





抽象的
专利加入Riociguat与肺动脉高压患者西地那非组合的安全性和有效性。
接受西地那非(20mg,每日3次)的患者被随机分配到安慰剂或riociguat(高达2.5 mg,每日3次),持续12周。主要结果是给药4小时内仰卧位收缩压(SBP)较基线的最大变化。次要目标包括额外的血压、心率和探索性疗效变量,以及安全性。患者可进入长期延长期(LTE),所有患者均接受利奥昔加西地那非。
在riociguat(n = 12)之间的4小时内,仰卧的SBP的最大变化没有差异(平均值±sd基线:-20.2±15.3毫米汞柱;第12周:-20.7±18.0 mmHg)和安慰剂组(n=6)(分别为-7.6±3.9和-20.2±12.9 mmHg)。站立收缩压和仰卧或站立舒张压的变化也无差异。联合治疗对包括血流动力学和运动能力在内的探索性临床参数没有显示出良好的效果。在LTE中,有很高的低血压停药率和3例(18%)死亡(研究者不认为研究药物相关)。
西地那非加利昔瓜特存在潜在的不良安全信号,没有证据表明其有利/风险比。因此,riociguat与磷酸二酯酶-5抑制剂同时使用是禁忌的。
抽象的
西地那非+riociguat用于PAH:没有证据表明其有利/风险比和潜在的不利安全信号http:///wly/hbmfh.
介绍
肺动脉高压(PAH)是一种慢性和危及生命的疾病,其特征在于由于逐步血管重塑导致的肺血管阻力(PVR),最终可能导致右心力衰竭和死亡[1- - - - - -3.].目前对PAH的处理包括磷酸二酯酶(PDE)-5抑制剂,内皮素受体拮抗剂(ERAS)和前列腺素[2].然而,尽管这些PAH治疗方法可用,但在治疗这一疾病方面仍有显著的医疗需求未得到满足,PAH患者的死亡率仍然很高(1年:15%;3年:32%)[4].
肺动脉高压与内皮功能障碍、一氧化氮(NO)合成受损和NO -可溶性鸟苷环化酶(sGC) -环鸟苷单磷酸(cGMP)通路刺激不足有关[5- - - - - -7].Riociguat是一个名为SGC刺激者的新型治疗剂的第一个成员[6- - - - - -9].它具有双重作用模式,通过稳定NO - sGC结合和直接刺激sGC使sGC对内源性NO敏感通过不同的结合位点,独立于否,从而导致CGMP的产生增加[6,7,10].在枢轴期III肺动脉高血压可溶性胍基环化酶 - 刺激试验(专利)-1中,Riociguat耐受良好,并且改善了治疗患者的治疗患者的运动能力和一系列次要终点 - 接受预处理的患者用时代或非抗体前列腺素[11].因此,Riociguat最近批准用于治疗症状PAH,为PAH提供了新的特定药物治疗[2,12].
PDE-5抑制剂,如西地那非和达拉非,防止CGMP降解,但取决于CGMP合成和NO的存在[1,6].PDE-5抑制剂已被证明是一种有效的治疗PAH患者。因此,我们研究了西地那非与riociguat联合治疗PAH患者时可能的协同作用,riociguat作为sGC的NO-independent刺激物,通过刺激sGC增加cGMP,同时抑制其分解。然而,伴随PDE-5的抑制和sGC的刺激也可能是不耐受的。因此,PATENT PLUS研究评估了riociguat或安慰剂联合西地那非12周对症状性PAH患者收缩压(SBP)的影响。
方法
研究对象
患有症状PAH的患者接受稳定(≥90天)西地那非治疗(每日三次批准剂量20毫克)有资格进行研究。包含标准包括:18-75岁,6分钟步行距离(6MWD)> 150米,PVR> 300 DYN·S·CM5,平均肺动脉压(MPAP)≥25mmHg,SBP≥95mmHg和心率≤105击球率·分钟−1在服用西地那非后2小时。接受其他PDE-5抑制剂、非特异性PDE抑制剂、ERAs、前列腺素类或NO供体治疗的患者不包括在内,尽管支持性肺动脉高压治疗是允许的。
学习规划
PATENT PLUS是一项随机、双盲、安慰剂对照、多中心研究,于2010年8月至2013年5月在捷克共和国、德国、意大利、西班牙和英国的11个中心进行(www.clinicaltrials.gov标识符号码NCT01179334).该研究评估了riociguat (1-2.5 mg,每日3次)与西地那非(20 mg,每日3次)同时给药对症状性PAH患者服用研究药物后4小时血压的影响。根据Bayer Randomisation Management提供的计算机生成的随机代码,患者以2:1的比例随机接受riociguat或安慰剂,使用交互式语音响应系统或交互式网络响应系统。鉴于本研究的探索性,没有进行正式的样本量计算。该研究由一个独立的非盲数据监测委员会(DMC)监督;DMC和发起人有权在任何时候终止研究。
根据以往的临床研究[11,13],在8周剂量调整阶段,根据收缩压和症状的出现,分别调整了riociguat的剂量,从起始剂量1 mg,每日3次,每2周0.5 mg增加。然而,出于安全原因,在治疗阶段的任何时间点都允许减少riociguat的剂量。然后患者在4周的维持期(总治疗时间:12周)继续服用他们的最佳剂量。
完成最初12周随机研究并对研究药物耐受良好的患者有机会参与一个开放标签长期延长(LTE),该研究评估了riociguat/西地那非组合的长期安全性和耐受性。在为期8周的LTE剂量调整阶段,主要阶段的riociguat患者继续使用他们个人“最佳”剂量的riociguat,并进行假剂量调整,而最初研究中的安慰剂患者则进行了riociguat的盲法个体剂量调整(1-2.5 mg,每日3次)。按照研究主要阶段使用的相同方案。在8周的滴定期之后,患者在剩下的研究中接受了最佳剂量的开放标签治疗。
PATENT PLUS是根据良好临床实践指南和赫尔辛基宣言实施的。该研究方案得到了所有参与中心的伦理委员会的批准,所有患者都给予了书面的知情同意。2012年,拜耳医疗保健制药公司(伍普塔尔,德国)与首席研究员一致终止了这项研究,原因是在非受控LTE阶段,因低血压事件、严重不良事件(SAEs)和死亡而终止。再加上在随机研究阶段缺乏良好临床效果的信号。因此,PATENT PLUS研究的第二部分被取消,该研究旨在评估riociguat和西地那非的联合剂量高于批准剂量。
程序
在治疗前随访时首先评估血压和心率。分别于给药前30、15分钟,给药后15、30、45、60分钟,后3小时半小时记录平卧、站立血压和心率。在随后的治疗访问中,同时给予西地那非和瑞昔哥特或安慰剂后,对血压和心率的评估遵循类似的日程。这些时间点是根据初步药效学数据选择的,设计用于捕获给药后血压和心率的最大变化。
总体研究基线收缩压曲线是随机分组前,在研究第1天使用西地那非时的曲线。第一次就诊后,患者在滴定期第2、4、6和8周就诊,在维持期第12周就诊。主要终点是在给药4小时内卧位收缩压与基线相比的最大变化。每次就诊时与基线相比的最大变化被定义为给药后4小时内患者内与基线相比的最大降幅(如果基线低于该曲线中所有随后的收缩压测量值,则为零)。每个基线是研究给药前30分钟及30分钟内记录的最后一次收缩压。
次要变量包括:站立SBP,仰卧和站立舒张压(DBP)的最大变化,以及从研究药物4小时内从基线中仰卧和仰卧和静止的HR;效果曲线下的面积从基线改变,仰卧SBP,4小时内的仰卧SBP,DBP和心率在4小时内。安全变量包括不良事件,生命体征和实验室参数。6MWD,世界卫生组织(世卫组织)功能阶级,脑Natiretic Peptide(NT-probnp),Borg呼吸困难评分,临床恶化和血液表达(通过右心导管术后)的初步分析,12周后临床恶化和血液学患者也进行了。
在LTE滴定阶段,患者使用与最初12周研究相同的计划记录仰卧和站立血压和心率。对于剩余的LTE,每3个月进行一次安全性评估,但没有严格的血压评估(在计划的随访中,在研究药物摄入前和2-3小时后测量站立血压)。
分析
在本探索性研究中,所有变量均采用描述性分析,并没有计划进行正式的统计学检验。数据显示(意味着±sd除非另有说明)除非在各自访问的观测值下计算,缺少数据没有避阻。不良事件包括从施用第一剂研究药物的施用时开始的所有事件,直至给予最后一次剂量后2天。
结果
12周的随机研究
耐心
18名符合条件的患者进入了最初的12周随机研究,12名患者随机接受riociguat治疗,6名患者随机接受安慰剂治疗(图1).
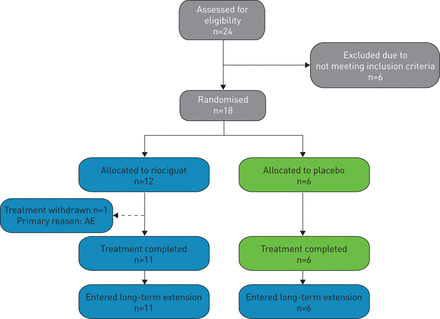
PATENT PLUS患者的性格。AE:不良事件。
人口统计学在基线的群体之间得到良好平衡;50%的患者具有特发性PAH,大多数是世卫组织职能II级或III(89%)(表格1).1例(8%)riociguat患者因不良事件(视力模糊)退出最初12周的随机研究。12周时,64%的患者接受了利奥瓜特2.5 mg每日3次,27%接受2 mg每日3次,9%接受1.5 mg每日3次。
血压和心率
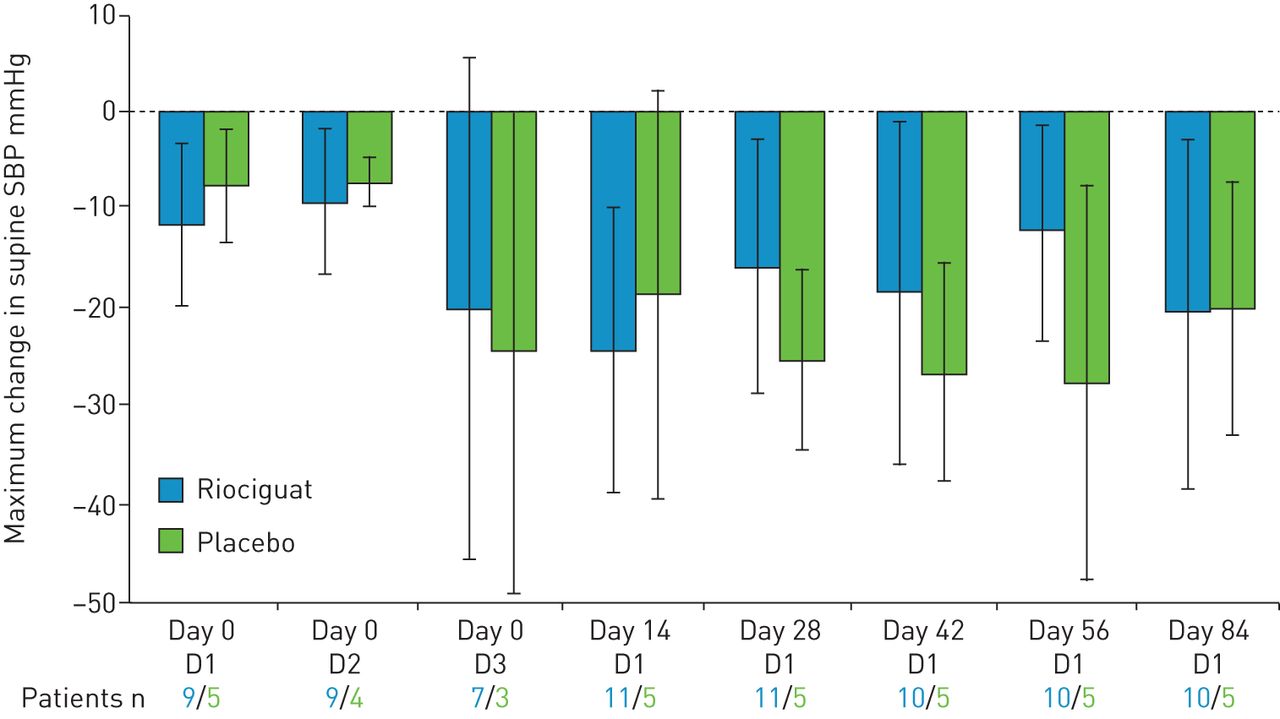
riociguat组和安慰剂组在主要终点,即服用研究药物后4小时内仰卧位收缩压与基线值的最大变化没有差异(图2).均值±sd在RIOCIGUAT组中的每周12(n = 10),在整个研究基线(n = 11)的整个研究基线(n = 11)的最大变化从-20.2±15.3mmhg增加到-20.7±18.0mmhg,从-7.6±3.9 mmhg(n =5)安慰剂组中至20.2±12.9 mmHg(n = 5)。基线(n = 12)的平均绝对静脉SBP为130.2±17.6mmHg,在Riociguat组的第12周(N = 11),与127.0±17.9mmHg(n = 6)和127.0±10.3mmHg(n = 11),117.1±13.5mmHg(n = 5)分别在安慰剂组中(图3).
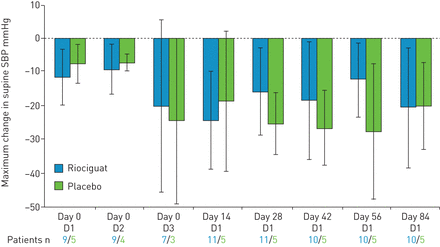
初始12周随机研究中仰卧位收缩压(SBP)的平均最大变化(给药后4小时内)。误差条表示标准差。D:剂量。
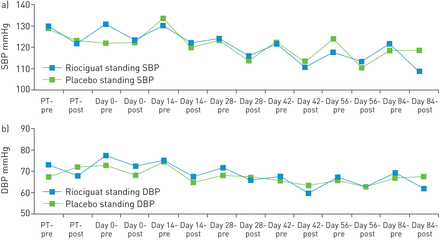
在最初的12周随机研究中,绝对收缩压(SBP)和舒张压(DBP)随时间的变化。在特定的时间点,所显示的值是仅对那些有数据的患者计算的平均值。第0天显示的值是给药前和给药后1。PT:预处理;给药前:给药前30分钟测压;后:给药4小时后测压。
从基线到12周,利西瓜特组的站立收缩压平均最大变化从-22.7±13.9下降到-18.0±16.5 mmHg,而安慰剂组从-9.2±3.7上升到-16.8±10.6 mmHg (图2).riociguat组基线时平均绝对站立收缩压为124.1±17.4 mmHg,第12周时平均站立收缩压为108.8±12.5 mmHg,而安慰剂组分别为122.8±20.5和119.0±9.8 mmHg (图3).
在Riociguat和安慰剂组之间的仰卧或静止的DBP的平均最大变化没有差异。在第12周,在安慰剂组中,苏内华组的平均最大变化为-13.7±11.7mmHg,安慰剂组中的-13.8±12.9mmHg(平均基线的绝对仰卧DBP:73.1±8.6和68.8±11.0 mmhg;周12:66.3±11.3和64.6±9.7 mmHg)(图3).常设DBP的平均最大变化分别为-14.4±10.6和-14.2±10.0 mmHg(平均直接DBP,分别为72.4±10.0和68.0±13.9 mmHg;第12周:62.1±9.7和67.6±13.4 mmHg,分别) (图3).在一项潜在异常值分析中,riociguat组有32例低收缩压(<85 mmHg)或DBP (<45 mmHg)事件(n=12)。与安慰剂组24例(n=6)。
第12周,平均最大卧位心率变化为5.9±7.1次·min−1riociguat组(n=10)与9.6±5.5比·分钟−1在安慰剂组(n=5),平均最大站立心率变化为6.5±7.2和10.4±6.5 beats·min−1, 分别。在常设或仰卧SBP,DBP或心率(表S1-S3)的效果曲线下,该面积之间的组差异无间差异(表S1-S3)。
探索变量
从基线到第12周,riociguat组和安慰剂组的探索性临床参数变化无差异。riociguat组的平均6MWD增加了7±48 m(基线:359±122 m),安慰剂组增加了30±56 m(基线:426±66 m),而WHO功能等级改善/稳定在18%/82%的riociguat组和20%/80%的安慰剂组。NT-proBNP增加834±1858 pg·mL−1(基线:748±715 pg·ml−1)在Riociguat组中,减少162±259 pg·ml−1(基线:278±330 pg·毫升−1)。肺血流动力学,包括PVR、系统性血管阻力(SVR)、mPAP和心脏指数,组间未见差异(数据未显示)。
长期的扩展
安全
所有完成最初12周随机研究的患者(riociguat, n=11;安慰剂,n=6)进入LTE。平均总治疗时间为305天。17例LTE患者均报告了不良事件,最常见的是低血压(41%)、头晕(24%)、外周水肿(24%)、呕吐(24%)和贫血(24%)。8例(47%)患者共报告了9例特别感兴趣的不良事件(表4.).9例(53%)患者报告了19例SAEs:低血压(n=2;急性右心室衰竭、心绞痛、房颤、心脏骤停、慢性右心室衰竭、结肠炎(药物相关)、便秘、丹毒、肺炎、关节脱位、硬膜下血肿、椎间盘突出、意识丧失、晕厥、急性呼吸衰竭、缺氧和间质性肺病(所有n=1)。
6例(35%)患者因不良事件(表4.),这些都被研究者认为与研究药物有关。LTE患者中有3例(18%)死亡(由于跌倒后硬膜下血肿、心脏骤停和右心衰),研究者认为这些死亡与药物无关(表4.).在跌倒后发生致命硬膜下血肿的患者没有报告在跌倒前出现头晕、晕厥前或晕厥。然而,研究者认为,不能肯定地排除在跌倒前由尿瓜引起的全身低血压。riociguat的剂量与停药或不良事件的发生率之间没有关系。
2012年,拜耳医疗保健制药公司(Bayer HealthCare Pharmaceuticals)与首席研究员一致,基于LTE阶段的低血压事件、多次停药和3例死亡,以及在随机研究阶段缺乏有利临床效果的信号,决定终止该研究。DMC并不建议在任何时候提前终止试验。停用riociguat后的最佳治疗方案是由治疗医师根据患者个人情况根据目前的PAH治疗指南决定的。在90天的随访期间收集进一步的数据。
讨论
PATENT PLUS研究的目的是探索riociguat与稳定剂量的西地那非(sildenafil)联合治疗有症状PAH患者的耐受性和潜在的有利协同效应。选择一个安全结果作为研究的主要终点,即。与单独使用西地那非相比,在稳定的西地那非治疗后的4小时内,利昔瓜特给药后仰卧收压从基线值的最大变化。在最初的12周随机研究中,在西地那非的基础上添加利昔瓜特或安慰剂,也导致了类似的血压变化。然而,与安慰剂相比,riociguat组在12周给药后站立收缩压降低了大约10毫米汞柱(图3).此外,探索性疗效变量没有明显的益处,包括6MWD,血液动力变量和职业职业阶级。由于两组之间的基线值差异,无法解释NT-ProbNP级别的变化。在非抵抗LTE阶段,Sildenafil和Riociguat的组合与由于低血压和三种死亡导致的高停药率。因此,决定在2012年12月停止(RIOCIGUAT撤离),3个月随访。总体上,潜在的安全信号与Sildenafil Plus Riociguat,没有积极效益/风险比的证据。因此,恰好地使用具有PDE-5抑制剂的RIOCIGUAT(例如Sildenafil,Tadalafil和Vardenafil)是禁忌的。
在研究的随机阶段,联合治疗对血压没有明显的急性影响,但LTE患者频繁(35%)因低血压而停药的潜在原因仍有待确定。PDE-5抑制剂和sGC刺激剂的组合可能导致过度加性效应,使细胞内cGMP大幅增加,导致体循环中明显的血管舒张。与安慰剂组相比,riociguat组患者在12周时站立收缩压的减少可能是由于一种潜在的累积效应,目前的研究数据没有直接证实这一效应。此外,我们不能排除长期延长期间伴随药物(如利尿剂)剂量的变化可能是导致低血压发生的原因。由于目前官方的禁忌症,这方面和其他伴随因素对这两种联合血管扩张剂的临床低血压的作用难以进一步探讨。
药代动力学相互作用不太可能解释PATENT PLUS中观察到的结果。西地那非主要通过细胞色素P450 (CYP)3A(主要途径)和CYP2C9(次要途径)清除[14],而Riociguat及其主要代谢产物不是主要CYP同种型或运输扣的抑制剂或诱导剂体外以治疗血浆浓度[15].相反,水龙主要通过CYP1A1、CYP3A、CYP2C8和CYP2J2代谢清除[16],虽然西地那非是某些CYP亚型(包括CYP3A4)的弱抑制剂,但预计它不会在临床相关浓度下影响作为这些CYP酶底物的化合物的药代动力学[14,17].虽然在专利加研究中未调查药代动力学,但七位PAH患者的小型血液动力学相互作用研究发现RIOCIGUAT和Sildenafil之间没有药代动力学相互作用(www.clinicaltrials.gov标识符nct00680654.).
虽然riociguat和西地那非联合使用的益处/风险比并不有利,但关键的patel -1研究数据显示,当riociguat与ERAs和非静脉前列腺素(prostanoids)联合使用时,可以提供额外的临床疗效[11].此外,无论是在研究的随机对照阶段,还是在LTE, PATENT-2期间,这些组合都具有良好的耐受性[18].
由于不同的研究设计和纳入的患者数量,PATENT PLUS和PATENT之间的直接比较需要谨慎。然而,在12周的PATENT-1研究中,92%的2.5 mg-最大值的riociguat患者(n=131)接受了ERAs或非静脉前列腺素治疗,出现了治疗紧急不良事件,15%的患者发生了SAE (表5.)[11],与在本研究中分别为100%和17%。然而,在PATENT-2 LTE研究中,同时接受riociguat和其他PAH治疗的患者的停药率和药物相关SAEs比在PATENT plus LTE研究中接受riociguat加西地那非的患者更低[19].同时使用西地那非和瑞昔胍(平均治疗时间305天)可导致较高的不良事件停药率(35%)与8%和17%,药物相关的Saes(18%)与8%和0%)和不良事件相关的死亡(18%)与2%和4%)比riociguat + ERAs (n=161;平均治疗时间409天)或前列腺样物质(n=23;平均治疗时间412天)(以前未发表的数据)。低血压率(41%与7%)和由于低血压引起的停止(24%与0%)的专利PLUS LTE也高于专利2 [19].因此,刺激与西地那非的Riociguat的No-SGC-CGMP途径的刺激不如患有Riociguat单药治疗,或者与时代或前列素组合的耐受性。
在PATENT PLUS研究中,长期使用riociguat联合西地那非与PATENT-2中长期使用riociguat联合前列腺素和内皮素受体拮抗剂(ERAs)的安全性比较
来自PATENT PLUS小样本的关于疗效的有限信息并不表明该药物组合有额外的临床益处。尽管在这项小型探索性研究中数据的可变性很高,但安慰剂组的6MWD较基线有更大的增加,PVR较基线有更大的减少与riociguat组,尽管mPAP、SVR和WHO功能级别有相似的变化。将小型探索性临床研究的结果外推到更大的人群中应始终谨慎进行。
专利加研究有几个限制。这是一个相对较小的研究,没有计划正式的统计测试。尽管如此,由于LTE研究中的多种中断导致的清晰安全信号促使预先取消计划的其他研究阶段。
总之,初期12周专利加入研究中BP的短期添加到SildboAFIL的短期增加导致了对BP的显然相似的变化,并且在探索性疗效变量中没有明确的临床益处临床益处。在专利加LTE研究中,由于低血压,SAES和死亡,与西地那非组合的与西地那非联合的长期治疗与Sildenafil联合的高速度相关。因此,具有Sildenafil Plus Riociguat的可能性不利的安全信号,没有积极效益/风险比的证据。因此,恰当地使用具有PDE-5抑制剂(例如西地那非,塔达拉非)的rIociguat的禁忌。
脚注
对于编辑评论看欧元和J2015;45:1211-1213 [DOI:10.1183 / 09031936.00032715]
这篇文章的补充材料可从www.qdcxjkg.com
临床试验:本研究注册于www.clinicaltrials.gov标识号NCT01179334.
支持声明:本研究由拜耳医疗保健制药公司(柏林,德国)支持。编辑协助由阿德尔菲通信有限公司(Bollington,英国)提供,由拜耳医疗保健制药赞助。本文的资金信息已存入乐趣.
利益冲突:披露可以在本文的在线版本旁边找到www.qdcxjkg.com
- 收到了2014年6月10日。
- 接受2014年12月17日。
- 版权所有©2015











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}