





文摘
患者肺动脉高血压患病率的增加胰岛素抵抗。我们旨在确定代谢缺陷与骨形态发生蛋白受体2型(Bmpr2)突变小鼠,是否这些可能导致肺血管疾病的发展。
代谢表型出现进行诱导表达的转基因老鼠Bmpr2突变,R899X。在Bmpr2表型外显率R899X在高脂肪饮食评估胰岛素抵抗模型。改变糖皮质激素反应在小鼠肺微血管内皮细胞和Bmpr2评估R899X老鼠用地塞米松治疗。
控制相比,Bmpr2R899X老鼠体重增加和增加了胰岛素抵抗的稳态模型评估胰岛素抵抗(1.0±0.4与2.2±1.8)和脂肪积累在骨骼肌和耗氧量降低。Bmpr2R899X高脂肪饮食的老鼠在肺动脉高压强劲增长外显率(7 11与三11)。在细胞培养和在活的有机体内实验中,Bmpr2突变导致本构糖皮质激素受体的激活和不敏感的组合。
胰岛素抵抗存在Bmpr2突变小鼠的早期特征。通过高脂肪饮食恶化肺表型加剧胰岛素抵抗,暗示可能在疾病因果作用。受损的糖皮质激素的反应可能导致代谢缺陷。
肺动脉高压(PAH)是一种破坏性疾病的特点是进步的肺血管阻塞,右心衰和死亡(1,2]。尽管治疗的进步,死亡率在PAH仍然很高3]。高频率的胰岛素抵抗和葡萄糖耐受不良被描述在PAH患者(4,5]。肺在PAH患者葡萄糖代谢增加的证据6)和血脂异常也被描述在多环芳烃7]。目前还不清楚这些代谢异常是由于多环芳烃或如果他们发挥决定性作用。
有许多研究表明肥胖之间的联系,骨形态发生蛋白受体2型(BMPR2)和血管功能。突变的遗传PAH BMPR2是最常见的原因(8,9)和抑郁BMPR2表达式或突变已被证明在其他形式的多环芳烃10,11]。单核苷酸多态性在BMPR2与肥胖相关联12),最近的研究表明BMPR2表达式在脂肪组织的增加超重和肥胖的人类。此外,糖皮质激素敏感性与肥胖(13- - - - - -15糖皮质激素敏感性[]和BMPR2强烈调节16- - - - - -18]。肥胖动物模型如Zucker肥鼠和载脂蛋白e- / -老鼠被证明有肺血管功能障碍(19- - - - - -21]。
我们曾表明,转基因小鼠表达突变形式的Bmpr2开发肺动脉高压和有更大的总比同窝出生仔畜身体质量控制(22,23]。尽管BMPR2的基本和人力上的数据交互,肥胖和PAH表明BMPR2突变可能导致肺血管疾病的发展,目前未经证实的这种潜在的因果关系。诱导,普遍表达突变Bmpr2老鼠提供了一个机会来研究人类遗传的系统性代谢影响多环芳烃Bmpr2突变。我们提出的普遍表达式Bmpr2突变会与早期体重和胰岛素抵抗有关,在肺血管疾病的发展。我们进一步假设,恶化的体重增加会加重肺血管表型由于收购了糖皮质激素的变化信号,在Bmpr2突变,这可能构成这些代谢障碍。
方法
所有动物程序批准的机构动物保健和使用委员会,范德比尔特大学医学院(美国纳什维尔,TN)。
转基因小鼠
我们使用了Rosa26-rtTA2 x TetO7-Bmpr2R899X如前所述(FVB / N老鼠24,25),称为Rosa26-Bmpr2R899X为简便起见(23]。转基因的表达发生之后才开始强力霉素。
重量曲线
Rosa26-Bmpr2 Rosa26-only还是五个R899X断奶后小鼠体重每天直到达到15克,然后chow转向doxycycline-containing chow (200 mg·公斤−1强力霉素,Bio-Serv S3888;Bio-Serv Frenchtown,新泽西,美国),然后重3、7和10天,之后每周。
高脂肪饮食
所有的动物都开始强力霉素常规饮食或猪油为主,高脂肪的饮食强力霉素(60%千卡,Bio-Serv定制chow F6290;Bio-Serv)在72 - 74天的年龄。重量记录的强力霉素和每周6周。血糖记录每周6周。超声心动图和右心室收缩压(RVSP)进行6周(如前所述)(23),和动物牺牲组织的收获。
地塞米松治疗小鼠
男性Rosa26-only或Rosa26-Bmpr2R899X老鼠鉴于chow强力霉素和地塞米松在1毫克公斤−1·天−1或车辆(PBS)。7天后,老鼠吸入异氟烷麻醉,称重。尾静脉血液被评估的葡萄糖,白细胞计数和微分细胞计数。
代谢研究
代谢研究是由范德比尔特大学鼠标代谢表型中心(MMPC;美国纳什维尔TN)一周内在雄性老鼠发起强力霉素。量热法、食物摄取监测和动物活动测量使用Oxymax /蛤系统(哥伦布仪器,哥伦布,哦,美国)7 - 14天。身体成分分析是由核磁共振光谱学使用力量的Minispec(美国Billerica的)。血液是胰岛素和葡萄糖测量5 - 6 h后快速和水平被MMPC化验在2 - 3周的强力霉素。稳态模型评估胰岛素抵抗(HOMA-IR)计算如前所述[26]。
超声心动图
二维超声心动图进行使用体内770高分辨率图像系统(VisualSonics,多伦多,加拿大)。超声心动图包括表、M-mode和频谱多普勒图像得到一天之前牺牲在异氟烷的审美。速度时间积分和心率测量升主动脉,和直径测量在同一位置。中风量(SV)使用公式推导:SV =[π(主动脉直径)2/ 4)×(主动脉速度时间积分)。心输出量(CO)使用公式推导:公司= SV(钙)×心率27,28]。
组织学
腓肠肌肌肉当时从右后腿分离的牺牲和临时冻结。10-μM部分被切割和安装在幻灯片。油红O染色进行如前所述[29日]。微管蛋白进行了免疫组织化学(Ab15246;Abcam,剑桥,英国)和糖皮质激素受体(Ab 16510;Abcam)如前所述25]。
糖皮质激素反应element-luciferase
A7R5 BMPR2突变的血管平滑肌细胞稳定转染如前所述[30.]。突变都是源自人类病人家庭和包括T354G(或C118W)的配体结合域,一个C994T(或R332X)突变激酶结构域,和一个2570 - 2580解决转移,导致胞质尾截断。与10 mg·板细胞是暂时性的转染−1糖皮质激素响应element-luciferase SuperFect转染试剂(试剂盒,日耳曼敦,医学博士,美国),并允许在一夜之间就恢复。每个好然后用PBS,媒体被DMEM取代含10%胎牛血清。2 h后,三个井的每个细胞类型1毫米地塞米松处理,三个处理。6小时后,细胞与PBS洗一次,然后在被动裂解细胞溶解一次缓冲区(200毫升·−1光度计)进行分析。
数据分析
统计测试是单向或双向方差分析与因果费舍尔最显著的差异,除特殊说明外,或使用GraphPad棱镜执行+(5.0版本;美国圣地亚哥GraphPad CA)。
结果
在Rosa26-bmpr2体重增加R899X老鼠
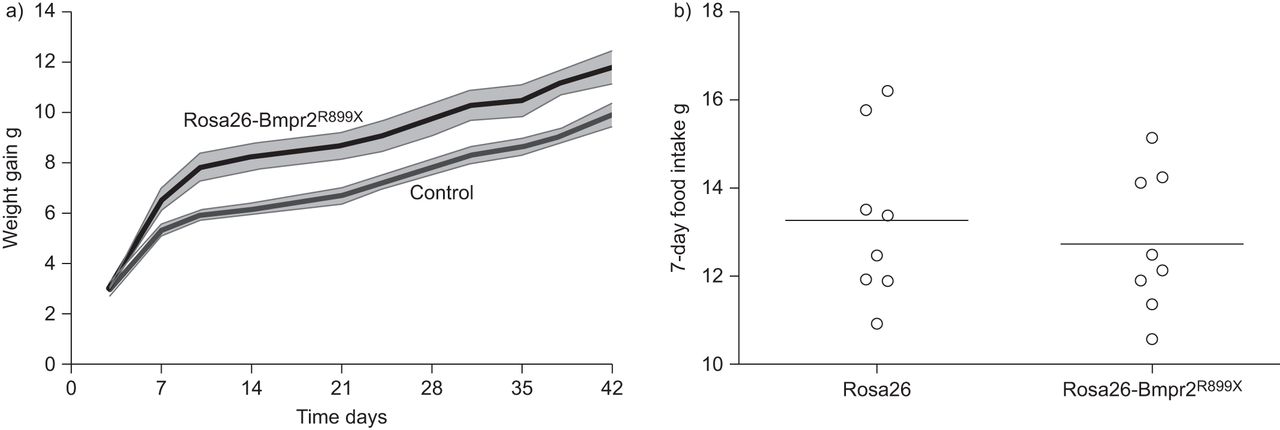
我们之前提到的增加我们的体重增加在几个Bmpr2突变模型与强力霉素诱导后22,23,31日]。为了量化的动态体重增加,年轻的老鼠(15克)与环球(Rosa26) Bmpr2的强力霉素诱导表达R899X转基因(25]或Rosa26-only控制转基因与强力霉素诱导。Rosa26-Bmpr2R899X小鼠体重比控制时明显增加,但增加的体重主要发生在前两周的感应(图1一个)。在Rosa26-Bmpr2多余的体重R899X老鼠发生尽管消费同样数量的食物(图1 b)。
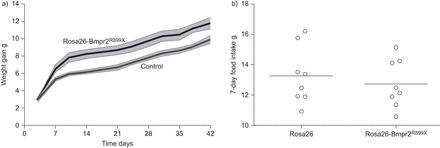
体重增加与强力霉素诱导后。所有的老鼠开始∼15克重量,达到能力天的年龄,每组五个老鼠。阴影区域扫描电镜。曲线是不同的重复测量方差分析,p < 0.01。b) 7天食物摄入量在野生型和转基因小鼠(Rosa26-Bmpr2 (Rosa26)R899X)显示食物摄入量的两组没有区别。单杠代表平均值。n = 8每组。
能量平衡和身体Rosa26-Bmpr2组成R899X老鼠
我们发现统计上无法区分两组的脂肪和无脂质水平,但自由流体减少,增加骨骼和结缔组织的质量与通用Bmpr2集团R899X表达式(表1和图2)。因为减少肥胖动物的活动可能造成体重增加,我们在三个平面测量动物的活动。我们没有发现在Bmpr2减少活动R899X动物,这一趋势增加活动在清醒(夜间)时间普遍Bmpr2突变表达R899X(X活动表明,Y和Z活动是类似的;数据未显示)图3)。间接执行量热法测量能量消耗。我们发现Bmpr2R899X氧气消耗比例∼10%低于控制动物,尽管这一趋势增加活动(图3 b)。
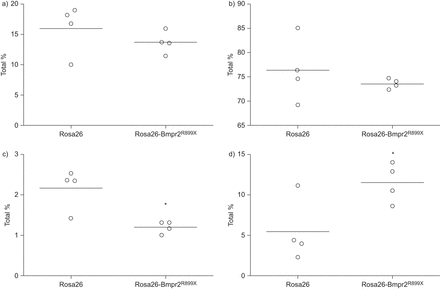
身体成分分析核磁共振在野生型和转基因小鼠(Rosa26-Bmpr2 (Rosa26)R899X)在脂肪),b)肌肉,c)自由流体和d)结缔组织。相似的两组中拥有大量的肌肉和脂肪,但有一个更高比例的结缔组织,包括骨骼、毛发和皮肤的转基因小鼠相比,控制和自由流体的比例较低。单杠代表平均值。n = 4 /组。*:p < 0.05 Rosa26与Rosa26-Bmpr2R899X。
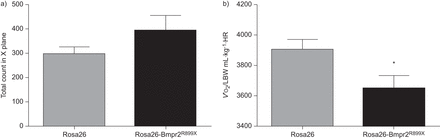
沿着x轴)夜间活动在野生型和转基因小鼠(Rosa26-Bmpr2 (Rosa26)R899X)显示转基因组夜间活动增加的趋势。然而,这没有达到统计学意义(p = 0.19)。Y和z轴活动是类似的(数据未显示)。b)能源消耗分析间接量热法在野生型和转基因小鼠。休息时耗氧量(V′O2)/瘦体重(激光焊)Rosa26-Bmpr2低R899X集团与同窝出生仔畜控制相比,尽管趋势增加活动。人力资源:心率。n = 8每组。*:p < 0.05 Rosa26与Rosa26-Bmpr2R899X。
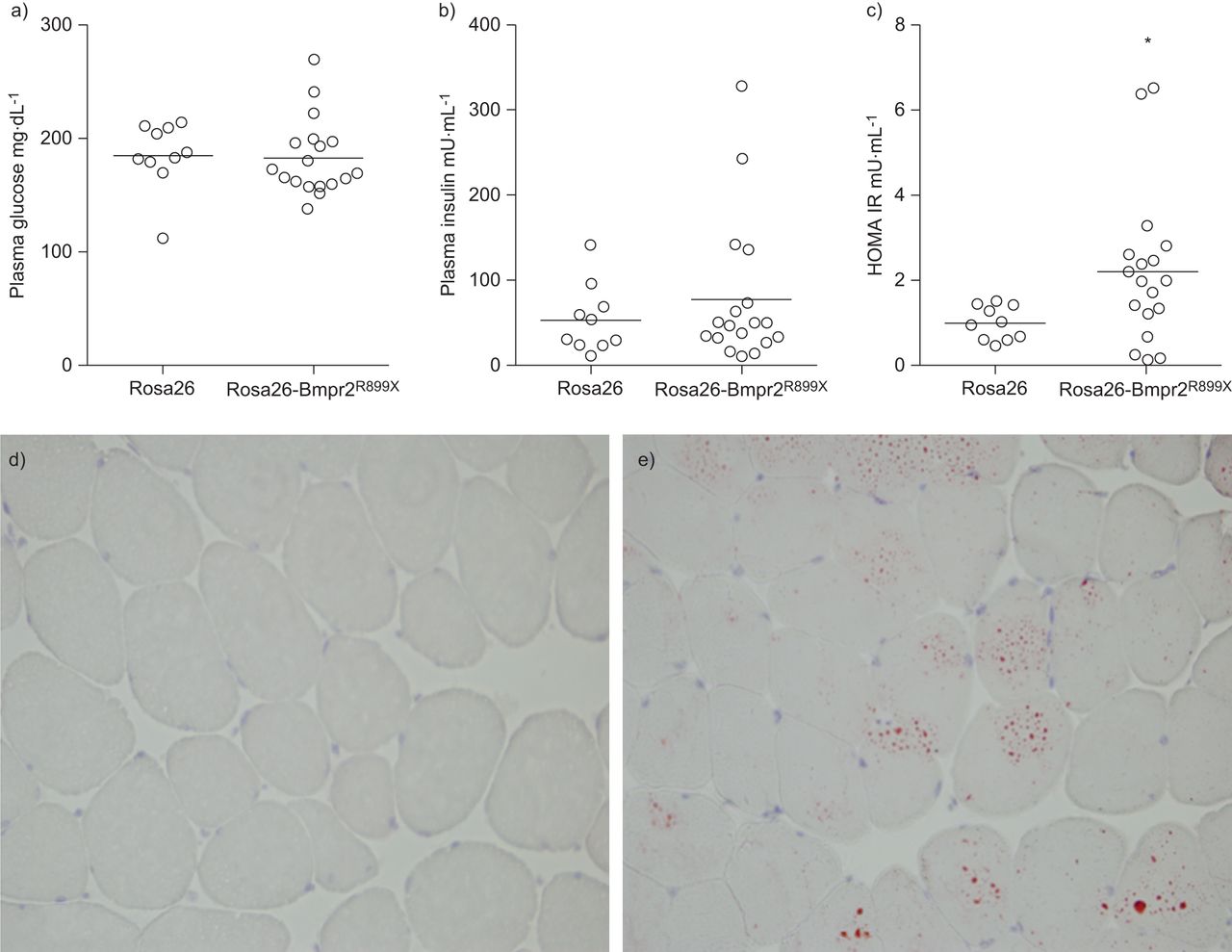
为了确定如果存在胰岛素抵抗的肥胖Rosa26-Bmpr2R899X,我们测量空腹胰岛素和葡萄糖。我们发现更大的可变性在转基因小鼠的血糖水平,以及数值更高意味着在Rosa26-Bmpr2胰岛素水平R899X老鼠(77.5±85.4与53.9±40.2μU·毫升−1)。使用综合和敏感程度的胰岛素抵抗,HOMA-IR,七18(39%)与Rosa26-Bmpr2老鼠R899X是胰岛素抵抗的门槛之上,虽然没有Rosa26小鼠胰岛素抵抗(±意味着什么sdRosa26 1±0.4与2.2±1.8 Rosa26-Bmpr2R899X)(图4)。因为骨骼肌脂质积累与胰岛素抵抗密切相关(32- - - - - -34),我们的彩色冻结部分腓肠肌肌脂质使用油红o .我们发现的证据Rosa26-Bmpr2细胞内脂滴R899X老鼠不存在控制动物(图4 d和e)。
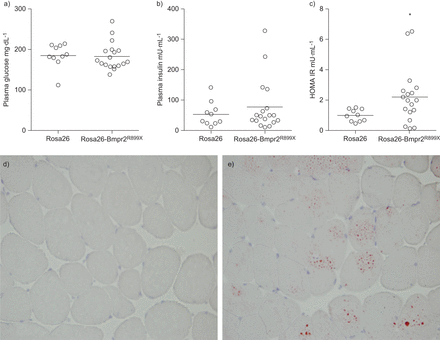
胰岛素抵抗(IR)是存在于Rosa26-Bmpr2R899X老鼠。a - c)指数的葡萄糖处理在野生型和转基因小鼠(Rosa26-Bmpr2 (Rosa26)R899X)。有更大的方差在转基因小鼠的血糖和胰岛素水平,但意思是没有统计学上高于控制。计算红外通过稳态模型评估(HOMA-IR)展示了更大程度的IR在转基因小鼠。单杠表明的意思。n = 10 - 18每组。*:p < 0.05。d, e)红外与intramyocyte脂质积累。冰冻切片的腓肠肌肌肉从d)野生型(Rosa26)和e)转基因小鼠(Rosa26-Bmpr2R899X)。积极的脂质染色是在红色。放大×60。
高脂饮食增加Rosa26-Bmpr2疾病外显率R899X老鼠
测试是否增加胰岛素抵抗是一个旁观者,或重要的疾病发展,我们测试了高脂肪饮食对疾病的影响在Rosa26-Bmpr2外显率R899X老鼠。高脂肪饮食与胰岛素抵抗增加有关人类[32,34,35),以及模型系统(36]。
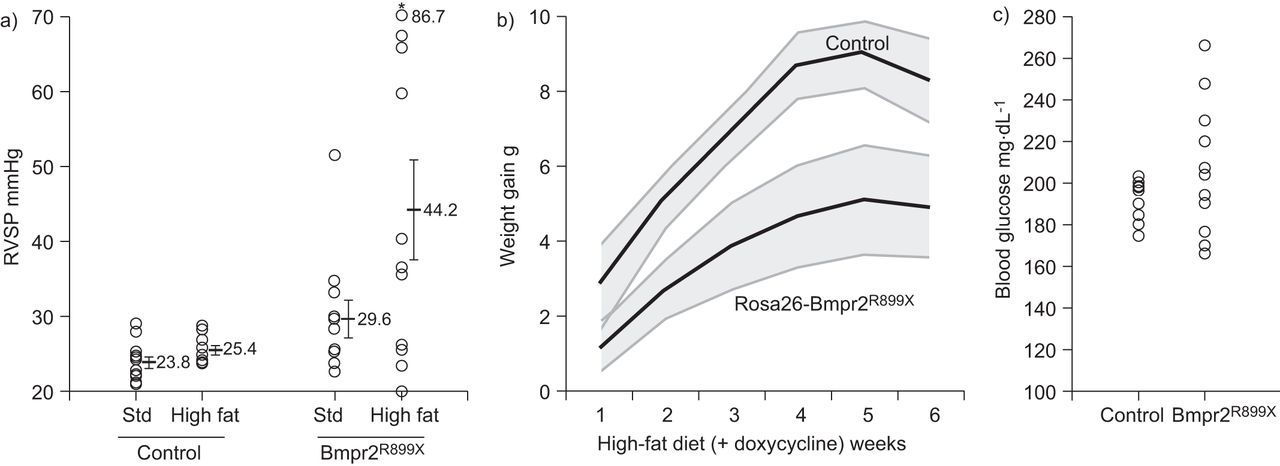
成人(72 - 74天的旧)控制或Rosa26-Bmpr2R899X小鼠随机标准饮食(∼5%的脂肪)或高脂肪的饮食(60%的脂肪),都含有200毫克公斤−1强力霉素激活转基因表达。高脂饮食导致Rosa26-Bmpr2 RVSP强劲增长R899X老鼠,增加平均RVSP从29.8至44.2毫米汞柱,比例与RVSP > 30毫米汞柱从三11 - 7 11 (图5)。心输出量在对照组平均7.7 - -7.9毫升·分钟−1;这个趋势小幅下降到7.5毫升·分钟−1在标准饮食和7.2毫升·分钟−1在高脂肪饮食Bmpr2突变组。高脂肪饮食没有显著影响RVSP控制老鼠。苏木精和伊红染色显示增加有核细胞的肺小动脉美联储Bmpr2高脂肪饮食R899X组与对照组(图S1)。

)高脂肪的饮食会加剧增加右心室收缩压(RVSP) Rosa26-Bmpr2R899X老鼠。每个符号代表RVSP测量从一个鼠标。心输出量略(∼8%)而不是突变组显著降低。n =每组9 - 11。基因型效应:p = 0034;饮食的影响:双向方差分析,p = 0388年性病:标准。*:p < 0.05,变异老鼠用高脂肪饮食喂养RVSP相比有了显著提高正常饮食,费舍尔的显著差异。b)高脂肪饮食控制和Rosa26-Bmpr2迅速增胖的原因R899X老鼠,但Rosa26-Bmpr2降低增益R899X。阴影区域扫描电镜的重量。每组小鼠n = 9 - 11。p < 0001年重复测量方差分析。c)血糖控制和Rosa26-Bmpr2不是明显不同R899X老鼠(平均193.5和207.2 mg·dL−1分别),但血糖差异增加的标准差9 - 32的标准差。n =每组9 - 11。意义:p = 0005据Bartlett测试相同的方差和p = 0059根据列文的测试相同的方差(Bartlett假设常态,列文不)。
与我们的结果在正常的食物,Rosa26-Bmpr2R899X小鼠高脂饮食体重比Rosa26-only控件(慢图5 b)。此外,血糖是相当稳定的跨越时间在所有的老鼠,但几乎三倍Rosa26-Bmpr2可变性R899X老鼠(图5度)。实验组之间没有差异,在收缩期系统性动脉压力或左心室质量(图S2),或者重大的冠状动脉粥样硬化病理证据的任何组。相结合,这些数据可能表明,胰岛素抵抗可能是重要的疾病发展,绝对体重、体重和血糖就不是。
Bmpr2突变会导致异常的糖皮质激素受体易位
基因表达分析在人类曾认为Bmpr2糖皮质激素敏感性的一个强大的监管机构(16),最有可能通过调节细胞骨架元素(37]。正如我们先前已证明有缺陷的细胞骨架在Bmpr2突变细胞(25糖皮质激素受体穿梭],我们想测试是否与Bmpr2突变缺陷细胞中。我们以前使用的特征(31日小鼠肺微血管内皮细胞(PMVEC)来源于野生型老鼠或两个不同的Bmpr2突变的老鼠。
使用糖皮质激素受体的免疫细胞化学,我们发现细胞质尾(R899X)和激酶结构域(delx4 +)突变PMVEC似乎本构核本地化糖皮质激素受体的相对不敏感,加入地塞米松(图6)。我们还发现添加Bmp4配体除了地塞米松似乎把糖皮质激素受体的原子核在所有细胞类型;在野生型而不是突变细胞糖皮质激素受体与细胞骨架结构。

骨形态发生蛋白(BMP)和BMP 2型受体(Bmpr2)调节糖皮质激素受体突变易位。一)免疫细胞化学染色法对糖皮质激素受体(红色)在小鼠肺微血管内皮细胞(PMVEC)显示正常的糖皮质激素受体与1毫米地塞米松(Dex)易位野生型(WT)细胞,破坏的ng 50毫升−1BMP4。Bmpr2突变,似乎有弱本构glucocortocoid受体激活,不大大影响了地塞米松。添加BMP4仍然块糖皮质激素受体核易位,虽然本地化在细胞质中出现改变。b)增加50 ng·毫升−1BMP4也块核易位的糖皮质激素受体在不同的细胞类型,A7R5大鼠血管平滑肌细胞(SMC)。c) A7R5稳定转染质粒表达Bmpr2突变的细胞在胞质域(CD),细胞外的域(ED)或激酶结构域(KD)地塞米松都有钝化反应的荧光素酶报告实验评估由糖皮质激素的反应元素。突变效应:p < 0001;地塞米松效应:p = 0018,通过双向方差分析(p < 0.0001)。Tx:治疗。*:p < 0.05为差异与地塞米松原生细胞;#:p < 0.05为差异从没有治疗本地,据费舍尔最显著的差异。
因为暂时的困难使转染PMVEC,我们搬到了A7R5血管平滑肌细胞糖皮质激素受体易位量化的差异。我们发现A7R5稳定转染细胞与细胞外的领域,激酶结构域或胞质域BMPR2突变表现出完整的糖皮质激素受体的反应,和BMP-sensitive (图6 b)。当瞬变与糖皮质激素反应element-luciferase转染,所有BMPR2突变细胞相对比本地细胞(地塞米松不敏感图6 c)。只有激酶结构域突变体复制本构激活和不敏感的组合出现在两类突变PMVEC,虽然都有相对不敏感。小鼠PMVEC和A7R5之间的差异可能是由于不同的细胞类型或引入BMPR2突变的方法。
因为之前的数据显示改变细胞骨架函数Bmpr2突变的细胞(25),我们提出了糖皮质激素受体本地化模式可能是由于损伤的细胞骨架的功能。我们没有Bmpr2 PMVECs和染色R899X微管蛋白,糖皮质激素受体或两者(图7)。与完整的细胞骨架,野生型细胞糖皮质激素受体存在于细胞核。在Bmpr2R899XPMVEC细胞,基线微管结构改变和糖皮质激素受体存在于细胞核周围的时尚,与核小礼物本身。这些数据表明,细胞骨架功能受损与改变糖皮质激素受体的上下文中本地化Bmpr2突变。
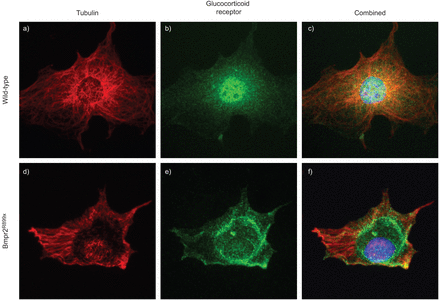
糖皮质激素受体减少核易位与受损在成骨细胞骨架相关蛋白受体2型(Bmpr2)突变。野生型和肺微血管内皮细胞(PMVECs) Bmpr2突变(Bmpr2R899X)与微管蛋白抗体染色(红色;a和d),糖皮质激素受体(绿色;b和e)或(c和f)证明改变微管结构与染色Bmpr2突变的糖皮质激素受体的微管在细胞核周围的时尚。
Bmpr2突变引起异常的糖皮质激素的反应在活的有机体内
接下来,我们试图确定这些改变糖皮质激素反应也见过在活的有机体内在老鼠身上。首先,我们进行了回顾性分析已知的糖皮质激素信号目标表达式(37在数组发表在整个肺1周的转基因Rosa26-Bmpr2激活R899X老鼠与控制(25]。我们发现许多已知的糖皮质激素受体激活目标(图8),包括胰岛素样生长因子1 (Igf1),弹性蛋白(Eln)、赖氨酰化氧(Lox)和disintegrin-like metallopeptidase域蛋白质12 (Adam12)和血小板反应蛋白2 (Adamts2)图案。我们还发现糖皮质激素受体抑制抑制已知的目标,包括CCAAT /δ(C / EBPd)和增强子结合蛋白抑制剂κ光多肽基因的增强剂在b细胞,激酶β(Ikbkb)。一些糖皮质激素受体的目标肺未受影响。例如,没有改变任何表面活性剂的蛋白质(没有显示)。此外,转基因小鼠强劲,但变量抑制糖皮质激素受体本身的表达与控制相比,可能导致反馈其本构激活。

一)Rosa26-Bmpr2R899X老鼠的组成性激活糖皮质激素受体(GR)从微阵列分析评估目标的表达,与降低GR表达本身。误差线代表扫描电镜。结果表示为叠化而控制。Igf1:胰岛素样生长因子1;民族解放军:弹性蛋白;液态氧:赖氨酰化氧;Adam12: disintegrin-like和metallopeptidase域蛋白12;Adamts2: disintegrin-like metallopeptidase域蛋白质和血小板反应蛋白图案2;Cebpd: CCAAT /增强子结合蛋白三角洲;Ikbkb:在b细胞抑制剂κ光多肽基因的增强剂,激酶β。b)体重增加了地塞米松(Dex)在野生型老鼠但在Rosa26-Bmpr2下降R899X老鼠。n = 3 - 8每组。敏捷的微分效应在野生型和突变小鼠被双向方差分析p = 0.0005。从内部*:p < 0.05为差异基因型车辆控制费舍尔最显著的差异。c)血糖增加敏捷在Rosa26-Bmpr2野生型老鼠但不是R899X老鼠。敏捷的微分效应在野生型和突变小鼠被双向方差分析p = 0125。*:p < 0.05。d)体重对血糖关系非常密切。灰色象征代表控制和黑色符号代表敏捷。相关性:0.75,p < 0.0001相关性z检验(JMP程序;SAS、卡里、数控、美国)。e)白细胞计数增加敏捷在野生型老鼠但在Rosa26-Bmpr2下降R899X老鼠。敏捷的微分效应在野生型和突变小鼠被双向方差分析p = 016。从内部*:p < 0.05为差异基因型车辆控制费舍尔最显著的差异。
测试Rosa26-Bmpr2的能力R899X老鼠反应合成糖皮质激素地塞米松,控制或突变的老鼠开始强力霉素和地塞米松或车辆1周,然后检测标记的类固醇的回应。根据体重增加,血糖及血白细胞计数,Bmpr2突变小鼠类固醇的缺失或矛盾的反应。野生型老鼠体重增加了强劲的类固醇,如预期,而突变小鼠体重没有增加以应对地塞米松管理局(图8 b)。同样,野生型小鼠强劲增长与类固醇非空腹血糖,但Bmpr2突变体抑制增长的非空腹血糖(图8 c)。血糖和体重密切相关(图8 d)。最后,白细胞计数增加预计在野生型小鼠,但如果有的话,在突变小鼠(被压抑了图8 e)。这些变化主要是由于中性粒细胞计数(没有显示),如预期。
讨论
中央发现本文激活Bmpr2突变在活的有机体内与早期胰岛素抵抗和血糖稳态功能障碍(图4),胰岛素抵抗可能导致疾病进展和可能不仅仅是一个旁观者或肺血管疾病的标志(图5)。具体来说,胰岛素抵抗杀虫剂肺血管疾病的发展,并通过高脂肪饮食恶化加剧了肺血管表型。我们还研究了体重和次要成分Bmpr2突变(无花果1和2),表现为Bmpr2耗氧量降低突变体与控制相比,尽管他们趋势更大的活动(图3)。这匹配先前确定的耗氧量降低肺动脉内皮细胞从特发性肺动脉高压病人,可能引起的能量代谢的变化(6]。最后,我们表明,Bmpr2突变导致异常的糖皮质激素受体贩卖,在PMVEC和血管平滑肌细胞(无花果6和7),这些受损的糖皮质激素反应也见过在活的有机体内(图8)。
先前的研究已经表明代谢综合征(38],胰岛素抵抗[5),增加血红蛋白A1c (4)和其他代谢异常(6,39),在大多数情况下,在特发性肺动脉高压病人。这些研究主要是联想;目前尚不清楚这些攻击是否独立危险因素或疾病的后遗症。目前的研究表明,在部分特发性或遗传PAH归因于BMPR2突变,这些临床代谢发现可能与底层相关基因突变(图4)。它曾被认为最特发性多环芳烃和遗传的多环芳烃有共同的分子病因学40];因此,该协会也可能适用于特发性多环芳烃没有Bmpr2突变。我们的研究显示,在这两个人类突变Bmpr2-associated PAH和转基因小鼠模型,有变量和不完全外显率的表现型肺动脉高压(大约20% Bmpr2突变携带者发展多环芳烃)和胰岛素抵抗与胰岛素抵抗(39%影响小鼠研究)(无花果4和5)[41]。额外修改因素和基因可能扮演了一个重要的角色在调节患者BMPR2突变的表型和遗传易感性可能通过其他基因会有胰岛素抵抗添加剂影响胰岛素抵抗和肺动脉高压的表型。然而,不管胰岛素抵抗的病因学,如果肺血管疾病可以通过增加胰岛素抵抗,恶化也许可以被纠正改善胰岛素抵抗。
几项研究已经发现证据指向线粒体功能缺陷多环芳烃(30.,42,43]。虽然这通常被认为是一个简单的转向有氧糖酵解(Warburg效应),最近的数据表明,可能有一个更复杂的代谢表型涉及反循环连接脂肪酸氧化和糖酵解44]。然而,BMPR2突变与代谢的机制,或胰岛素抵抗,特别是了解甚少。我们的研究提出了一些初始数据支持这里列出一个假设。Bmpr2突变,R899X,树叶规范BMP通路信号(通过SMAD转录因子)完好无损23),但干扰信号通过细胞骨架元素包括监管cofilin [25,45- - - - - -47),是为了调节糖皮质激素受体激活(48]。在这里,我们显示功能,糖皮质激素都极其不正常的反应在体外(无花果6和7),在活的有机体内(图8),包括本构激活和不敏感的证据。长期糖皮质激素激活导致胰岛素抵抗和代谢综合征(49]。因此,是BMPR2是似是而非的代谢缺陷引起的变异是由缺陷引起的细胞骨架调节糖皮质激素受体易位。
最近的工作由P大米et al。(50在肺动脉高压的野百合碱模型表明,扩展地塞米松政府改善了肺血管疾病的抑制和逆转正常Bmpr2记录中发现这个模型。我们的研究中,关于地塞米松管理,专注于早期的系统性影响糖皮质激素的上下文中Bmpr2突变。因为肺动脉高压是很少出现在我们的转基因模型在两周时间点,我们没有评估肺血液动力学比较与野百合碱糖皮质激素在这个模型的影响。然而,增加表达突变Bmpr2由于地塞米松管理在我们的模型中可能不会改善肺血管疾病。
在葡萄糖稳态有重要的鼠标应变差异(51]。的背景我们转基因菌株FVB / N,这已被证明在应对胰岛素分泌受损高血糖症与其他常用的菌株(相比51,52]。我们已经开始穿越我们的基因转移到C57BL / 6株,F1代的初步实验表明,代谢表型将在C57BL / 6背景强应变与更严重的胰岛素抵抗。然而,我们能够证明FVB / N Rosa26胰岛素抵抗R899X鼠标使用的敏感指标,HOMA-IR于一体的葡萄糖和胰岛素水平。最后,虽然我们有多种类型的干预研究(Bmpr2突变、高脂肪饮食和地塞米松),我们正在研究的系统非常复杂,我们的一些观测结果必然是相关的。例如,肌肉内的脂质积累可能是胰岛素抵抗的原因,而不仅仅是它的存在的标志,但我们的数据不允许测定intramyocyte脂的作用是在肺血管疾病表型(34]。
我们的研究集中在雄性老鼠。然而,它是可能的,性激素可能与葡萄糖体内平衡调节肺血管表型。雌激素和睾丸激素,很好地描述与代谢功能和调节胰岛素抵抗[53,54]。雄性老鼠的好处在相似年龄通常是统一的性激素水平,但性激素的交互的进一步研究,代谢功能和肺血管疾病是十分必要的。此外,进一步的研究的影响与严重程度的胰岛素抵抗和相关性肺动脉高压的线索在胰岛素抵抗的潜在作用肺脉管系统。尽管胰岛素抵抗的高脂肪饮食是一个证据确凿的模型(55,56),胰岛素抵抗程度的相关性通过血糖euglycemic夹将允许这个协会更好的量化。
本研究为理解人类疾病的潜在影响。首先,是BMPR2的影响表明,突变可能是系统性的。其次,它表明代谢缺陷在PAH患者可能对疾病的分子病因学积分胰岛素抵抗可能加剧肺血管疾病。最后,它可以提供分子的开始解释了这些缺陷,在异常的糖皮质激素受体易位。了解肺血管疾病的分子病因学找到有效的治疗方法,是至关重要的,目前的研究表明这样的一个目标可能是胰岛素抵抗。
确认
范德比尔特大学的代谢评估进行了老鼠代谢表型出现中心(美国纳什维尔,TN),由美国国立卫生研究院的资助(DK59637)。
脚注
可以从本文的补充材料www.www.qdcxjkg.com
支持声明
本研究的资助美国国立卫生研究院的(HL82694和HL95797 U24 DK59637, K08 HL093363)。
感兴趣的语句
利益冲突的信息可以发现与本文的在线版本www.www.qdcxjkg.com
- 收到了2012年2月21日。
- 接受2012年7月9日。
- ©2013人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}