





Abstract
肺动脉高血压患者increased prevalence of insulin resistance. We aimed to determine whether metabolic defects are associated with bone morphogenic protein receptor type 2 (Bmpr2) mutations in mice, and whether these may contribute to pulmonary vascular disease development.
Metabolic phenotyping was performed on transgenic mice with inducible expression of Bmpr2 mutation, R899X. Phenotypic penetrance in Bmpr2R899Xwas assessed in a high-fat diet model of insulin resistance. Alterations in glucocorticoid responses were assessed in murine pulmonary microvascular endothelial cells and Bmpr2R899Xmice treated with dexamethasone.
Compared to controls, Bmpr2R899Xmice showed increased weight gain and demonstrated insulin resistance as assessed by the homeostatic model assessment insulin resistance (1.0±0.4versus2.2±1.8) and by fat accumulation in skeletal muscle and decreased oxygen consumption. Bmpr2R899Xmice fed a high-fat diet had strong increases in pulmonary hypertension penetrance (seven out of 11versusthree out of 11). In cell culture andin vivoexperiments, Bmpr2 mutation resulted in a combination of constitutive glucocorticoid receptor activation and insensitivity.
Insulin resistance is present as an early feature of Bmpr2 mutation in mice. Exacerbated insulin resistance through high-fat diet worsened pulmonary phenotype, implying a possible causal role in disease. Impaired glucocorticoid responses may contribute to metabolic defects.
- Dexamethasone
- glucocorticoid
- homeostatic model assessment of insulin resistance
- insulin resistance
- mouse model
- pulmonary hypertension
Pulmonary arterial hypertension (PAH) is a devastating disease characterised by progressive pulmonary vascular obstruction, right heart failure and death [1,2]。Despite advances in treatment, mortality in PAH remains high [3]。A high frequency of insulin resistance and glucose intolerance has been described in PAH patients [4,5]。The lungs in PAH patients have evidence of increased glucose metabolism [6] and abnormal lipid profiles have also been described in PAH [7]。It is not presently known whether these metabolic abnormalities are a consequence of PAH or if they play a causative role.
There are many studies suggesting connections between obesity, bone morphogenic protein receptor type 2 (BMPR2) and vascular function. Mutations of BMPR2 are the most common cause of heritable PAH [8,9] and depressed BMPR2 expression or mutations have been demonstrated in other forms of PAH as well [10,11]。Single nucleotide polymorphisms in BMPR2 are associated with obesity [12] and recent studies have demonstrated increased BMPR2 expression in adipose tissues of overweight and obese humans. Furthermore, glucocorticoid sensitivity has been linked to obesity [13–15] and BMPR2 strongly regulates glucocorticoid sensitivity [16–18]。Animal models of obesity such as the Zucker fatty rat and ApoE-/-mice have been shown to have pulmonary vascular dysfunction [19–21]。
We have previously shown that transgenic mice expressing mutant forms of Bmpr2 develop pulmonary hypertension and have greater total body mass than their littermate controls [22,23]。Although basic and human data on the interactions of BMPR2, obesity and PAH suggest that BMPR2 mutation may contribute to pulmonary vascular disease development, this potential causal relationship is presently unproven. The inducible, universally expressed mutant Bmpr2 mouse offers an opportunity to study the systemic metabolic effects of a human heritable PAH BMPR2 mutation. We hypothesised that universal expression of Bmpr2 mutation would be associated with early weight gain and insulin resistance, prior to development of pulmonary vascular disease. We further hypothesised that exacerbation of weight gain would worsen pulmonary vascular phenotype due to acquired alterations in glucocorticoid signalling, in the context of Bmpr2 mutation, which may underlie these metabolic impairments.
METHODS
All animal procedures were approved by the Institutional Animal Care and Use Committee, Vanderbilt University School of Medicine (Nashville, TN, USA).
Transgenic mice
We used the Rosa26-rtTA2 x TetO7-Bmpr2R899XFVB/N mice as previously described [24,25], called Rosa26-Bmpr2R899Xfor brevity [23]。Expression of transgene occurs only after initiation of doxycycline.
Weight curve
Five Rosa26-only or five Rosa26-Bmpr2R899Xmice were weighed daily after weaning until reaching 15 g, then chow switched to doxycycline-containing chow (200 mg·kg−1doxycycline, Bio-Serv S3888; Bio-Serv, Frenchtown, NJ, USA) and were then weighed at 3, 7 and 10 days, and weekly thereafter.
High-fat diet
All animals were started on the doxycycline regular diet or the lard-based, high-fat doxycycline diet (60% Kcal, Bio-Serv custom chow F6290; Bio-Serv) at 72–74 days of age. Weights were recorded at the start of doxycycline and weekly for 6 weeks. Blood glucose was recorded weekly for 6 weeks. Echocardiogram and right ventricular systolic pressure (RVSP) were performed at 6 weeks as previously described [23], and the animals were sacrificed for harvesting of tissues.
Dexamethasone treatment in mice
Male Rosa26-only or Rosa26-Bmpr2R899Xmice were given doxycycline in chow and either dexamethasone at 1 mg·kg−1·day−1or vehicle (PBS). After 7 days, mice inhaled isoflurane anaesthesia and were weighed. Tail vein blood was drawn for assessment of glucose, white blood cell count and differential cell count.
Metabolic studies
Metabolic studies were performed by the Vanderbilt University Mouse Metabolic Phenotypic Center (MMPC; Nashville, TN, USA) within 1 week on initiation of doxycycline in male mice. Calorimetry, food intake monitoring and animal activity were measured using the Oxymax/CLAMS system (Columbus Instruments, Columbus, OH, USA) for 7–14 days. Body composition analysis was performed by nuclear magnetic resonance spectroscopy using Bruker's Minispec (Billerica, MA, USA). Blood was drawn for insulin and glucose measurements after a 5–6 h fast and levels were assayed by the MMPC at 2–3 weeks of doxycycline exposure. Homeostatic model assessment insulin resistance (HOMA-IR) was calculated as previously described [26]。
Echocardiography
Two-dimensional echocardiography was performed using a Vivo 770 high-resolution image system (VisualSonics, Toronto, Canada). Echocardiograms including B-mode, M-mode and spectral Doppler images were obtained the day prior to sacrifice under isoflurane aesthetic. Velocity time integral and heart rate were measured in the ascending aorta, and diameter was measured in the same location. Stroke volume (SV) was derived using the formula: SV=[π (aortic diameter)2/4]×[aortic velocity time integral]. Cardiac output (CO) was derived using the formula: CO=SV(calc)×heart rate [27,28]。
Histology
Gastrocnemius muscle was isolated from the right hind leg at the time of sacrifice and snap frozen. 10-μM sections were cut and mounted on slides. Oil red O staining was performed as previously described [29]。Immunohistochemistry was performed for tubulin (Ab15246; Abcam, Cambridge, UK) and glucocorticoid receptor (Ab 16510; Abcam) as previously described [25]。
Glucocorticoid response element–luciferase
A7R5 vascular smooth muscle cells were stably transfected with BMPR2 mutations as previously described [30]。All mutations were derived from human patient families and included T354G (or C118W) in the ligand binding domain, a C994T (or R332X) mutation in the kinase domain, and a 2570–2580delT frameshift, resulting in a cytoplasmic tail truncation. Cells were transiently transfected with 10 mg·plate−1glucocorticoid response element–luciferase with SuperFect transfection reagent (Qiagen, Germantown, MD, USA) and allowed to recover overnight. Each well was then washed with PBS, and media was replaced with DMEM containing 10% stripped fetal bovine serum. After 2 h, three wells of each cell type were treated with 1 mM dexamethasone and three were treated with vehicle. After 6 h, cells were washed once with PBS and then lysed once in passive lysis buffer (200 mL·well−1) for analysis in a luminometer.
Data analysis
Statistical tests were either one-way or two-way ANOVA with post-hoc Fischer's least significant difference, except where noted, or performed using GraphPad Prism Plus (version 5.0; GraphPad, San Diego, CA, USA).
RESULTS
Weight gain in Rosa26-bmpr2R899Xmice
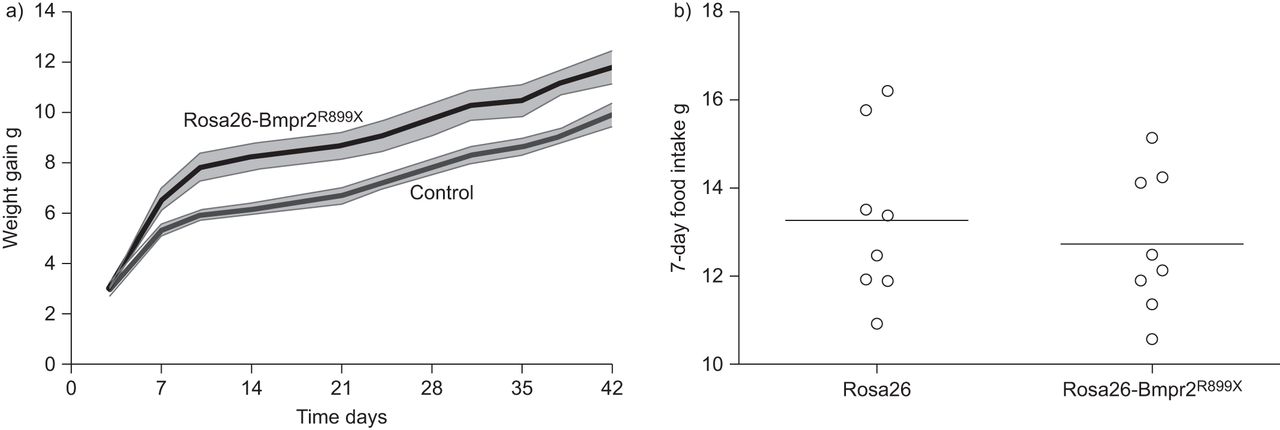
We had previously noted increased weight gain in several of our Bmpr2 mutant models after induction with doxycycline [22,23,31]。In order to quantify the dynamics of the weight gain, young (15 g) mice with universal (Rosa26) doxycycline inducible expression of the Bmpr2R899Xtransgene [25] or Rosa26-only controls had transgene induced with doxycycline. Rosa26-Bmpr2R899Xmice gained significantly more weight than controls, but with the increased weight gain primarily occurring in the first 2 weeks of induction (fig. 1a)。The excess weight gain in Rosa26-Bmpr2R899Xmice occurred despite consumption of the same amount of chow (fig. 1b)。
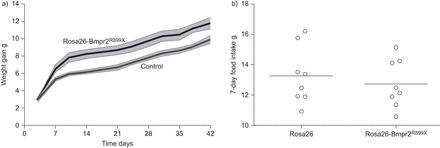
a) Weight gain after induction with doxycycline. All mice were started at ∼15 g weight, achieved at 24–30 days of age, with five mice per group. Shaded areas aresem. Curves are distinct at p<0.01 by repeated measures ANOVA. b) 7-day food intake in wild-type (Rosa26) and transgenic mice (Rosa26-Bmpr2R899X) showing no difference in food intake in the two groups. Horizontal bars represent mean values. n=8 per group.
Energy balance and body composition of Rosa26-Bmpr2R899Xmice
We found statistically indistinguishable levels of fat and fat-free mass in the two groups, but decreased free fluid, and increased mass of bone and connective tissue in the group with universal Bmpr2R899Xexpression (table 1andfig. 2)。Because decreased activity may underlie weight gain in obese animals, we measured animal activity in three planes. We found no decrease in activity in Bmpr2R899Xanimals, and a trend to increased activity during waking (nocturnal) hours in the universally expressed mutant Bmpr2R899X(X activity shows that Y and Z activity were similar; data not shown) (fig. 3a)。Indirect calorimetry was performed to measure energy expenditure. We found that the Bmpr2R899Xconsumed proportionally ∼10% less oxygen than control animals, despite a trend to increased activity (fig. 3b)。
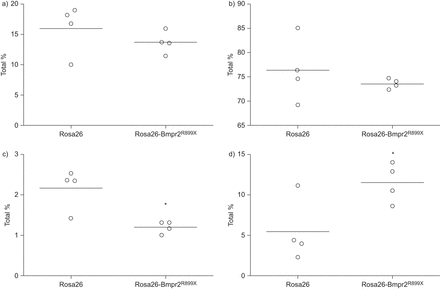
Body composition analysis by nuclear magnetic resonance in wild-type (Rosa26) and transgenic mice (Rosa26-Bmpr2R899X) in a) fat, b) muscle, c) free fluid and d) connective tissue. Similar amounts of muscle and fat were present in both groups, but there was a higher proportion of connective tissue including bone, fur and skin in the transgenic mice compared with controls and a lower proportion of free fluid. Horizontal bars represent mean values. n=4 per group. *: p<0.05 Rosa26versusRosa26-Bmpr2R899X.
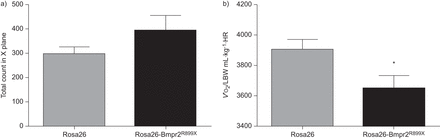
a) Nocturnal activity along the X-axis in wild-type (Rosa26) and transgenic mice (Rosa26-Bmpr2R899X) showing a trend towards increased nocturnal activity in the transgenic group. However, this did not meet statistical significance (p=0.19). The Y- and Z-axis activity was similar (data not shown). b) Energy consumption analysis by indirect calorimetry in wild-type and transgenic mice. Resting oxygen uptake (V′O2)/lean body weight (LBW) was lower in the Rosa26-Bmpr2R899Xgroup compared with littermate controls despite trends to increased activity. HR: heart rate. n=8 per group. *: p<0.05 Rosa26versusRosa26-Bmpr2R899X.
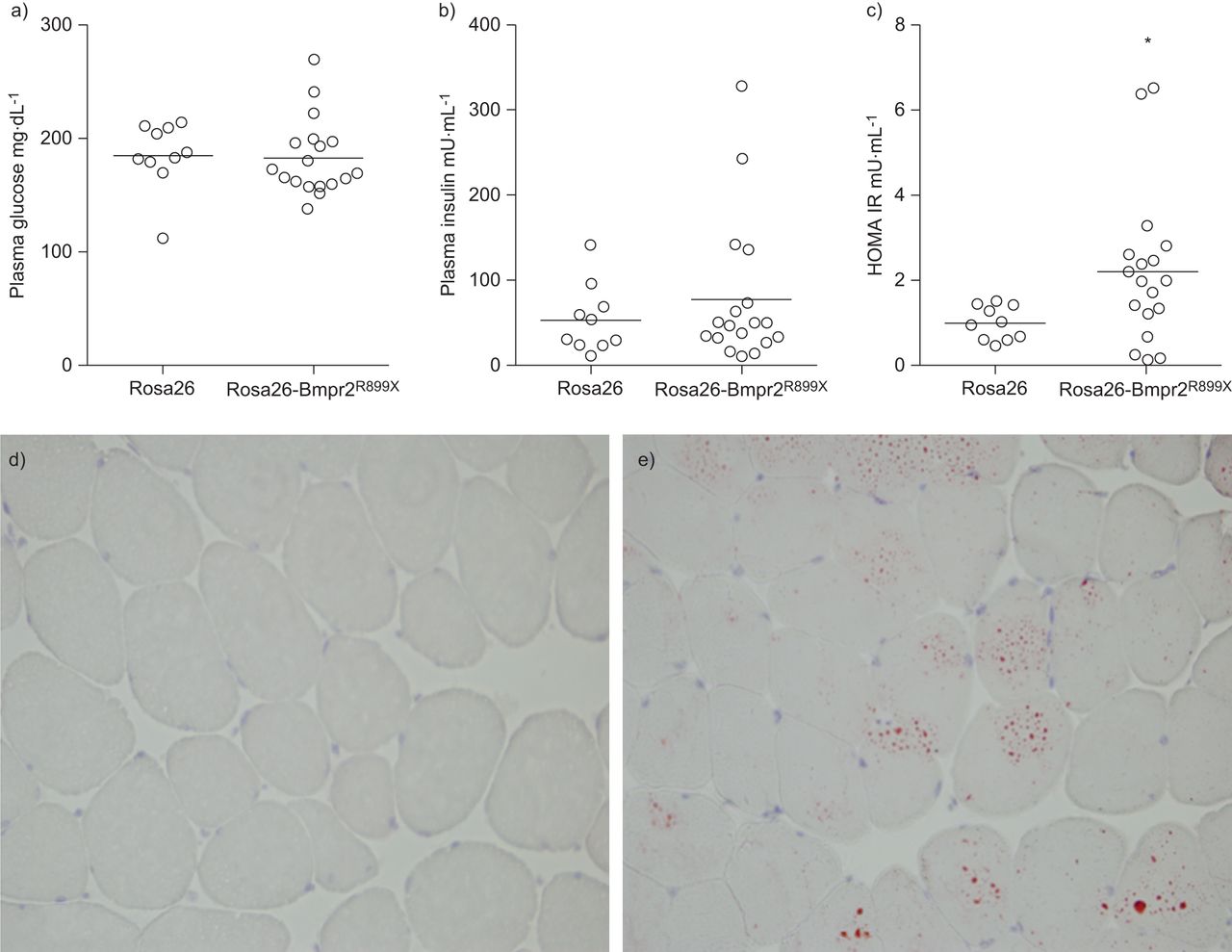
In order to determine if insulin resistance is present in the obese Rosa26-Bmpr2R899X, we measured fasting insulin and glucose. We found greater variability in glucose levels in transgenic mice, as well as numerically higher mean insulin levels in Rosa26-Bmpr2R899Xmice (77.5±85.4versus53.9±40.2 μU·mL−1)。Using the integrative and sensitive measure of insulin resistance, HOMA-IR, seven out of (39%) 18 mice with Rosa26-Bmpr2R899Xwere above the threshold for insulin resistance, while no Rosa26 mice were insulin resistant (mean±sdRosa26 1±0.4versus2.2±1.8 Rosa26-Bmpr2R899X) (fig. 4)。Because skeletal muscle lipid accumulation closely correlates with insulin resistance [32–34], we stained frozen sections of the gastrocnemius muscle for lipid using Oil Red O. We found evidence of lipid droplets within myocytes from Rosa26-Bmpr2R899Xmice that were not present in control animals (图4 d和e)。
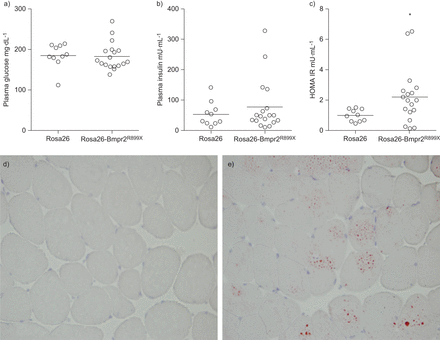
Insulin resistance (IR) is present in Rosa26-Bmpr2R899Xmice. a–c) Indices of glucose handling in wild-type (Rosa26) and transgenic mice (Rosa26-Bmpr2R899X)。There was a greater variance in glucose and insulin levels in transgenic mice, but the mean was not statistically higher than in controls. Calculation of IR through homeostasis model assessment (HOMA-IR) demonstrated greater degrees of IR in transgenic mice. Horizontal bars indicate mean. n=10–18 per group. *: p<0.05. d, e) IR is correlated with intramyocyte lipid accumulation. Frozen sections of gastrocnemius muscle from d) wild-type (Rosa26) and e) transgenic mice (Rosa26-Bmpr2R899X)。Positive lipid staining is demonstrated in red. Magnification ×60.
A high-fat diet increases disease penetrance in Rosa26-Bmpr2R899Xmice
To test whether this increased insulin resistance was a bystander, or important to disease development, we tested the effect of a high-fat diet on disease penetrance in Rosa26-Bmpr2R899Xmice. A high-fat diet has been associated with increased insulin resistance in humans [32,34,35], as well as model systems [36]。
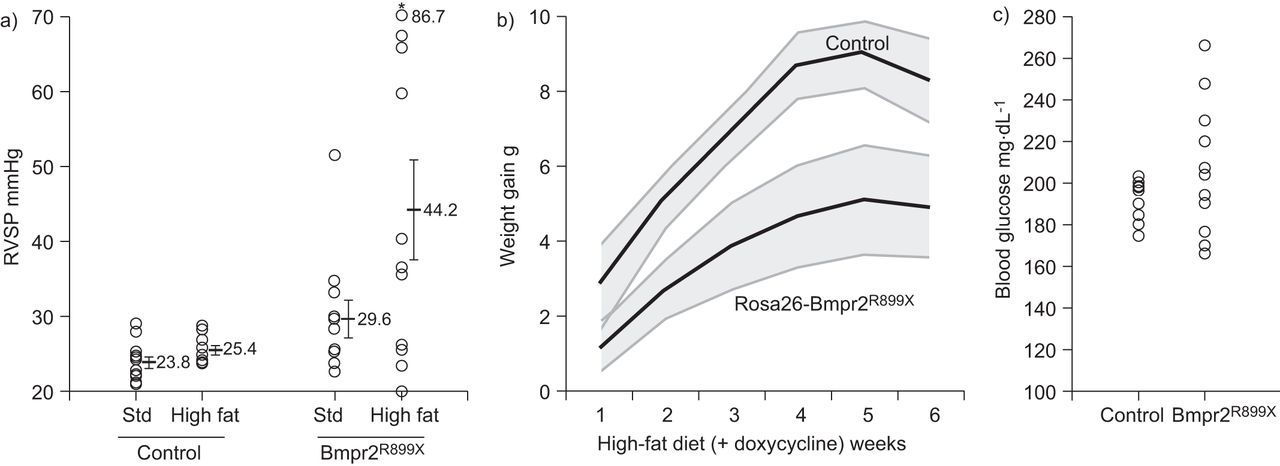
Adult (72–74-day old) control or Rosa26-Bmpr2R899Xmice were randomised to standard diet (∼5% fat) or high-fat diet (60% fat), both containing 200 mg·kg−1doxycycline to activate transgene expression. A high-fat diet caused a strong increase in RVSP in Rosa26-Bmpr2R899Xmice, increasing average RVSP from 29.8 to 44.2 mmHg, and the proportion with RVSP >30 mmHg from three out of 11 to seven out of 11 (fig. 5a)。Cardiac output in control groups averaged 7.7–7.9 mL·min−1; this trended down slightly to 7.5 mL·min−1in standard diet and 7.2 mL·min−1在高脂肪饮食Bmpr2突变groups. A high-fat diet did not have a significant effect on RVSP in control mice. Haematoxylin and eosin staining showed an increase in nucleated cells in the pulmonary arterioles of the high-fat diet fed Bmpr2R899Xgroup compared with controls (fig. S1).

a) A high-fat diet exacerbates an increase in right ventricular systolic pressure (RVSP) in Rosa26-Bmpr2R899Xmice. Each symbol represents the RVSP measured from an individual mouse. Cardiac output was slightly (∼8%) but not significantly decreased in both mutant groups. n=9–11 per group. Effect of genotype: p=0034; effect of diet: p=0388 by two-way ANOVA, Std: standard. *: p<0.05, mutant mice fed a high-fat diet had significantly increased RVSP compared to normal diet by Fischer's least significant difference.b) The high-fat diet causes rapid weight gain in both control and Rosa26-Bmpr2R899Xmice, but with reduced gain in Rosa26-Bmpr2R899X. Shaded areas aresemfor weight. n=9–11 per group mice. p<0001 by repeated measures ANOVA. c) Blood glucose is not significantly different in control and Rosa26-Bmpr2R899Xmice (averaging 193.5 and 207.2 mg·dL−1, respectively), but variance in blood glucose is increased from a standard deviation of 9 to a standard deviation of 32. n=9–11 per group. Significance: p=0005 according to Bartletts tests of equal variance and p=0059 according to Levene’s tests of equal variance (Bartlett assumes normality, Levene does not).
In contrast to our results on normal chow, Rosa26-Bmpr2R899Xmice on a high-fat diet gained weight more slowly than Rosa26-only controls (fig. 5b)。Furthermore, blood glucose was fairly stable across time in all mice, but there was almost three times as much variability in Rosa26-Bmpr2R899Xmice (fig. 5c)。There were no differences between the experimental groups in systolic systemic arterial pressure or left ventricular mass (fig. S2), or gross pathological evidence of coronary atherosclerosis in any of the groups. In combination, these data may suggest that while insulin resistance may be important to disease development, absolute weight, weight gain and blood glucose alone are not.
Bmpr2 mutation causes aberrant glucocorticoid receptor translocation
在人类基因表达分析identified Bmpr2 as a strong regulator of glucocorticoid sensitivity [16], most likely through regulation of cytoskeletal elements [37]。As we have previously demonstrated defective cytoskeleton in Bmpr2 mutant cells [25], we wished to test whether glucocorticoid receptor shuttling was defective in cells with Bmpr2 mutation. We used previously characterised [31] murine pulmonary microvascular endothelial cells (PMVEC) derived from wild-type mice or mice with two different Bmpr2 mutations.
Using immunocytochemistry for the glucocorticoid receptor, we found that both cytoplasmic tail (R899X) and kinase domain (delx4+) mutations in PMVEC appeared to have constitutive nuclear localisation of the glucocorticoid receptor, with relative insensitivity to added dexamethasone (fig. 6a)。We also found that adding the Bmp4 ligand in addition to dexamethasone appears to drive the glucocorticoid receptor out of the nucleus in all cell types; in wild-type but not mutant cells the glucocorticoid receptor appears to associate with a cytoskeletal structure.

Bone morphogenic protein (BMP) and BMP receptor type 2 (Bmpr2) mutations modulate glucocorticoid receptor translocation. a) Immunocytochemistry staining for glucocorticoid receptor (red) in murine pulmonary microvascular endothelial cells (PMVEC) shows normal glucocorticoid receptor translocation with 1 mm dexamethasone (Dex) in wild-type (WT) cells, disrupted by the addition of 50 ng·mL−1BMP4. With Bmpr2 mutation, there appears to be weak constitutive glucocortocoid receptor activation, not greatly affected by the addition of dexamethasone. Addition of BMP4 still blocks glucocorticoid receptor translocation to the nucleus, although it's localisation in the cytoplasm appears altered. b) The addition of 50 ng·mL−1BMP4 also blocks nuclear translocation of the glucocorticoid receptor in a different cell type, A7R5 rat vascular smooth muscle cells (SMC). c) A7R5 cells stably transfected with plasmids expressing Bmpr2 mutations in the cytoplasmic domain (CD), extracellular domain (ED) or kinase domain (KD) all have blunted response to dexamethasone as assessed by luciferase reporter assay driven by the glucocorticoid response element. Mutation effect: p<0001; dexamethasone effect: p=0018, by two-way ANOVA (p<0.0001). Tx: treatment. *: p<0.05 for difference from native cells with dexamethasone;#: p<0.05 for difference from no treatment native, according to Fischer's least significant difference.
因为暂时的困难使转染ing PMVEC, we moved to A7R5 vascular smooth muscle cells to quantitate the differences in glucocorticoid receptor translocation. We found that A7R5 cells stably transfected with either extracellular domain, kinase domain or cytoplasmic domain BMPR2 mutations exhibited intact glucocorticoid receptor response, and were BMP-sensitive (fig. 6b)。When transiently transfected with glucocorticoid response element-luciferase, all BMPR2 mutant cells were relatively dexamethasone insensitive compared to native cells (fig. 6c)。Only the kinase domain mutants reproduced the combination of constitutive activation and insensitivity seen in both classes of mutation in the PMVEC, although all had relative insensitivity. The difference between murine PMVEC and A7R5 could be because of the difference in cell type or the method of introducing the BMPR2 mutation.
Because of prior data showing altered cytoskeletal function in cells with Bmpr2 mutations [25], we hypothesised that altered glucocorticoid receptor localisation patterns may be due to impairment of cytoskeletal function. We stained PMVECs with and without Bmpr2R899Xfor tubulin, glucocorticoid receptor or both (fig. 7)。In wild-type cells with intact cytoskeleton, glucocorticoid receptor is highly present in the nucleus. In Bmpr2R899XPMVEC cells, the baseline microtubular structure is altered and the glucocorticoid receptor is present in a perinuclear fashion, with little present in the nucleus itself. These data suggest that impaired cytoskeletal function correlate with altered glucocorticoid receptor localisation in the context of Bmpr2 mutation.
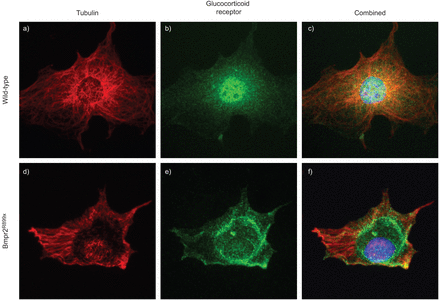
Decreased glucocorticoid receptor nuclear translocation is associated with impaired cytoskeleton in bone morphogenic protein receptor type 2 (Bmpr2) mutations. Wild-type and pulmonary microvascular endothelial cells (PMVECs) with Bmpr2 mutation (Bmpr2R899X) were stained with antibodies against tubulin (red; a and d), glucocorticoid receptor (green; b and e) or both (c and f) demonstrating altered microtubular structure in Bmpr2 mutation with staining of glucocorticoid receptor at the end of microtubules in a perinuclear fashion.
Bmpr2 mutation causes aberrant glucocorticoid responsein vivo
Next, we attempted to determine if these alterations in glucocorticoid response were also seenin vivoin mice. First, we performed a retrospective analysis of known glucocorticoid signalling target expression [37] in published arrays in whole lung from 1 week of transgene activation in Rosa26-Bmpr2R899Xmice compared with controls [25]。We found activation of many known glucocorticoid receptor targets (fig. 8a), including insulin-like growth factor 1 (Igf1), elastin (Eln), lysyl oxidase (Lox), and disintegrin-like and metallopeptidase domain proteins 12 (Adam12) and with thrombospondin motifs 2 (Adamts2). We also found suppression of known glucocorticoid receptor suppression targets, including CCAAT/enhancer binding protein delta (C/EBPd) and inhibitor of κ light polypeptide gene enhancer in B-cells, kinase β (Ikbkb). Some glucocorticoid receptor targets in the lung were unaffected. For instance, there were no changes in any of the surfactant proteins (not shown). In addition, transgenic mice had strong but variable inhibition of expression of the glucocorticoid receptor itself compared with controls, possibly as feedback resulting from its constitutive activation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
a) Rosa26-Bmpr2R899Xmice have constitutive activation of the glucocorticoid receptor (GR) as assessed by expression of targets from microarray analysis, with decreased expression of the GR itself. Error bars representsem. Results are expressed as fold-change compared to controls. Igf1: insulin-like growth factor 1; Eln: elastin; Lox: lysyl oxidase; Adam12: disintegrin-like and metallopeptidase domain proteins 12; Adamts2: disintegrin-like and metallopeptidase domain proteins with thrombospondin motifs 2; Cebpd: CCAAT/enhancer binding protein delta; Ikbkb: inhibitor of κ light polypeptide gene enhancer in B-cells, kinase β. b) Body weight is increased by dexamethasone (Dex) in wild-type mice but decreased in Rosa26-Bmpr2R899Xmice. n=3–8 per group. The differential effect of Dex in wild-type and mutant mice was p=0.0005 by two-way ANOVA. *: p<0.05 for difference from within genotype vehicle control by Fischer's least significant difference. c) Blood glucose is increased by Dex in wild-type mice but not in Rosa26-Bmpr2R899Xmice. The differential effect of Dex in wild-type and mutant mice was p=0125 by two-way ANOVA. *: p<0.05. d) Body weight correlates strongly to blood glucose. Grey symbols represent control and black symbols represent Dex. Correlation: 0.75, p<0.0001 by correlation z-test (JMP program; SAS, Cary, NC, USA). e) The white blood count is increased by Dex in wild-type mice but decreased in Rosa26-Bmpr2R899Xmice. The differential effect of Dex in wild-type and mutant mice was p=016 by two-way ANOVA. *: p<0.05 for difference from within genotype vehicle control by Fischer's least significant difference.
To test the ability of Rosa26-Bmpr2R899X老鼠对合成糖皮质激素dexamethasone, control or mutant mice were started on doxycycline and given dexamethasone or vehicle for 1 week, and then tested for markers of steroid response. Based on weight gain, blood glucose and white cell blood count, Bmpr2 mutant mice had an absent or paradoxical response to steroids. Wild-type mice showed strong weight gain with steroids, as expected, while mutant mice failed to gain weight in response to dexamethasone administration (fig. 8b)。Similarly, wild-type mice had strong increases in non-fasting blood glucose with steroids, but Bmpr2 mutants had suppression of the increase in non-fasting blood glucose (fig. 8c)。Blood glucose closely correlated with weight gain (fig. 8d)。Finally, white blood cell count increased as expected in the wild-type mice but, if anything, was suppressed in mutant mice (fig. 8e)。These changes were primarily due to neutrophil count (not shown), as expected.
DISCUSSION
The central finding of this paper is that activation of Bmpr2 mutationin vivois associated with early insulin resistance and blood glucose homeostasis dysfunction (fig. 4), and that insulin resistance may contribute to disease progression and is probably not just a bystander or marker of pulmonary vascular disease (fig. 5)。具体来说,胰岛素抵抗也刷新了development of pulmonary vascular disease, and exacerbation through high-fat diet worsens the pulmonary vascular phenotype. We also examined body weight and composition secondary to Bmpr2 mutation (figs 1and2) and showed decreased oxygen consumption in Bmpr2 mutants compared to controls, even though they trended to greater activity (fig. 3)。This matches the decreased oxygen consumption previously identified in pulmonary artery endothelial cells from idiopathic PAH patients, probably caused by a shift in energy metabolism [6]。Finally, we showed that Bmpr2 mutation results in aberrant glucocorticoid receptor trafficking, both in PMVEC and in vascular smooth muscle cells (figs 6and7) and that these impaired glucocorticoid responses are also seenin vivo(fig. 8)。
Previous studies have shown metabolic syndrome [38], insulin resistance [5], increased haemoglobin A1c [4] and other metabolic abnormalities [6,39], for the most part, in idiopathic PAH patients. These studies were primarily associative; it was unclear whether these were independent risk factors or sequelae of disease. The present study suggests that in the portion of idiopathic or heritable PAH attributable to BMPR2 mutation, these clinical metabolic findings may be associated with the underlying genetic mutation (fig. 4)。It has previously been suggested that most idiopathic PAH and heritable PAH share common molecular aetiology [40]; therefore, this association may also be true in idiopathic PAH without Bmpr2 mutation. As the our study shows, in both human mutant Bmpr2-associated PAH and our transgenic mouse model, there is variable and incomplete penetrance of the phenotype of both pulmonary hypertension (estimated 20% Bmpr2 mutation carriers develop PAH) and insulin resistance (39% affected with insulin resistance in the mice studies) (figs 4and5) [41]。Additional modifying factors and genes probably play important roles in regulating the phenotype of patients with a BMPR2 mutation and it is possible that a genetic predisposition to insulin resistance through other genes would have an additive effect on the phenotype of both insulin resistance and pulmonary hypertension. However, regardless of the aetiology of insulin resistance, if pulmonary vascular disease can be worsened by increasing insulin resistance, perhaps it can be improved by correcting insulin resistance.
Several studies have found evidence pointing to defects in mitochondrial function in PAH [30,42,43]。While this is often thought of as a simple shift to aerobic glycolysis (Warburg effect), recent data suggests that there may be a more complicated metabolic phenotype involving Randle's cycle connecting fatty acid oxidation and glycolysis [44]。However, the mechanism linking BMPR2 mutation to metabolism broadly, or to insulin resistance in particular, is poorly understood. Our study presents some initial data supporting one hypothesis outlined here. The Bmpr2 mutation used, R899X, leaves canonical BMP pathway signalling (through SMAD transcription factors) intact [23], but disrupts signalling through cytoskeletal elements including regulation of cofilin [25,45–47], which has been previously shown to regulate glucocorticoid receptor activation [48]。Herein, we show that functionally, glucocorticoid responses are profoundly abnormal bothin vitro(figs 6and7) andin vivo(fig. 8) including evidence of constitutive activation and insensitivity. Chronic glucocorticoid activation leads to insulin resistance and metabolic syndrome [49]。Thus, it is plausible that the metabolic defects caused by BMPR2 mutation are caused by defective cytoskeletal regulation of glucocorticoid receptor translocation.
Recent work by Priceet al.[50] in the monocrotaline model of pulmonary hypertension has demonstrated that extended dexamethasone administration ameliorates pulmonary vascular disease and reverses the suppression of normal Bmpr2 transcript found in this model. Our study, regarding dexamethasone administration, focused on the early systemic effects of glucocorticoid in the context of Bmpr2 mutation. Because pulmonary hypertension was rarely present in our transgenic model at the 2-week time-point, we did not assess pulmonary haemodynamics to compare the effects of glucocorticoids in this model with monocrotaline. However, increased expression of mutant Bmpr2 due to dexamethasone administration in our model may not ameliorate pulmonary vascular disease.
有重要的gl鼠标应变的差异ucose homeostasis [51]。The background of our transgenic strain is FVB/N, which has been shown to have impaired insulin secretion in response to hyperglycaemia compared with other commonly used strains [51,52]。We have started crossing our transgenics onto the C57BL/6 strain, and preliminary experiments in the F1 generation suggest that the metabolic phenotype will be stronger on a C57BL/6 background strain with more severe insulin resistance. We are, however, able to demonstrate insulin resistance in the FVB/N Rosa26R899Xmouse using the sensitive metric, HOMA-IR, which integrates glucose and insulin levels. Finally, while we have multiple types of interventions in this study (Bmpr2 mutation, high-fat diet and dexamethasone), the system we are studying is sufficiently complex that some of our observations are necessarily correlative. For instance, the intramuscular lipid accumulation may be a cause of insulin resistance and not just a marker of its presence but our data do not allow determination of the role that intramyocyte lipid is playing in the pulmonary vascular disease phenotype [34]。
Our study focused on male mice. However, it is possible that sex hormones may interact with glucose homeostasis to modulate pulmonary vascular phenotype. Oestrogens, as well as testosterone, are well described to interact with metabolic function and modulate insulin resistance specifically [53,54]。雄鼠在相似年龄的好处是基因rally uniform sex hormone levels, but further study of the interactions of sex hormones, metabolic function and pulmonary vascular disease is warranted. Additionally, further studies of the effect of degree of insulin resistance and correlation with severity of pulmonary hypertension would shed further light on the potential role of insulin resistance in the pulmonary vasculature. Although the high-fat diet is a well-documented model of insulin resistance [55,56], correlation of degree of insulin resistance through the hyperinsulinemic euglycemic clamp would allow this association to be better quantified.
This study has potential implications for the understanding of human disease. First, it demonstrates that the effects of BMPR2 mutation may be systemic. Secondly, it suggests that the metabolic defects seen in PAH patients may be integral to the molecular aetiology of disease as insulin resistance potentially exacerbates pulmonary vascular disease. Finally, it may provide the beginning of a molecular explanation for these defects, in the aberrant glucocorticoid receptor translocation. Understanding the molecular aetiology of pulmonary vascular disease is critical to finding effective treatments, and the present study suggests one such target may be insulin resistance.
Acknowledgments
The metabolic assessments were carried out by the Vanderbilt University Mouse Metabolic Phenotyping Center (Nashville, TN, USA), funded by a National Institutes of Health grant (DK59637).
Footnotes
This article has supplementary material available fromwww.www.qdcxjkg.com
Support Statement
This study was funded by grants from theNational Institutes of Health(HL82694 and HL95797, U24 DK59637, K08 HL093363).
Statement of Interest
Conflict of interest information can be found alongside the online version of this article atwww.www.qdcxjkg.com
- ReceivedFebruary 21, 2012.
- AcceptedJuly 9, 2012.
- ©ERS 2013
REFERENCES










