主反应对细菌的局部清除阻止了感染和炎症在整个肺部和全身的分布[2]。另一方面,尽管宿主和病原体之间的战斗持续不断,但必须保证宿主的有效氧合,从字面上讲,每分钟都是如此。
大多数社区获得性肺炎病例是由于感染链球菌引起的肺炎(肺炎双球菌)[3.]。toll样受体(TLR)2、TLR4和TLR9的激活似乎参与了肺炎球菌先天免疫应答的激活[4- - - - - -6]。这些受体启动细胞因子的强烈释放,导致肺部多形核白细胞的特征性浸润[2]。尽管平衡的炎症反应对肺炎至关重要,但人们对如何控制肺炎炎症知之甚少。
krueppel样转录因子(KLFs)是哺乳动物中表达的21种含锌指转录因子的一个亚类[7]。KLF4首次被确定为皮肤屏障建立的重要因素[8]。最近的研究表明KLF4参与了白细胞中细胞因子产生的调控[9,10]。
通过使用肺炎链球菌我们分析了抗炎白细胞介素(IL)-10的表达调控。我们报道了KLF4以细菌DNA - TLR9 - src依赖的方式诱导并调节IL-10的表达,将TLR9检测细菌DNA与抗炎反应的控制联系起来。
材料与方法
材料
src激酶抑制剂PP2及其对照品PP3购自Calbiochem (Merck, Darmstadt, Germany)。ODN M362和ODN TTAGGG购自InvivoGen (San Diego, CA, USA)。使用的所有其他化学品均为分析级,并从商业来源获得。
细菌菌株和细菌产物
小鼠肺炎是由感染肺炎链球菌血清3型PN36 (NCTC 7978)。肺炎链球菌R6x和R6xΔ厚度(溶血素(Ply)缺陷)菌株用于在体外研究是血清2型菌株D39的未封装衍生物。将肺炎球菌置于血琼脂板上过夜,然后在添加5%酵母提取物的Todd Hewitt肉汤中培养至600 nm光密度(OD)600) 0.3),离心收获,按照推荐的方法在细胞培养基中重悬。R6x在56℃下热失活1小时。为分离细菌DNA,采用体外培养法培养肺炎球菌(OD)6001.0),离心收获,重悬于N-三(羟甲基)甲基-2-氨基乙烷磺酸,用溶菌酶、变酵素、RNase、pronase E和sarcosyl裂解和处理。提取DNA,用苯酚、乙酸钠和2-丙醇沉淀。Ply是T. Mitchell(生物医学研究中心,格拉斯哥大学,格拉斯哥,英国)的礼物。
人类细胞系和DNA
转化的人支气管上皮细胞(BEAS)-2B细胞和人胚胎肾(HEK)293购自American Type Culture Collection (Rockville, MD, USA)。人原代小气道上皮细胞(SAECs)来自Clonetics/Cambrex (Taufkirchen, Germany)。按照前面描述的方法培养和感染细胞[11]。按照制造商的说明,使用QIAampDNA迷你试剂盒(Qiagen, Hilden, Germany)制备BEAS-2B细胞的人DNA。
小鼠肺炎模型
所有动物手术都是根据《赫尔辛基动物使用公约》进行的,并得到柏林(德国)国家卫生和社会事务办公室的批准。6周龄雌性C57BL/6小鼠在指定时间点麻醉、感染后处死。肺部细菌负荷60小时达到最大,感染性传播开始~ 24小时,小鼠在感染后60 - 120小时死亡[12]。切除的肺在液氮中冷冻,并进行支气管肺泡灌洗(BAL)。
il - 10 ELISA
通过商用ELISA (Becton Dickinson GmbH,海德堡,德国)评估上清液或BAL中IL-10的产生。为了实现更灵敏的IL-10检测,我们生成了3.9 ng至250 ng的标准稀释度在体外研究。对于所有ELISA数据,通过测量相同上清液中的乳酸脱氢酶来确认细胞活力(数据未显示)。
西方墨点法
按指示刺激细胞,清洗两次并收获。老鼠的肺在液氮中冷冻并粉碎。细胞或肺匀浆在含有NP40的缓冲液中裂解,并进行Western blotting。在4°C下,将膜暴露于IL-10 (Abcam, Cambridge, MA)、KLF2、KLF4、TLR9、MyD88、细胞外信号调节激酶(ERK)2、肌动蛋白(所有Santa Cruz Biotechnology, Inc., Heidelberg, Germany)、环氧化酶(COX)2、局点粘附激酶(Upstate Biotechnology, Lake Placid, NY, USA)的抗体中过夜,随后与二抗(IRDye 800标记的抗小鼠或抗山羊,或cy5.5标记的抗兔)孵育。蛋白质检测采用奥德赛红外成像系统(LI-COR Inc., Bad Homburg, Germany)。
质粒和转染程序
按照制造商的说明书(Clonetech, Palo Alto, CA, USA),用磷酸钙沉淀法共转染HEK293细胞。依赖于KLF4 (Z-Y)的荧光素酶报告基因。按照指示使用IL-10 (C. Wehner, KKG Entzuendliche Lungenerkrankungen der GSF, Gauting,德国)(各0.2 μg)或0.1 μg pRL-TK (Promega GmbH, Mannheim,德国)质粒。使用Fugene6 (Roche Applied Science, Mannheim, Germany)转染BEAS-2B细胞。萤火虫,Renilla采用双荧光素酶报告基因检测系统(Promega GmbH, Mannheim, Germany)测定荧光素酶活性。
RNA干扰
用AtuFECT01生成小干扰RNA (siRNA)脂质体,以每10个2 μg siRNA转染(Silence Therapeutics AG, Berlin, Germany)6细胞(13]。siRNA序列购自Ambion (Ambion Ltd, Huntington, Cambridge, UK),见表S1。
染色质免疫沉淀反应
按照指示刺激BEAS-2B细胞,按照前面描述的方法进行染色质免疫沉淀(ChIP) [4,6,14用…il10启动子特异性引物对(正向:tcgaggcgaccgcgacagt;反向:ggagcagcgcgtcgctga)。采用抗KLF4和RNA聚合酶II抗体(Santa Cruz Biotechnology)进行免疫沉淀。等量的输入DNA通过凝胶电泳控制。
统计数据
数据以平均值±表示扫描电镜对于至少三个独立的重复实验(在体外)或五次(在活的有机体内)。数据分析采用单因素方差分析,主效应比较采用Bonferroni后验。图1一个和5 f]采用非配对t检验进行分析。
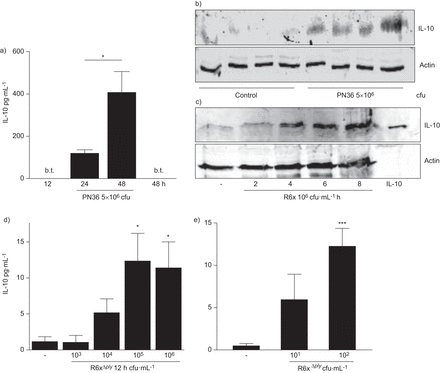
肺炎球菌诱导的白细胞介素-10表达。小鼠感染链球菌引起的肺炎诱导IL-10表达在活的有机体内a)支气管肺泡灌洗(BAL),每组5只小鼠;b)小鼠肺组织(4只对照小鼠或感染5×10的小鼠)6cfu PN36作用48 h),分别通过a) ELISA和b) Western blot检测。R6x(10)感染c)支气管上皮细胞(BEAS)-2B细胞6cfu·毫升−1;d)小气道上皮细胞(SAECs)和e)人胚胎肾(HEK)293与R6xΔ厚度d) 12 h或e) 24 h导致IL-10产量增加在体外。在b)中,共加载5ng重组IL-10作为阳性对照。Western blots代表三个实验中的一个。肌动蛋白检测显示蛋白质负载相等。低于临界值。*: p < 0.05;* * *: p < 0.001。
结果
肺炎球菌诱导IL-10表达
我们在BAL (图1一个)以及肺组织(图1 b)感染肺炎球菌的小鼠。此外,R6x肺炎球菌诱导了BEAS-2B细胞中IL-10的表达(图1 c和图S1)。同样,ply缺陷突变体(R6xΔ厚度诱导人原代SAECs中IL-10的表达(图1 d)和HEK293细胞(图1 e)。
肺炎球菌诱导的KLF4表达在体外和在活的有机体内
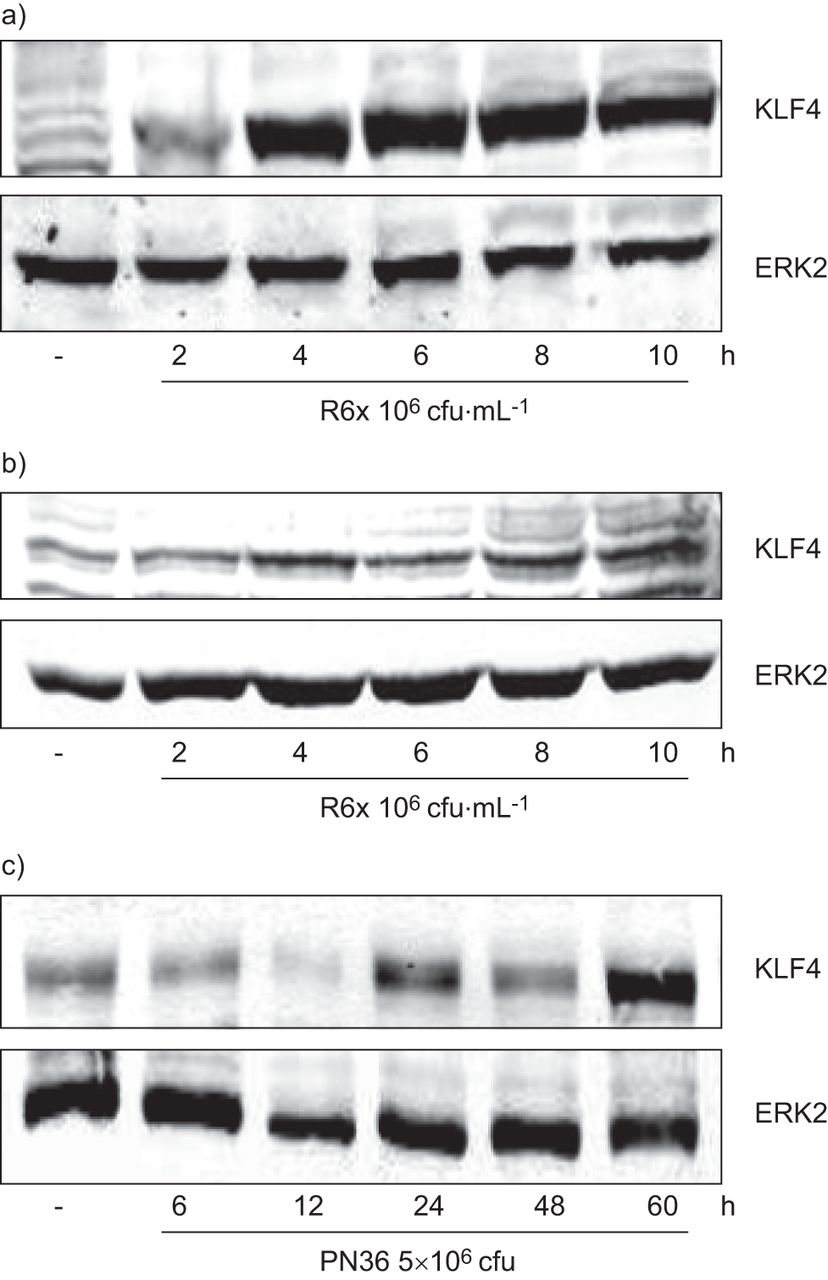
BEAS-2B细胞或SAEC暴露于肺炎球菌诱导的时间- (图2一个b)和剂量依赖性(图S2a和b) KLF4的表达在体外。此外,感染小鼠肺中也检测到KLF4表达升高(图2 c)。
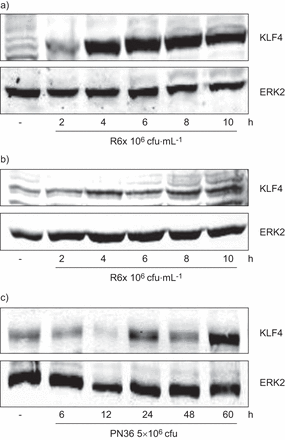
肺炎球菌诱导的krueppel样因子(KLF)4表达。a)支气管上皮细胞(BEAS)-2B和b)小气道上皮细胞以及c) PN36 (5×10)中KLF4蛋白表达的时间依赖性诱导6图示小鼠肺部感染的菌落形成单位。一种抗细胞外信号调节激酶(ERK)2的抗体被用作负载对照。从三个独立实验中选出一个具有代表性的斑点。
IL-10的klf4依赖性表达
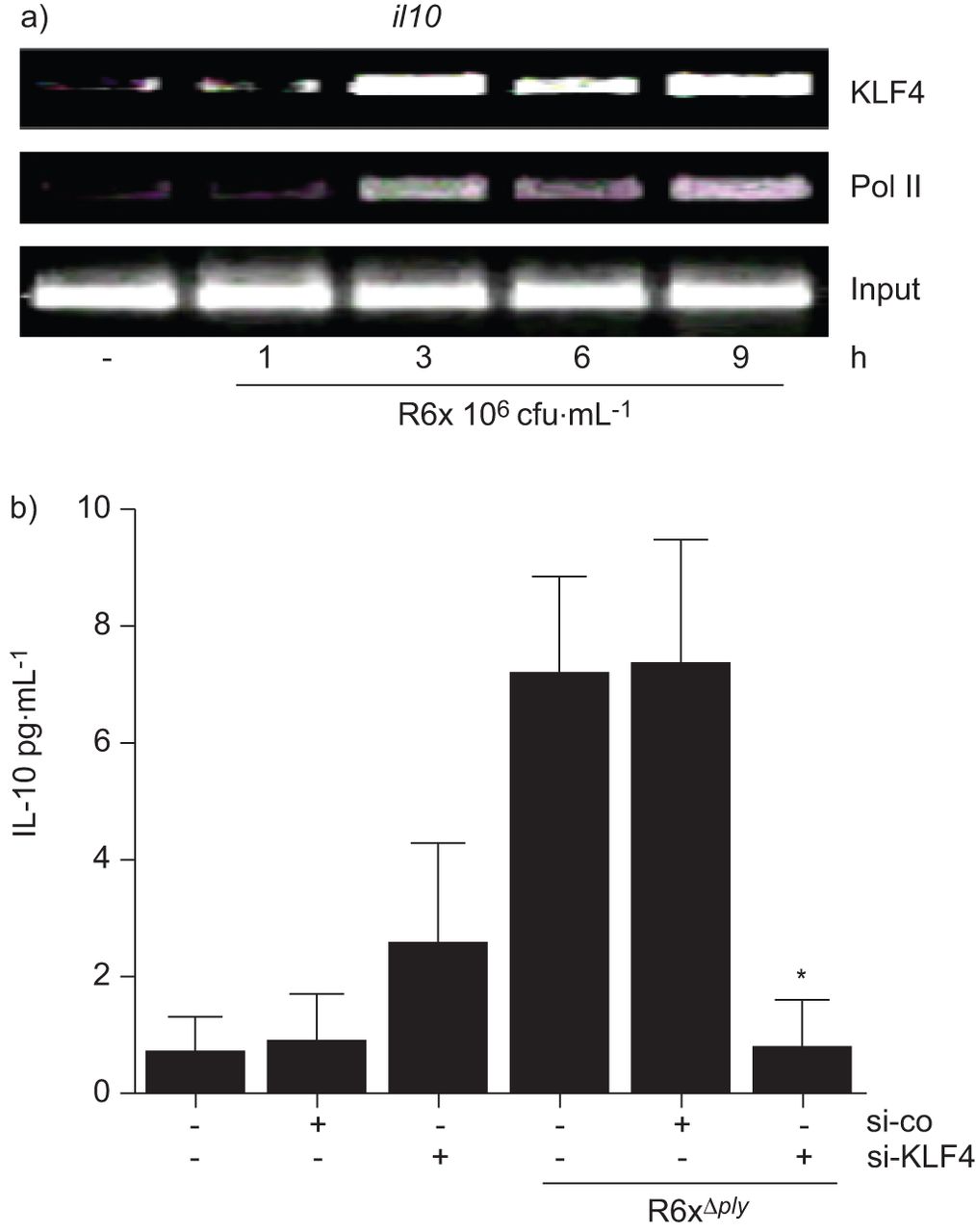
通过ChIP实验验证了IL-10在肺炎球菌感染的人肺细胞中受KLF4调控的假设。对肺炎球菌感染的BEAS-2B细胞的分析显示,KLF4和RNA聚合酶II在细胞中募集il10催化剂(图3)。特异性siRNA消融KLF4表达(图S3)可消除肺炎球菌诱导的IL-10分泌(图3 b)。
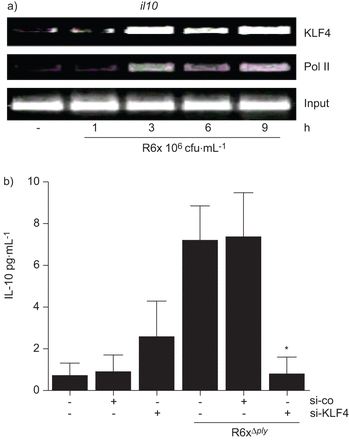
krueppel样因子(KLF)4调控白细胞介素(IL)-10的表达。a) KLF4和RNA聚合酶II (Pol II)的时间依赖性募集il10R6x刺激后支气管上皮细胞(BEAS)-2B细胞染色质免疫沉淀显示启动子。在三个独立的实验中,给出了一个具有代表性的凝胶。b)特异性小干扰RNA (si) (si) (si-KLF4)沉默KLF4表达(转染后96 h),而非对照siRNA (si-co),减少R6xΔ厚度(102菌落形成单位(cfu)·mL−1人胚胎肾(HEK)293细胞中IL-10相关表达。数据代表三个独立的实验,每个实验重复进行。*: p < 0.05。
KLF4在肺细胞中的表达受TLR9诱导
siRNA消除tlr相关接头分子MyD88降低了肺炎球菌相关的KLF4表达(图S4a和b)。ply缺陷肺炎球菌诱导HEK293细胞中KLF4蛋白表达(图S4c),已知HEK293细胞对TLR4刺激无反应[6]。此外,在BEAS-2B细胞中,Ply和脂多糖(LPS)未能诱导KLF4表达,但分别刺激了COX2或KLF2的表达(图S4d和e)。这些结果表明,Ply和TLR4的激活都没有促进KLF4在肺上皮细胞中的表达。此外,BEAS-2B细胞暴露于热灭活肺炎球菌后,虽然不能诱导KLF4表达,但仍能诱导COX2表达(图S4f),这表明需要活的肺炎球菌才能触发肺细胞中KLF4的表达。假设杀死的肺炎球菌仍能被TLR2识别[11]以及细菌诱导的tlr9依赖性信号通过热失活而减少[5],我们测试了TLR9可能是KLF4诱导的受体。
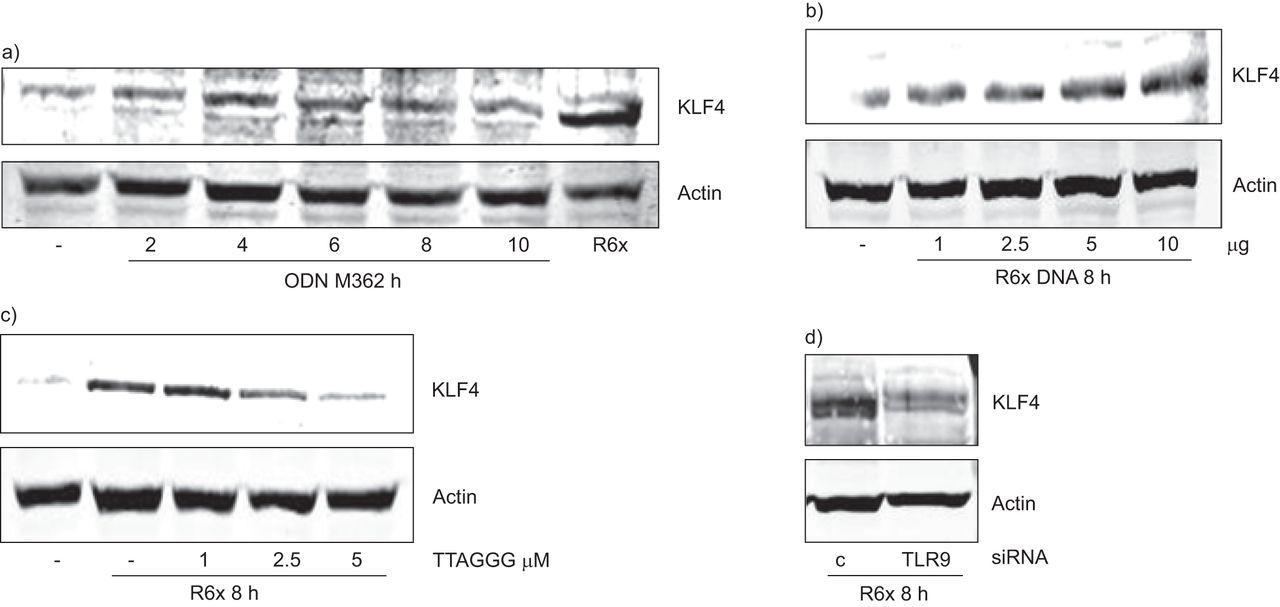
TLR9激动剂ODN M362 (图4)及肺炎球菌DNA (图4 b)诱导BEAS-2B细胞中KLF4的表达,而添加抑制CpG基序TTAGGG则阻断了肺炎球菌依赖的KLF4表达诱导(图4摄氏度)。最后,通过siRNA降低TLR9表达(图S4g), BEAS-2B细胞中与肺炎球菌相关的KLF4表达被消除(图4 d)。
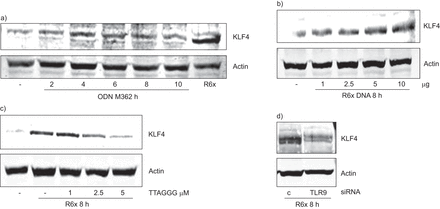
Krueppel-like factor (KLF)的表达a) ODN M362 (1 μg·mL)刺激支气管上皮细胞(BEAS)-2B−1b)用不同浓度的R6x DNA刺激(8 h)诱导KLF4表达。c)用TTAGGG预先孵育BEAS-2B,在R6x刺激后8小时降低了KLF4蛋白水平,d)用TLR9 siRNA转染BEAS-2B 96小时也降低了KLF4蛋白水平。所有Western印迹都是三个独立实验中的代表性印迹。肌动蛋白作为加载对照。c:控制。
肺炎球菌dna诱导的tlr9依赖性IL-10表达的识别
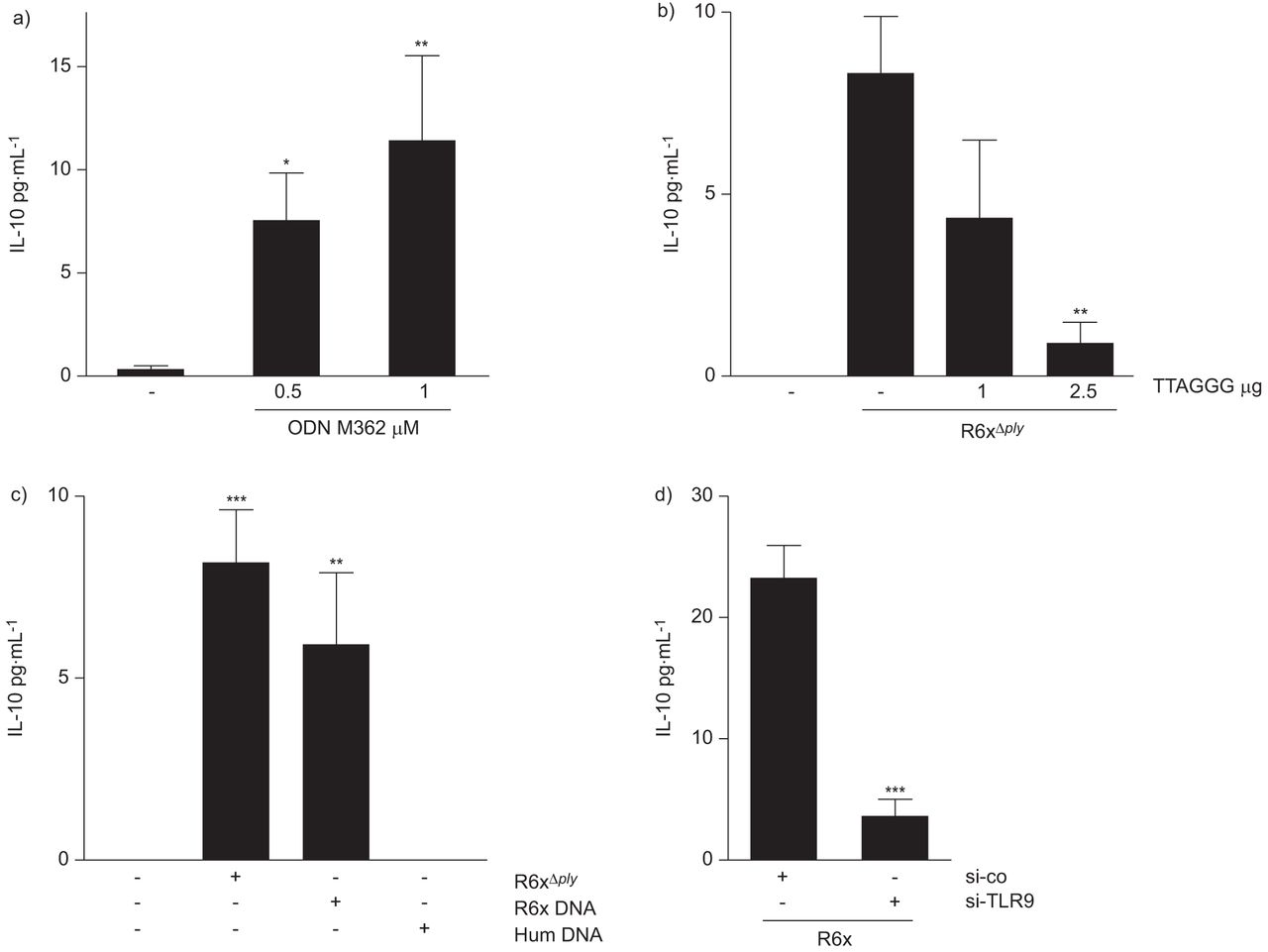
鉴于BEAS-2B和HEK293细胞表达TLR9(图S5a和b),而HEK293细胞不表达TLR2或TLR4 [15],这些细胞暴露于TLR9激动剂ODN M362后,IL-10的产生增加(图5和图5c)。ODN M362拮抗剂TTAGGG预先孵育上皮细胞,阻断HEK293细胞中肺炎球菌相关IL-10的表达。图5 b)。使用肺炎球菌DNA进行的实验显示,HEK293细胞中IL-10的表达增加,而人类DNA则没有明显的影响(图5度)。sirna介导的MyD88(图S5D)或TLR9(图5d)消融可消除肺炎球菌相关IL-10释放。图5 d)。
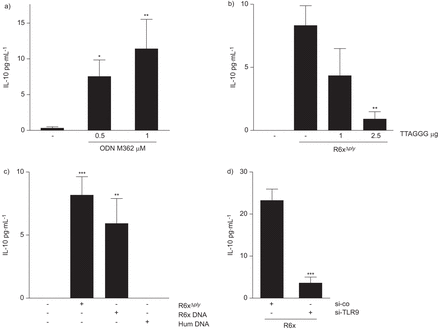
toll样受体(TLR)9依赖性分泌白介素(IL)-10。a)人胚胎肾(HEK)293暴露于0.5 μM和1 μM ODN M362诱导IL-10表达(24 h)2菌落形成单位(cfu)·mL−1R6xΔ厚度1 μM和2.5 μM的TTAGGG寡核苷酸抑制IL-10的表达。c) HEK293细胞暴露于102cfu·毫升−1R6xΔ厚度或1 μg·mL−1R6x DNA作用24 h,但不能达到1 μg·mL−1诱导IL-10的释放。d) TLR9表达的沉默降低了R6xΔ厚度(102cfu·毫升−1)介导的IL-10表达。每个图表代表三个独立实验的总结,重复进行。si-co:对照siRNA;si-TLR9:小干扰TLR9;*: p < 0.05;* *: p < 0.01;* * *: p < 0.001。
KLF4的诱导和IL-10的表达受Src激酶调控
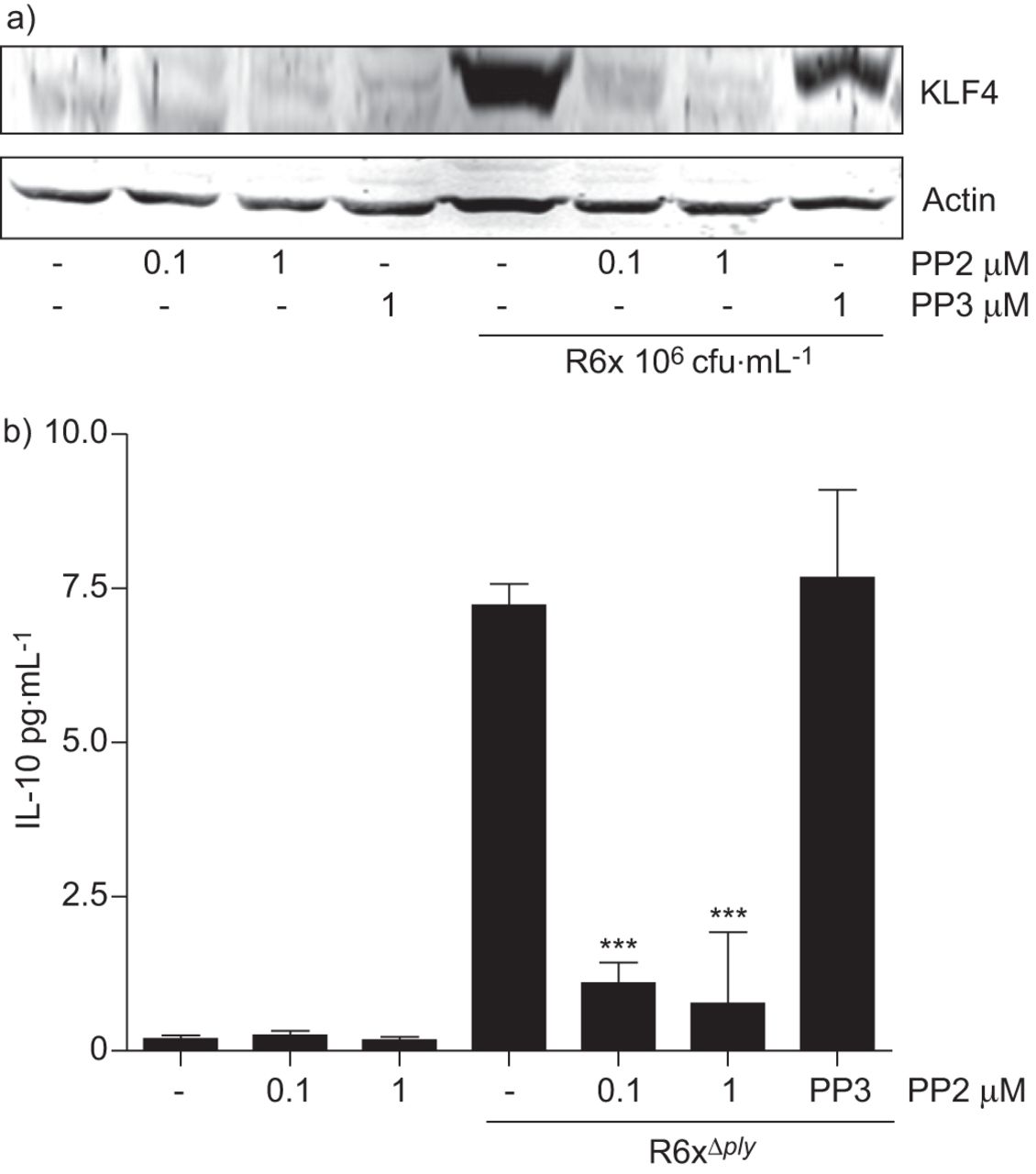
Src抑制剂PP2(而非其无活性对照化合物PP3)在r6x感染细胞中预先孵育1小时,可阻断KLF4的表达(图6)。因此,PP2(而非PP3)抑制肺炎球菌感染的HEK293细胞中IL-10的表达(图6 b)。
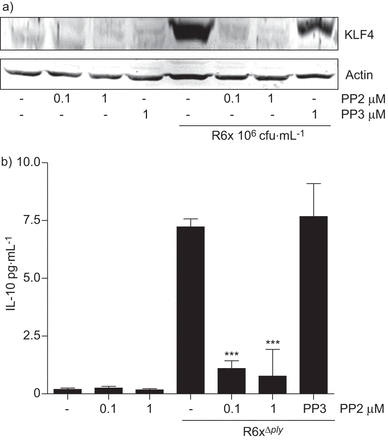
Src激酶抑制降低了a)克鲁珀尔样因子(KLF)4诱导和b)白细胞介素(IL)-10表达。将支气管上皮细胞(BEAS)-2B细胞与0.1-1 μM Src激酶抑制剂PP2(而非其无活性对照PP3的1 μM)共孵育1小时,感染前可降低人胚胎肾(HEK)293细胞中a) KLF4的表达和b) IL-10的产生。从三个独立实验中选出一个具有代表性的斑点。数据代表平均值±扫描电镜三个独立实验重复排列,差异表示如下:***:与抑制剂特异性效果比较p<0.001。
讨论
在此,我们证明了细菌dna依赖的KLF4表达随后在人肺上皮细胞中诱导抗炎IL-10表达。细菌DNA或cpg识别通过TLR9激活Src激酶依赖性信号,随后增加KLF4表达。因此,激活tlr9 - src - klf4依赖通路可能对控制炎症反应很重要。
炎症反应的控制和终止对维持组织完整性很重要,但与那些启动先天免疫反应的机制相比,对炎症控制机制的重视要少得多。在抗生素治疗后,可以推测,持续的强先天免疫反应可能对宿主弊大于利。此外,选择性操作抗炎和解决可能允许在严重感染,如肺炎辅助治疗的发展。
一些报告表明肺炎球菌可被宿主识别通过TLR2, TLR4和TLR9。绒毛是一种重要的毒力因子肺炎链球菌至少部分地激活宿主细胞,通过TLR4的刺激[4,6]。LPS-TLR4通路可诱导巨噬细胞中KLF4的表达[9,10]。本研究显示,TLR4配体(LPS或Ply)刺激并没有促进肺上皮中KLF4的表达。此外,ply缺陷肺炎球菌诱导KLF4表达,这些突变体也在HEK293细胞中诱导这种表达,而HEK293细胞对TLR4配体无反应[6]。这表明TLR4不参与肺炎球菌介导的KLF4在人肺上皮中的诱导。值得注意的是,热灭活肺炎球菌也未能诱导KLF4表达。肺炎球菌的热失活可导致Ply的破坏[4]和对TLR9信号的敏感性降低[5],尽管它们仍然能够激活细胞通过TLR2 [5,11]。鉴于Pam3Cys对TLR2的刺激也未能诱导胚胎干细胞中KLF4的表达[16],我们排除了该受体可能是KLF4诱导剂。相反,模拟细菌DNA和肺炎球菌DNA的cpg寡核苷酸诱导KLF4,可被拮抗寡核苷酸TTAGGG抑制。根据这些数据,TLR9或参与TLR9信号级联的MyD88的敲低[17],也减少了KLF4诱导。因此,KLF4的表达似乎以高度依赖于细胞和环境的方式调节。总之,这些数据表明肺炎球菌DNA在人肺上皮细胞中以tlr9依赖的方式诱导KLF4蛋白表达。
Src蛋白激酶家族的成员被认为参与tlr9相关的信号传导[18,19]。根据这一建议,我们观察到PP2抑制Src可阻断肺炎球菌感染细胞中KLF4蛋白的表达。因此,在肺炎球菌刺激的肺细胞中,KLF4的诱导依赖于肺炎球菌DNA-TLR9-MyD88-Src激酶途径。然而,TLR9刺激似乎并不仅仅具有抗炎作用,因为腹腔内应用CpG寡脱氧核苷酸可诱导小鼠肺部炎症反应[20.]。然而,观察到TLR9缺陷小鼠,而不是TLR1、TLR2、TLR4或TLR6敲除小鼠,在肺炎球菌性肺炎中表现出更高的死亡率,这表明TLR9在该疾病中总体上是有益的[14]。
一旦诱导,KLF4被证明与il10siRNA敲低KLF4可阻断IL-10在肺炎球菌感染细胞中的表达。与我们的数据一致,L国际单位等。[10]报道KLF4调控巨噬细胞中IL-10的表达。我们观察到IL-10在肺炎球菌感染小鼠的BAL和肺组织以及感染的人上皮细胞中的表达增加。此外,CpG寡核苷酸和肺炎球菌DNA诱导IL-10表达,而抑制TTAGGG寡核苷酸或TLR9-和MyD88敲低则降低IL-10的表达。这些观察结果与MyD88敲除细胞在CpG DNA刺激后产生较少的IL-10一致[21]。
最后,PP2对Src的抑制也降低了肺炎球菌相关IL-10的产生。这些观察结果证实了TLR9-Src-KLF4通路在诱导IL-10 (图7)。它可能是假设的在活的有机体内IL-10反应是由TLR激活引发的炎症反应触发的,包括TLR9。然而,尽管小鼠中TLR9的缺失减少了细菌对肺炎球菌的杀伤,但它并不导致细胞因子的产生增加[14]。众所周知,IL-10在患有严重感染(包括肺炎)的人体内会增加,但其作用仍有争议。早期将IL-10应用于暴露于LPS的人导致整体炎症反应降低[22]。实验研究表明,il - 10降低吞噬细胞的抗微生物能力,因此,在肺炎等疾病的早期阶段,高IL-10水平可能会增加病原体传播的风险[23]。人类IL-10-1082基因启动子多态性(与早期和高IL-10水平相关)增加肺炎有害结局的风险进一步表明,早期强IL-10释放可能是不利的[24]。在重症肺炎和肺炎相关败血症中,IL-6和IL-10水平均高于非并发症肺炎。一种假设是,在这些患者中,增加的IL-10水平反映了一种平衡机制,以避免压倒性的炎症。因此,尽管IL-10的广泛表达可能会降低宿主在肺炎早期对病原体作出适当反应的能力,但在疾病过程中减少炎症相关的肺损伤可能很重要。由于本文所描述的klf4相关的IL-10反应发生延迟,因此可以将其视为后期阶段平衡IL-10反应的一部分。
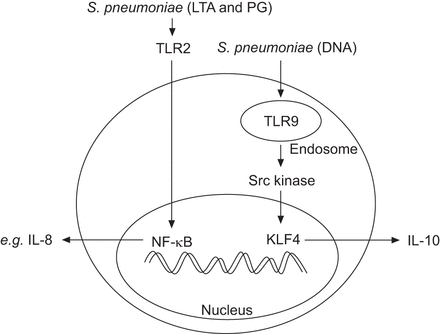
肺上皮细胞的促炎性和抗炎性信号。肺上皮细胞的刺激链球菌引起的肺炎导致不同信号通路的激活。TLR2识别细菌细胞壁成分,如脂质胆酸(LTA)和肽聚糖(PG),激活典型核因子(NF)-κB通路,随后释放促炎细胞因子和趋化因子,如白细胞介素(IL)-8 [6]。通过TLR9识别细菌DNA,随后激活Src激酶并将KLF4与细胞结合,可以触发一条不同的途径il - 10启动子,最终导致抗炎细胞因子IL-10的释放(本研究)。
在许多器官中,除了病原体本身造成的损伤外,炎症总是承担着由宿主反应引起的有害组织损伤的风险。分析KL4在…中的作用是很有意义的在活的有机体内肺炎球菌肺炎模型。然而,由于传统的KLF4敲除小鼠表现出致死表型[8],条件KLF4敲除小鼠可用于这些研究[25]。KLF4似乎参与了免疫细胞的免疫调节[9,10]。此外,例如,单核细胞在肺组织修复机制中非常重要[26,27]。这就提出了关于上皮细胞作用的问题与髓系KLF4在肺炎球菌性肺炎中的表达。此外,随访KLF4在肺炎肺实质细胞和髓源性细胞凋亡调控中的作用也很重要。通过使用细胞/组织特异性条件KLF4敲除模型或例如,互惠骨髓嵌合小鼠,这些有趣的问题将在进一步的研究中得到解决。
综上所述,细菌dna介导的TLR9-Src激酶激活可诱导KLF4表达,从而启动中枢抗炎介质IL-10的表达。因此,除了激活先天免疫反应外,非典型tlr相关信号通路可能对控制严重感染中的炎症反应很重要。
致谢
我们感谢J. Hellwig、A. k
脚注
本文的补充资料来自www.www.qdcxjkg.com
支持声明
这项工作得到了德国研究基金会(DFG) (SFB-TR84 to J. Zahlten, B. Schmeck, A.C. Hocke, M. Witzenrath, N. Suttorp和S. Hippenstiel),德国联邦建设和研究基金会(forss - partner F-KZ 0315256, HMWK (LOEWE-UGMLC)和DLZ to B. Schmeck)的支持;进展到N. Suttorp, B. Schmeck和S. Hippenstiel)和EU-Network CAREPNEUMO(到S. Hammerschmidt和S. Hippenstiel)。
利益声明书
没有宣布。
- 收到了2011年11月11日。
- 接受2012年4月2日。
- ©2013人队










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Pro- and anti-inflammatory signalling in lung epithelial cells. Stimulation of lung epithelial cells with Streptococcus pneumoniae leads to activation of different signalling pathways. Recognition of bacterial cell wall components like lipoteichoic acids (LTA) and peptidoglycan (PG) by TLR2 results in activation of the canonical nuclear factor (NF)-κB pathway and subsequent release of, for example, pro-inflammatory cytokines and chemokines, such as interleukin (IL)-8 [6]. A distinct pathway is triggered by recognition of bacterial DNA by TLR9, followed by activation of Src kinase and binding of KLF4 to the il-10 promoter, which finally leads to the release of the anti-inflammatory cytokine IL-10 (this study).](http://www.qdcxjkg.com/content/erj/41/2/384/F7.large.jpg?width=800&height=600&carousel=1){kind=link}
{kind=link}