





文摘
肺动脉高压(PH)是一个严重的和进步的疾病的特点是高肺动脉压力,通常以右心衰。目前的治疗方法在PH值主要提供症状缓解而预后率低是由于缺乏特异性靶分子和发展的几个因素的参与博士众多研究表明基质金属蛋白酶(MMP)轴的至关重要的作用在发展和疾病,特别是对细胞外基质重塑和血管内稳态。MMP的活动增加已经证明在PH值的实验动物模型,和MMP的抑制减弱或增强血管重塑。此外,几项研究强调,恢复生理管制基质金属蛋白酶MMP /基质金属蛋白酶组织抑制剂比率将加强逆转重塑博士基质金属蛋白酶在本文将突出的病理生理学作用,特别是血管重塑和博士的建立,我们将把重点放在MMP的表达和调控的肺血管和肺血管重塑。我们还将概述近期的临床与实验研究及其对实现最大逆转的PH值的影响,以及当前的问题和未来的观点。188bet体育备用网址
肺动脉高压
肺动脉高压(PH)不是一种疾病本身而是一个病理生理参数定义为平均肺动脉压力(P巴勒斯坦权力机构)超过正常上限,即。25 mmHg静止(1]。PH值出现在各种临床情况和与广泛的组织模式和异常。一个新的临床分类PH值,修改最近,指定的五类中独特的临床表现,诊断结果和对治疗的反应。其中,第1组包含一组不同的疾病称为肺动脉高血压(PAH)。PH值的其他四个主要临床群体包括肺部静脉阻塞疾病(组1),PH值由于心脏病(组2),PH值由于肺部疾病(组3),慢性血栓栓塞肺动脉高压(4组)和PH值与不清楚或多因子的目的(5)组(2]。SER我E年代“MATRIX METALLOPROTEINASES IN LUNG HEALTH AND DISEASE”Edited by J. Müller-Quernheim and O. EickelbergNumber 9 in this Series
多环芳烃的特点是内皮损伤、肺动脉持续的血管收缩和闭塞的重塑,随后减少肺微脉管系统的横截面积和增加血管阻力。随后的海拔P巴勒斯坦权力机构进一步增加右心室后负荷导致右心室肥厚和心力衰竭(3]。数十年的研究表明肺内皮细胞功能障碍的核心作用在肺血管重塑的发生和发展,开始在生产中不平衡的血管活性的介质(如一氧化氮(NO)、内皮、内皮素和其他人)。PAH的进步阶段包括内膜的形成和丛状的损伤,内皮细胞凋亡,内侧增厚,外膜增厚和增加细胞外基质(ECM)营业额,以及ECM蛋白质积累。这些事件最终导致毛细管rarification由于肺小动脉的闭塞远端肺血管和微血管变性[3- - - - - -5]。上述多个血管异常参与PAH, ECM合成和降解失衡导致病理改造过程的复杂性和恶化的治疗效果6,7]。
在肺血管结构改变仲裁通过激活血管细胞,获得和展示hyperproliferative迁移和入侵能力。这些表型异常可能促进ECM降解酶在不同的病理刺激(3]。
ECM的脉管系统
基质分子的血管ECM是一个复杂的网络,以精心策划的方式组装,导致血管的结构和功能完整性。ECM提供机械强度、弹性和压缩血管。此外,ECM还提供了一个复杂的微环境促进信息和cell-ECM相声来调节细胞迁移、增殖和分化,最终维持血管内稳态(8]。包含动脉腔的血管壁内膜层组成的三层体系结构,中模和外膜(图1)[9]。此外,肺血管床包括不同的细胞类型,提供结构和功能的完整性,维持血管内稳态。
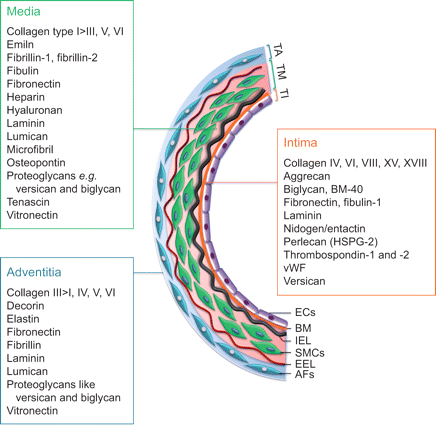
动脉壁的结构及其细胞外基质(ECM)的组件。血管壁有一个三层架构:内膜层(TI),中模(TM)和外膜(TA)。动脉壁组成细胞的主要元素成分的脉管系统(如内皮细胞、平滑肌细胞和外膜成纤维细胞(AFs))和ECM组件的容器,导致功能脉管系统的框架。电子商务:内皮细胞;BM:基底膜;IEL:内部弹性板;SMC:平滑肌细胞;鳗鱼:外弹性膜;HSPG-2:硫酸乙酰肝素蛋白聚糖2;vWF:血管性血友病因子。
最里面的一层,内膜层,是由一个连续的单层扁平的血管内皮细胞(ECs)行动脉腔的表面,形成严格监管透屏障(10]。通过细胞间连接复合体血管ECs相互连接(如紧密连接和缝隙连接粘合连接处并且)(11)和其他的基板基底膜(BM)。大英博物馆是由特定的超分子组装ECM组件之间的交互,如层粘连蛋白、IV型胶原蛋白,entactins (nidogens),蛋白聚糖、糖蛋白和不同的整合素和non-integrin受体(图1)[12]。众所周知,重要的是,ECs合成各种BM和内部弹性板(IEL)组件和一些血管活性的介质,如没有,环前列腺素、内皮素1 (ET),血清素和血栓素,这是很重要的调节血管张力(8,13]。在肌肉动脉,IEL可以认定为有孔的一层弹性组织(弹性)分离下的内膜和媒体sub-endothelial层也允许扩散(8]。肺动脉的内侧层是丰富的,由多个同心层平滑肌细胞(smc)和胶原纤维层之间插入中间有孔的弹性板。
一般来说,动脉smc静止,表现出分化收缩表型正常的生理条件下,构成了内侧层的主要细胞类型。最重要的是,smc保持发育可塑性,能够瞬时收缩之间的表型转换,合成或中间表型状态(14]。而收缩表型的smc展品低扩散率和合成,它呈现收缩血管和血管活性的调节血管张力的反应介质。相比之下,合成表型的特点是增加增殖,迁移和ECM营业额。青蒿酸和分泌不同的ECM组件中模的弹性蛋白纤维等纤维胶原蛋白(I型,III和V)、弹性蛋白、纤连蛋白、层粘连蛋白和蛋白聚糖(15]。这些不同的ECM组件已被证明发挥关键作用在维持SMC表型(16]。例如,弹性蛋白的主要成分之一,ECM内侧层(17),是一种有效的自分泌血管SMC活动的监管机构。除了呈现韧性和弹性动脉,动脉结构的弹性蛋白对稳定至关重要诱导静止收缩状态。几个在体外研究表明逆弹性蛋白表达之间的相关性和SMC增殖18- - - - - -20.]。外弹性膜的厚纤维层弹性纤维,划定媒体从动脉外膜和比IEL发现不太突出。动脉外膜较厚的肌肉相比,弹性动脉和血管的不同部分的厚度不同电路。外膜成纤维细胞外膜的主要细胞类型,被认为是血管壁的关键调节器功能在健康和疾病。成纤维细胞也调节合成和分泌的外膜ECM组件如胶原蛋白类型我和三世,构成的首席ECM内容动脉外膜(21]。除了呈现结构完整性的血管,动脉外膜居民免疫调节和祖细胞种群增长的影响因素和修复过程的血管壁22,23]。
矩阵METALLOPROTEASES, ADAMALYSINS,丝氨酸弹性蛋白酶及其抑制剂
ECM周转率控制之间的平衡蛋白水解酶,如基质金属蛋白酶(MMPs),丝氨酸弹性蛋白酶和内源性抑制剂。基质金属蛋白酶是一种家庭结构相关,zinc-dependent多功能可溶性或膜锚定的蛋白酶。基质金属蛋白酶属于一个更大的蛋白酶家族被称为metzincin总科(24]。
基质金属蛋白酶进行分类的方法。历史上基质金属蛋白酶检索根据他们的底物特异性。基于他们的底物特异性,基质金属蛋白酶或matrixins可以分为六组:1)间质胶原酶(金属蛋白酶- 1、MMP-8 MMP-13和MMP-18);2)IV型胶原酶、明胶酶(MMP-2和MMP-9);3)stromelysins (MMP-3、MMP-10 MMP-11和MMP-19);4)matrilysins (MMP-7和MMP-26);5)跨膜基质金属蛋白酶(膜型基质金属蛋白酶(MT): MMP-14, MMP-15, MMP-16, MMP-17, MMP-24和MMP-25);和6)其他基质金属蛋白酶(MMP-12、MMP-20 MMP-21, MMP-22, MMP-23, MMP-27和MMP-28) (24,25]。因为一些新的细胞外和non-extracellular矩阵基质金属蛋白酶的底物被发现,基质金属蛋白酶现在通常所指的数字名称(MMP-28金属蛋白酶- 1)。表S1提供了通用的和常用的组名,分类数,基质组合(矩阵和non-matrix)和细胞来源MMP的类型,到目前为止,尚未发现。然而,底物组合为特定基质金属蛋白酶可能不完整,由于积极的研究发生在这个区域。
基质金属蛋白酶通常由四个守恒的模块化结构:pro-peptide域;催化金属蛋白酶域;可变长度的链接器肽(也称为“铰链区”);和血液结合素域。例外是MMP-7 (matrilysin-1) MMP-26 (matrilysin-2)和MMP-23;所有这些都失踪链接器肽和血液结合素域。此外,MMP-23有着独特cysteine-rich域和一个immunoglobulin-like域后,金属蛋白酶域。而明胶酶MMP-2和MMP-9有三个重复的纤连蛋白II型图案的金属蛋白酶域。这纤连蛋白II型图案形式collagen-binding域允许绑定和IV型胶原蛋白或退化变性胶原蛋白(明胶)24,26]。
基质金属蛋白酶合成在一个不活跃的发酵菌形成的auto-inhibitory pro-peptide域(通常称为proMMPs)。这些潜在的基质金属蛋白酶的激活发生在一个协调的蛋白水解事件序列。MMP的催化域通过cysteine-Zn pro-peptide隐藏起来了2 +交互。MMP激活期间,蛋白水解NH的乳沟2终端pro-peptide域的序列导致分子的构象变化,因此暴露出锌2 +结合位点的催化域(27]。最初的乳沟pro-peptide域后,进一步的自溶的或外生分裂导致经常发生低分子量活性形式的MMP。在活跃的MMP包含锌催化领域2 +结合区域负责蛋白水解活性。血液结合素域授予底物特异性(26,27]。因此,基质金属蛋白酶降解ECM的不同组件在脉管系统可以调解范围广泛的基本生物过程包括正常的胚胎发育过程、血管生成、复制、骨重建和组织修复过程,还涉及到几种病理设置(24,28]。
在生理条件下,基质金属蛋白酶的活动是在转录水平的调节,平移和翻译后。MMP的活动的另一个关键控制点是通过一组激活酶的抑制内源性抑制剂称为组织基质金属蛋白酶抑制剂(TIMPs)。TIMPs严格控制基质金属蛋白酶的表达和活动,并提供一个平衡的机制来防止过度降解ECM (27]。
TIMP家庭由四个成员组成,通过TIMP-4 TIMP-1。TIMPs有N - c端域≈125和65个氨基酸,分别与每个包含三个守恒的二硫键。n端结构域折叠作为一个单独的单元,能够抑制基质金属蛋白酶(29日]。TIMPs可以形成配合物与基质金属蛋白酶1:1化学计量比通过协调的锌2 +的MMP活性部位的氨基和羰基组织TIMP氨基半胱氨酸残基。虽然TIMPs不显示任何特定的高特异性MMP有优惠绑定TIMP-2 MMP-2和TIMP-1 MMP-9。此外,TIMP-2, 3和4,但不是TIMP-1 MT-MMPs的有效抑制剂。TIMP-3是独特的在四个TIMPs由于其直接绑定到ECM蛋白质,从而提供一个意味着稳定MMP-TIMP复合物内的间隙空间(24,30.]。TIMP-3也被证明抑制亚当(disintegrin和金属蛋白酶),特别是ADAM-17 [31日]。然而,TIMPs内生MMP抑制剂并不是唯一的。事实上,α2巨球蛋白,显示了一个丰富的血浆蛋白,是主要的内源性抑制剂在血浆MMP的活动。尽管TIMPs可逆地抑制基质金属蛋白酶、α2巨球蛋白/ MMP的复合物通过清道夫受体介导内吞作用,从而发挥重要作用的不可逆转的间隙中基质金属蛋白酶(30.]。
的另一个成员metzincin总科密切相关基质金属蛋白酶是亚当,也被称为adamalysins或metalloproteinase-like disintegrin-like,半胱氨酸富有。Adamalysins /亚当斯属于zinc-dependent金属蛋白酶的总科,由两组:membrane-anchored亚当斯和分泌ADAMTSs。至少40亚当斯被描述,25是表达人类。其中,19显示蛋白水解活性(32]。亚当斯的域结构由pro-peptide域,metalloprotease域中,disintegrin域中,半胱氨酸-富领域,血管内皮生长因子(EGF)同域,跨膜域和胞质尾。像大多数蛋白酶,亚当斯最初是作为惰性酶前体蛋白质合成。这种不活跃的状态在大多数亚当斯是由于一个半胱氨酸残基之间的相互作用与锌pro-peptide域2 +在催化部位。对蛋白酶激活,这pro-peptide域是通过furin-like转化酶或自动催化作用,取决于特定的亚当(32,33]。旁边prodomain是金属蛋白酶域。metalloprotease域水解蛋白质基质,如细胞因子和生长因子,以及各自的受体。这个过程称为ectodomain脱落和亚当经常被称为“sheddases”。disintegrin域结合整合素受体如α9β1因此,协调和信息和cell-matrix交互。的确切功能cysteine-rich域和EGF-like域尚不清楚。然而,胞质尾含有磷酸化网站和SH3绑定域被认为是参与信号也可以用来组装一组胞质适配器分子(33]。然而,ADAMTS的特点是存在额外的血小板反应蛋白c端I型图案,而EGF-like、跨膜和胞质域缺乏(34]。
这些不同的领域组成多功能亚当和ADAMTS赋予这些蛋白质的活动,如增殖、迁移和血管生成。例如,EGF受体配体(amphiregulin和heparin-binding EGF) /流发布ADAM-17增强肿瘤细胞的细胞增殖和诱导血管生成35]。按照,改变表达特定的亚当斯一直与不同的疾病;他们最好的记录的作用是在癌症的形成和发展36]。然而,迄今为止,没有数据可用的角色,这些蛋白质在PH值的发病机理。
在其他蛋白酶降解ECM,丝氨酸弹性蛋白酶的被广泛的特征。丝氨酸蛋白酶是裂开的酶在蛋白质,肽债券中丝氨酸作为亲核活性部位的氨基酸。人类有六个弹性蛋白酶基因编码结构蛋白弹性蛋白酶1类似,2、2、2 b, 3 a、3 b (37]。在几个丝氨酸弹性蛋白酶,中性粒细胞弹性蛋白酶分泌中性粒细胞和23-kDa丝氨酸弹性蛋白酶被称为内源性血管弹性蛋白酶(夜)强烈与血管疾病。夏娃是由血管smc和降解弹性蛋白和其他几个ECM蛋白(38,39]。监管控制丝氨酸弹性蛋白酶活动是通过内源性抑制剂的伴随生产α1抗胰蛋白酶,α2巨球蛋白和弹力素,最相关的抑制剂在血管抑弹性酶蛋白(40]。
我们回顾了基质金属蛋白酶的生理和病理生理作用和丝氨酸弹性蛋白酶在脉管系统特定的专注于基质金属蛋白酶的影响和丝氨酸弹性蛋白酶的病机博士此外,使用MMP抑制剂的药理干预措施和结果在不同的临床前动物模型和将讨论未来的发展方向。
基质金属蛋白酶的表达和调控血管
血管细胞
不同的细胞类型,构成了血管的细胞成分,包括内皮、smc、成纤维细胞浸润先天免疫细胞浸润血管壁,被描述为成人血管的主要细胞来源的基质金属蛋白酶(41- - - - - -44)(表S1)。基质金属蛋白酶在脉管系统的不同功能主要取决于其表达式和活动。在生理和病理状态下,生物、机械、血液动力学的或神经激素的刺激调节血管特异性MMP的表达和描述活动(45]。
几项研究已经证实血管内皮基质金属蛋白酶的主要来源在血管生成或伤口愈合过程。ECs被证明持续表达检测金属蛋白酶- 1的水平,MMP-2, TIMP-1 TIMP-2,但不是MMP-9基底条件下(41]。虽然基底基质金属蛋白酶的表达很低/察觉ECs,几个pro-angiogenic因素、炎性介质和有机化合物(如。佛波醇酯)可以诱导MMP的表达和激活潜在的基质金属蛋白酶(46]。例如,血管内皮生长因子(VEGF)端依赖MMP-2活动的增加和大幅削减TIMP-1 TIMP-2水平观察微血管ECs。TIMP水平差别VEGF-induced对这些随后允许激活已有的胶原酶,在一起可以促进内皮入侵大英博物馆和间质矩阵(47]。
SMC-specific MMP的表达和活动明显调制由多种病理生理条件下生长因子和细胞因子。暴露的smc炎性分子像白介素(IL) 1α,肿瘤坏死因子(TNF) -α和氧化低密度蛋白显著增加MT1-MMPs的表达和活性,进而导致的激活增加proMMP-2 [43]。然而,另一项研究表明,自分泌转化生长因子的表达和激活(TGF) -β1抵销的血小板源生长因子(PDGF) bb (PDGF亚基为B /β多肽)全身upregulation MMP-2在smc表型调制(48),这表明TGF-β1促进收缩表型调制基质金属蛋白酶。日积月累,这些研究结果表明微分调节MMP的表达的不同的生长因子和细胞因子。
外膜成纤维细胞外膜的主要细胞类型和发现是活跃在增长,但通常静止在正常血管。MMP-2显著增加活跃,特别是TIMP-1 TIMP-2生产也观察到在低氧刺激相比,常氧成纤维细胞(44]。它已经表明,活性氧(ROS)全身的氧化应激增加MMP活性和潜在的调节成纤维细胞增殖和胶原蛋白的合成49]。
居民企业和免疫细胞浸润
除了血管细胞,浸润炎症细胞如单核/巨噬细胞系的细胞,树突状细胞,中性粒细胞,肥大细胞,淋巴细胞和淋巴细胞也构成pro-angiogenic蛋白酶的主要细胞来源的细胞因子和生长因子(50]。
单核细胞/巨噬细胞
不同的体外研究报道合成和金属蛋白酶- 1的分泌,MMP-2, MMP-3, MMP-7, MMP-9 macrophage-specific metalloelastase (MMP-12)培养的巨噬细胞体外在基底或刺激条件下(51]。此外,人类肺泡巨噬细胞的接触本地或变性胶原蛋白I型和III选择性刺激间质胶原酶的表达和TIMPs,表明ECM组件可以直接影响macrophage-mediated MMP的分泌(52]。单核细胞与健康个体的血表示可检测多种基质金属蛋白酶包括金属蛋白酶- 1的水平,MMP-2, MMP-3, MMP-9, MMP-10, MMP-19, MT1-MMP, MT4-MMP MT6-MMP基底条件下(50]。此外,当添加集落刺激因子结合TNF-α或IL-1β-induced金属蛋白酶- 1合成协同增强MMP-9 TIMP-1表达式。而MMP-9生产负受il - 4、il - 10, IFN-β,IFN-γTGF-β在单核细胞(50]。此外,在体外研究表明表达和活动增加MMP-2和MMP-9在单核细胞分化成巨噬细胞,它强调的重要性在单核细胞/巨噬细胞谱系分化的细胞状态(53]。同样,MMP-12表达在人类周围血液巨噬细胞是由一些细胞因子和生长因子诱导包括IL-1βTNF-α,csf、VEGF和PDGF-BB [54]。
白细胞和淋巴细胞
大量研究发现了一个独特的MMP的表达模式不同子集的循环白细胞和单核细胞在基底和刺激的条件下(55]。著名表达MMP-11 MMP-26和MMP-27 B -细胞,MMP-15, MMP-16, MMP-24 MMP-28 t细胞,和MMP-7 MMP-8, MMP-21 MMP-23表达式被发现在所有从健康人白细胞孤立的子集55]。然而,大多数基质金属蛋白酶包括金属蛋白酶- 1、MMP-2, MMP-3, MMP-9, MMP-10, MMP-14, MMP-17, MMP-19 MMP-25在单核细胞显著代表。这种截然不同的模式在单核细胞MMP的表达似乎提供了一种增加transmigratory能力跨血脑屏障模型,与B相比,t细胞(55]。此外,t细胞外渗到血管周围组织炎症是由感应和本地化的MMP-2 t细胞表面通过与血管细胞粘附molecule-1 ECs (56]。
中性粒细胞
中性粒细胞是广泛的潜在细胞来源的蛋白水解酶,参与不同的炎症过程通过释放保持酶的活性,中性粒细胞弹性蛋白酶和其他蛋白酶包括组织蛋白酶G,蛋白酶3,金属蛋白酶- 1,MMP-8, MMP-9 MMP-12 [57]。除了中性粒细胞的先天免疫功能,几项研究表明neutrophil-derived蛋白酶的作用trans-endothelial中性粒细胞迁移(58)的起始血管生成开关(59]。具体来说,neutrophil-derived MMP-9发挥重要作用在基板显示IV型胶原降解和在催化血管生成开关通过促进MMP-9-dependent VEGF动员和顺向pro-angiogenic信号(59]。
肥大细胞
肥大细胞释放多种因素包括基质金属蛋白酶、中性和丝氨酸蛋白酶,肝素,heparinase,类胰蛋白酶,chymase,组胺,和血管生成生长因子如碱性纤维母细胞生长因子(bFGF), VEGF和细胞因子(60]。肥大细胞衍生中性蛋白酶类胰蛋白酶和chymase可能调解激活潜在的基质金属蛋白酶在人类颈动脉和随后增强血管生成表型也降低BM的不同组件61年]。此外,在体外研究表明,细胞因子profibrotic TGF-β变弱工具包ligand-mediated感应MMP-9表达式的居民组织肥大细胞(62年]。具体来说,分析肺动脉主干部分显示本地化的肥大细胞外膜层和扩散的本地化模式免疫反应性的肥大细胞衍生的媒体和动脉外膜胶原酶肌肉肺动脉(63年]。
生理功能被基质金属蛋白酶切断的脉管系统
除了在脉管系统矩阵蛋白水解作用,基质金属蛋白酶在形态发生等多种生理过程中起关键作用,血管生成,组织改造和组织修复,以及调制基本细胞过程扩散、迁移、分化、凋亡、渗透率、宿主防御,释放ECM-bound趋化因子和趋化因子处理(27,64年]。
血管生成
生理血管生成是一个时空上策划了多步骤的过程,包括多种生长因子的相互作用调节的复杂信息和cell-matrix交互通过激活不同的蛋白水解系统在血管微环境。血管生成刺激,endothelial-derived基质金属蛋白酶的调节蛋白水解降解的内皮和信息和EC-matrix交互,促进ECs获得增殖和迁移或侵入性表型(65年]。特别是,血管生成因素分泌MMP-2, MMP-9和MT1-MMP血管ECs对EC扩散至关重要,迁移和血管ECM入侵与发芽血管生成(65年]。具体来说,明胶酶,MMP-2 MMP-9发挥重要作用在水解的家乡IV型胶原,BM促进内皮发芽的主要组成部分(66年]。此外,激活的焦蛋白水解作用因此解放ECM-sequestered pro-angiogenic因素,公开神秘pro-angiogenic整合素结合位点的血管ECM和还生成pro-angiogenic片段,而集体触发血管生成开关(67年- - - - - -69年]。
研究与白明胶酶和MT1-MMP-knockout小鼠基质金属蛋白酶参与的病理生理提供了强有力的证据表明血管再生。MMP-2缺陷小鼠显示肿瘤neovascularisation率降低,总血管面积,血管和肿瘤进展,以及胚胎发育正常的血管系统(70年]。然而,合并后的缺陷MMP-2 / MMP-9在肿瘤血管生成的实验模型显示一个受损的血管生成和减少侵入性的表型gelatinolytic活动;然而,血管生成和侵入性表型不受单一缺乏主机MMP-2 MMP-3或MMP-9 [71年]。这些研究在double-deficient老鼠阐明的在活的有机体内明胶酶及其在neovascularisation协同效应的重要性。同样,FGF-2-induced MT1-MMP缺陷小鼠缺乏血管生成反应,表明MT1-MMP起始血管生成(可能是重要的72年]。此外,研究MMP抑制剂显示强烈的抑制血管生成反应在体外和在活的有机体内(28]。
细胞增殖和迁移
能够很好的证明,基质金属蛋白酶可以直接和间接地调节增殖,迁移,血管ECs的入侵和细胞凋亡73年]和smc [74年)通过在体外和抑制剂的研究。MMP-2和协同MMP-9 MT1-MMPs被证明促进EC细胞迁移和管形成的蛋白水解改造的大英博物馆,通过执行焦点和控制解散ECM和通过释放从ECM ECM-bound趋化因子75年,76年]。neovessel形成期间,前沿的发展中neovessel EC扩散,随着MT1-MMP-dependent激活MMP-2 MT1-MMP-dependent焦collagenolysis,观察(77年]。
动脉损伤后内膜增生的重要的是包括SMC增殖、迁移和ECM重塑,这可能是由各种细胞因子和生长因子。PDGF-BB或ab (PDGF A和B亚基的异质二聚体/α和β多肽)全身迁移SMC的内侧外植体被MMP-2介导,而内侧SMC迁移bFGF的刺激效应是由MMP-2和MMP-9 [78年]。管理强有力的MMP抑制剂(BB94)剂量依赖性抑制PDGF-BB-induced迁移培养SMC和抑制动脉损伤后内膜增厚和中SMC增殖(74年]。Zymographic研究血管壁显示组成型表达的MMP的表达MMP-2 MMP-9在正常颈动脉,而激活形式观察balloon-injured颈动脉SMC增殖和迁移的阶段期间(74年]。此外,原位和在体外研究进行肺动脉和肺动脉平滑肌细胞(PASMCs)患者显示MMP的表达增加,表明基质金属蛋白酶的一个关键的角色在不同的血管重塑过程包括SMC增殖、迁移和内膜的增厚(74年,79年]。
细胞分化
血管smc表现出显著的表型可塑性,可以从静止的调制,收缩表型向合成表型在生理或病理血管重塑。增生性或迁徙的SMC表型与蛋白水解活性和ECM营业额增加,伴随着失去分化标记包括肌凝蛋白包和α-actin成分合成SMC表型特征(80年]。
在PDGF-BB介导招聘smc和对新生血管性芽;从收缩血管SMC去分化迁移表型,这一过程是伴随着MMP-2 upregulation [81年,82年]。此外,一项研究R伊辛et al。(48]证明了PDGF-BB-induced upregulation SMC的MMP-2不顾自分泌表达和激活TGF-β1导致显著延迟SMC表型转换。此外,另一项研究发现,TGF-β1治疗表达下调金属蛋白酶- 1和MMP-3生产调节TIMP-1 mRNA水平在人类子宫肌层的smc (83年]。总之,这些研究结果证实TGF-β促进收缩表型,部分通过调节MMP / TIMP比率。然而,明胶酶的活性增加,MMP-2 MMP-9,显示是分化的过渡需要SMC合成表型(84年]。此外,弹性蛋白肽由丝氨酸产生弹性蛋白酶和/或基质金属蛋白酶刺激基质糖蛋白纤连蛋白的生产,从而改变smc从收缩迁移表型。对成纤维细胞的稳态居民成纤维细胞和胶原蛋白矩阵之间的关系让他们在一个静止的、未分化的状态(3]。然而,正常成纤维细胞受到缺氧显示低氧诱导myofibroblasts表型转换,通过MMP-2 / TIMP介导途径(44]。TGF-β1,具体来说,PDGF tenascin-C (TN-C)、纤连蛋白和促有丝分裂的ET-1展览活动,诱发myofibroblast表型在体外和在活的有机体内(85年),可能通过调节MMP / TIMP的平衡。
血管通透性和细胞粘附
内皮屏障的完整性,血管通透性和静止EC表型,以内皮cell-matrix,和持续的交互,可以由pro-angiogenic介质如VEGF和组(73年]。具体来说,VEGF促进血管渗透性的解偶联inter-endothelial路口,诱导内皮膜孔的形成和增加修改的小窝73年]。最近的一项研究表明,低氧诱导血管渗漏在活的有机体内和相关的紧密连接蛋白occludin的重排及其表达式是由低氧诱导减少MMP-9激活。然而,VEGF抑制衰减血管渗漏,低氧诱导MMP-9激活和依赖gelatinolytic活动。同时,MMP的抑制减弱血管研究进展,防止差距形成和紧密连接重排(74年]。重要的是,整合素、钙粘着蛋白selectin immunoglobin家族的内皮细胞粘附分子已确定在信息发挥着至关重要的作用,cell-matrix交互和脉管系统在不同病理生理事件。特别是,αvβ3和αvβ5整合蛋白调节内皮细胞粘附和迁移描述在血管生成和血管生成(75年]。此外,αvβ3整合蛋白相互作用的数组ECM配体如vitronectin、纤连蛋白、胶原蛋白、层粘连蛋白、血管性血友病因子、纤维蛋白原、骨桥蛋白、血小板反应蛋白和RGD-containing肽,潜在MMP的基质(表S1) (75年]。此外,在内皮发芽,表面表达和激活MMP-2 MT1-MMP依赖ECs被发现与整合素αvβ3基底和TIMPs焦接触和调解焦ECM的降解(76年]。
整合蛋白、钙粘蛋白和基质金属蛋白酶已知扮演至关重要的角色在决定组织凝聚力,集体细胞迁移和重组期间ECM的入侵(77年]。研究在体外入侵模式表明,组织凝聚力与钙粘蛋白的表达。然而,中间组织凝聚力的细胞系和相对较高的MMP的表达表现出复杂的迁徙模式(78年]。MMP的抑制研究证实,减少钙粘蛋白的表达增加和IV型胶原酶活性(MMP-2和MMP-9)将增强分离肿瘤细胞和促进入侵(79年]。相比之下,ECs,增加VE-cadherin观察与表达下调MMP的生产和pericellular蛋白水解作用在稳定和成熟的neovessel86年]。最近的研究证实信息联系机制依赖监管pericellular蛋白水解作用在血管生成。这与发现MMP抑制剂治疗增强colocalisation钙粘着蛋白/β-catenin cadherin-mediated信息接触,推动了稳定的信息在成纤维细胞粘附暗示可能反馈回路之间MMP和钙粘蛋白/β-catenin系统81年]。
动员的生长因子和细胞因子处理
除了MMP-mediated崩溃的ECM障碍促进细胞迁移和入侵,MMP的过程中扮演着重要的角色在解放ECM-bound组件,通过直接消化的有丝分裂原矩阵的监管matrikines的形成和释放生物活性的生长因子从ECM87年,88年]。除了提供结构支撑的维管组织,ECM作为存储库等生物活性分子包括pro-angiogenic生长因子VEGF, bFGF,肝细胞生长因子、胰岛素样生长因子- 1,TGF-β1和结缔组织生长因子(CTGF) [26,69年]。MMP-dependent蛋白水解作用促进这些生长因子的释放的血管ECM从而影响细胞增殖和细胞迁移68年]。例如,基质金属蛋白酶调节VEGF的生物利用度。VEGF是被作为一种活动形式由ECM组件,如CTGF pleiotrophin,等。,导致生物利用度降低。几个基质金属蛋白酶(金属蛋白酶- 1、2、7、9,-16年和-19年)调节蛋白水解的乳沟抑制VEGF的复杂,提高生物利用度,这在各种病理生理过程中扮演着重要角色88年]。MMP-9,并在较小程度上MMP-2,有效降低硫酸乙酰肝素蛋白聚糖或perlecans和增加的动员BM-sequestered VEGF (89年]。小鼠靶向破坏MMP-9表现出减少肥厚性软骨vascularisation由于缺乏动员ECM-bound VEGF,确认MMP-9生物利用度的增长因素的重要作用[90年]。此外,在培养smc,丝氨酸弹性蛋白酶(夜)被证明调解弹性蛋白的降解,最终促进解放ECM-bound bFGF。
然而,MMP-dependent蛋白质水解matrikines ECM组件的版本。Matrikines是支离破碎的矩阵肽生物活性的调节结缔组织细胞活动。例如,MMP-mediated蛋白质水解的胶原蛋白和perlecan生成抗血管生成matrikines arrestin等canstatin, tumstatin, metastatin,血管内皮抑制素,neostatin, vastatin, restin endorepellin [69年]。然而,MMP-based蛋白质水解的纤连蛋白、层粘连蛋白、骨粘连蛋白和elastin-derived matrikines促进细胞增殖,迁移和血管生成(88年]。
重要的是,基质金属蛋白酶在细胞因子处理也发挥着至关重要的作用。TGF-β、多功能细胞因子在调节多种细胞过程中起着举足轻重的作用,如增殖、分化、迁移、血管细胞的生存和ECM合成(91年]。TGF-β亚型分泌和维护以潜在的形式通过绑定到弹性微纤维和基质。TGF-β活跃的生物利用度是由微纤维的降解,释放活性TGF-β,潜在的关联由不同的蛋白水解酶如MMP-9 protein-β1 MT1-MMP和血纤维蛋白溶酶91年]。除了调制的细胞因子活性和生物利用度,基质金属蛋白酶可以调节趋化因子处理,可以改变趋化因子的趋化现象的性质,因此影响炎症反应。米c问uibban等。(92年]发现MMP-2调解处理CCL7 / MCP-3生成裂解MCP-3,作为通用趋化因子拮抗剂和变弱趋化性和宿主炎性反应在活的有机体内。
失调的基质金属蛋白酶及其对多环芳烃发病机制的贡献
MMP的表达多环芳烃
一些证据显示干扰的基质金属蛋白酶和TIMPs平衡PH值和其他肺部疾病的发病机理。如前所述,基质金属蛋白酶促发多种病理生理过程,如ECM营业额、表型转换,增生,细胞迁移,细胞凋亡,暗示了至关重要的作用的基质金属蛋白酶在肺血管重塑和PAH的建立(图2)。
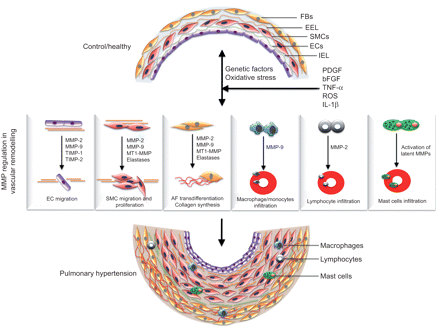
基质金属蛋白酶(MMPs)肺动脉高血压的发病机制(PAH)。各种生理、获得和/或外源性刺激如缺氧、氧化应激、炎症、感染和遗传因素导致血管损伤和随后促进异常生长因子和细胞因子的生产(如血小板源生长因子(PDGF)、表皮生长因子、成纤维细胞生长因子2,改变增长factor-β,细胞因子如白细胞介素(IL) 1β,和肿瘤坏死因子(TNF) -α)仲裁的病理生理改变血管调节MMP的表达和调控。放松管制的基质金属蛋白酶等病理生理过程牵涉到内皮细胞(EC)迁移,平滑肌细胞(SMC)迁移和增生,外膜成纤维细胞(AF) trans-differentiation,细胞外基质增加营业额和招聘的炎症细胞,从而促进肺血管重塑的发展过程和多环芳烃的建立。弗拉维奥-布里亚托利:;鳗鱼:外弹性膜;IEL:内部弹性板;bFGF:碱性纤维母细胞生长因子;ROS:活性氧;金属蛋白酶组织抑制剂TIMP:;MT1:膜1型。
一些研究调查了基质金属蛋白酶的表达谱(表1)和丝氨酸在人类PAH弹性蛋白酶及其潜在的作用。米atsuiet al。(98年)评价基质金属蛋白酶的表达和本地化不同血管病变的多环芳烃通过免疫组织化学和免疫荧光研究。这项研究发现了一个扩散和强烈反应MT1-MMP本地化myofibroblasts和洋葱皮损伤内皮细胞和细胞丛状的病灶。而只有少数myofibroblasts MMP-3这些病变阳性和MMP-7。此外,co-localisation MT1-MMP和MMP-2 myofibroblasts中发现和细胞网状内皮细胞损伤98年]。特别是,不连续IV型胶原蛋白和焦变薄在洋葱皮损伤和成熟的丛状的病灶。本研究强调,调制的血管细胞基质金属蛋白酶可能安排的关键病理事件参与血管病变的发展多环芳烃(98年]。值得注意的是,我epetitet al。(79年)确定MMP-2表达和活动增加PASMCs从特发性肺动脉高压病人。Co-localisation MMP-2和内侧gelatinolytic活动层的内弹性板的板在特发性肺动脉高压血管相比,控制标本观察(79年]。此外,显著降低MMP-3和增加在特发性多环芳烃PASMCs TIMP-1生产检测,有利于ECM积聚在特发性肺动脉高压(79年]。虽然这项研究特别关注PASMC-derived MMP的表达和活性,证实存在MMP / TIMP失衡和增加gelatinolytic人类PH值的活动场景。
此外,测量循环MMP的水平横向和纵向研究高血压的主题显示等离子体浓度显著升高MMP-9和TIMP-1基线血压正常的控制。这显著增加患者在基线循环MMP-9 PH值可以反映出更高的血管和心脏组织MMP-9水平和增加ECM营业额(96年]。
MMP的表达主要是研究在两个广泛使用的模型PH值:慢性低氧诱导的PH值和野百合碱(MCT)全身的PH值在啮齿动物。动物在慢性低氧诱导的PH值、接触normoxia或低比重的增加缺氧时间为2 - 3周通常会导致50%的意思P巴勒斯坦权力机构和右心室肥大。MCT-induced PH值的模型,一个皮下注射的MCT (pyrrolizidine生物碱的植物来源)3 - 4周后会导致严重的血管重塑,慢性PH值和肺心病(3,99年]。
微阵列基因表达分析MCT-treated肺透露微分表达多个基因与ECM的调控和细胞粘附。Upregulation不同的MMP亚型包括明胶酶(MMP-2和MMP-9)、中性粒细胞胶原酶(MMP-8) stromelysins (MMP-10 MMP-11)、巨噬细胞metalloelastase (MMP-12) MMP-20,平台(tPA)和SERPINB2被确认和验证,表明基质金属蛋白酶不可或缺的角色建立实验博士此外,这项研究表明基质金属蛋白酶的协会加强迁徙的反应体外培养smc源自MCT-PH动脉(One hundred.]。根据,重要upregulation MMP-2, MMP-9 TN-C以及增加gelatinolytic活动孤立肺动脉从MCT-treated动物被确认101年]。此外,转基因的表达白明胶酶B(人类MMP-9)导致巨噬细胞的广泛渗透,最终严重MCT-induced PH值、充实的病理意义增加MMP-9 PH值(102年]。有趣的是,最近的一项研究表明,macrophage-specific转基因小鼠模型的人类金属蛋白酶- 1的表达MCT-induced PH值导致胶原纤维沉积衰减,SMC增殖、浸润的巨噬细胞和因此内侧增厚(103年]。
不同的研究已经报道了解除血管基质金属蛋白酶的表达在两个经典的啮齿动物模型MCT -和低氧诱导的PH值(表2)。Frisdalet al。(110年)评估表达式,活动和本地化的明胶酶特别是肺血管在进步的多环芳烃在缺氧实验模型——和MCT-induced博士两种啮齿动物模型PH值显示强大的MMP-2表达整个肺脉管系统与控制。重要的是,增加gelatinolytic活动主要是在内侧层和与增加MMP-2表达MCT-induced PH肺动脉(110年]。然而,弥散分布的gelatinolytic活动被发现在低氧诱导博士可以归因于MMP-2 gelatinolytic活动增加,由于缺乏MMP-9表达在两个博士的模型重要的是,增加时间之间的相关性MMP-2 gelatinolytic观察活动和缺氧的PH值(110年]。溶胶原的活动增加也观察到在低氧诱导博士Hergetet al。(113年证实胶原蛋白分解产物的外观和溶胶原的活动增加由于低氧诱导的表达MMP-13缺氧大鼠的肺动脉。这些实验研究都表明一个重要的角色在ECM营业额和病理性血管重塑中基质金属蛋白酶的PH值。
与上面的结果涉及到MMP的活动增加严重性的PH值,一些研究表明一个重要的角色逆转重塑过程中基质金属蛋白酶。T在野阵营- v咏叹调et al。(114年)增加了瞬态间质胶原酶(金属蛋白酶- 1)的表达,stromelysin-1 (MMP-3)和明胶酶在常氧恢复从低氧诱导博士stromelysins和总蛋白水解,大幅提高溶胶原的gelatinolytic活动是主要的媒体和动脉外膜中发现肺动脉高血压血管在常氧复苏(114年]。这个观察蛋白酶活动增加与快速减少肺动脉胶原蛋白和弹性蛋白含量,从而表明相关性降低血管重塑和MMP的活动增加在逆转早期低氧诱导的PH值(114年,119年]。在类似的研究中,Tozziet al。(63年],增加激活肥大细胞来源的间质胶原酶被证明调解恢复在改建肺动脉血管结构通过促进胶原蛋白在早期恢复阶段的慢性缺氧。Poianiet al。(119年)也报道积累主肺动脉胶原蛋白和弹性蛋白的老鼠在慢性缺氧,在常氧恢复高血压血管与胶原蛋白和弹性蛋白含量下降有关。日积月累,这些实验研究表明一个重要的角色在血管的再吸收胶原蛋白基质金属蛋白酶de-remodelling过程发生在post-hypoxic恢复阶段(63年,114年,119年]。
除了基质金属蛋白酶,丝氨酸弹性蛋白酶被显示在PAH发病机制特异表达。实验模型的PH值显示早期增加elastinolytic活动前血管的发展变化和后面的增加与疾病进展相关。提高丝氨酸弹性蛋白酶活性和分散的弹性蛋白在肺动脉MCT-induced PAH的老鼠。丝氨酸弹性蛋白酶活动的增加,特别是内源性血管弹性蛋白酶(夜),之前MCT-induced PAH的发展以及随之而来的血管病变(120年]。在低氧诱导PAH,观察瞬态弹性蛋白酶活性的增加(121年]。最近,转基因小鼠模型overexpressing S100A4发达血管内膜损伤后注射的小鼠γ疱疹病毒68 (mhv - 68)和活动加剧了丝氨酸弹性蛋白酶和弹性蛋白降解的病毒感染后,重新激活(122年]。
影响多环芳烃的MMP的失调
异常ECM组件的生产
内稳态的内侧ECM组件,如弹性蛋白血管胶原蛋白、纤连蛋白,TN-C和他们的营业额基质金属蛋白酶起着至关重要的作用在调节SMC和相关的表型转换PASMC肥大,增生、迁移和内侧ECM营业额(123年]。胶原蛋白和弹性蛋白沉积的增加被认为是重要的决定因素的内侧增厚过程中多环芳烃(123年]。此外,证据也显示增加血管壁的胶原蛋白和弹性蛋白的积累的重要因素的恶化慢性低氧诱导的PH值(119年]。胶原蛋白积累导致肺动脉硬化,与PH值通过两种机制进展和右心室功能障碍:增加远端动脉循环应变损伤,促进SMC增殖;和近端波反射,增加右心室后负荷(124年]。
超微结构的评估肺动脉的PAH患者肺组织活检显示IEL的碎片(125年]。此外,缺口IEL PAH患者中也发现了和血管内膜损伤126年]。同意这一观点,动物研究显示,分裂的弹性蛋白前血管的发展变化。弹性蛋白肽由丝氨酸产生弹性蛋白酶和/或基质金属蛋白酶刺激基质糖蛋白纤连蛋白的生产,从迁徙的收缩变化smc表型(123年]。
MMP和弹性蛋白酶激活的另一个后果是增加产量的糖蛋白在PASMCs TN-C积极调节PASMC增殖。的增加表达TN-C PH值与进展相关的临床和实验(123年]。机关文化的支持,治疗与MMP-2或弹性蛋白酶抑制剂导致TN-C表达的抑制,和回归与PASMC凋亡有关的内侧肥大(111年]。后续研究培养PASMCs证明,TN-C放大了FGF-2和促有丝分裂的响应是一个先决条件EGF-dependent SMC增殖(127年]。
监管的PAH的关键分子
一些生长因子(PDGF、表皮生长因子等,FGF-2,骨形态形成蛋白(BMP)和TGF-β),作用于血管的内皮素等物质,细胞因子如IL-1βTNF-α、5 -羟色胺和ROS管制在演示了多环芳烃(128年]。这些因素仲裁增生等病理生理改变肥厚,血管细胞的迁移,表型调制,因此肺血管重塑7,80年,128年]。重要的是,一个潜在的这些pro-hypertensive分子之间的串扰和基质金属蛋白酶/丝氨酸弹性蛋白酶已被观察到。击倒的人类PASMCs BMPR1A MMP-2和MMP-9活动减少,减毒serum-induced扩散,受损PDGF-BB-directed迁移。此外,击倒MMP-2或MMP-9捕捉这些异常,支持BMP信号之间的功能互动,基质金属蛋白酶(129年]。基质金属蛋白酶和生长因子受体之间的直接关联。血清诱导PASMC弹性蛋白酶是显示信号通过激活酪氨酸激酶(130年]。然而,丝氨酸弹性蛋白酶降解ECM因此释放SMC bFGF等生长因子。这个函数可以被放大通过激活基质金属蛋白酶(38]。基质金属蛋白酶也积极调节EGF-dependent SMC增殖通过促进TN-C诱导α的集群vβ3整合素受体(111年]。此外,研究5 -羟色胺受体的基因或药物消融2 bR表明5 -2 bR-dependent弹性蛋白酶活动增加和MMP / TIMP失衡,随后导致潜在生长因子释放,包括TGF-β[105年,131年]。反之亦然,证据表明TGF-β1刺激表达式的pro-MMP-9 IL-1βPASMCs治疗。IL-1β,主要与PH值相关的细胞因子,是显示显著增加MMP-2和MMP-9白明胶酶活性(132年]。对内皮素系统,提升MMP-9活动中发现缺少endothelin-B受体的老鼠们注入了PH值,表明endothelin-mediated调节基质金属蛋白酶(117年]。
治疗使用MMP抑制剂PH值的实验模型
一些药理研究MMP的抑制主要表现在两个实验模型的PH值:缺氧,MCT -诱导PH值(表3)。Cowanet al。(111年]报道了弹性蛋白酶和MMP抑制剂的治疗效果MCT-induced PH值模型。这种效应与减少TN-C,抑制SMC增殖和诱导细胞凋亡。此外,选择性镇压TN-C(矩阵分子诱导基质金属蛋白酶)使转染肺动脉与反义/核糖酶结构细胞凋亡SMC和逮捕进步血管增厚但未能诱发疾病的回归。Vieillard- b阿伦et al。(6]表明,气管内的滴剂的adenovirus-mediated人类TIMP-1基因的超表达在肺部的老鼠暴露在MCT降低肺血管重塑,右心室肥大,白明胶酶活性和muscularisation外围肺动脉,表明平衡MMP / TIMP比率可以逆转疾病。
然而,混合结果MMP抑制低氧诱导的PH值模型。Hergetet al。(113年]表明,慢性缺氧诱导的表达鼠间质胶原酶(MMP-13)肺动脉和外围,因此,合成MMP抑制剂治疗batimastat减少muscularisation远端肺血管和明显的减毒诱导博士是这项研究强调溶胶原的活动的刺激是一个实质性的诱发因素在低氧诱导肺血管重塑和高血压的发病机理。此外,增加胶原蛋白和弹性蛋白的合成和积累被确认在主肺动脉缺氧老鼠在早期发展的PH值(119年]。根据一项由K犯错et al。(135年),使用胶原蛋白合成抑制剂(cis-4-hydroxy-L-proline)和交联(β-aminopropionitrile)大鼠暴露于慢性缺氧,减少了多余的血管胶原蛋白和衰减的博士然而,相比之下,Vieillard- b阿伦et al。(134年]表明,气管内的滴剂的adenovirus-mediated人类TIMP-1基因的超表达或政府广谱MMP的拦截器(强力霉素)的老鼠受到慢性缺氧与muscularisation和periadventitial胶原蛋白积累增加远端动脉。这项研究表明,抑制肺基质金属蛋白酶在老鼠受到慢性缺氧肺动脉重塑最终导致显著的恶化和PAH的恶化。
除了MMP的抑制,丝氨酸弹性蛋白酶抑制(M249314或ZD0892)显示减弱(136年)或反向MCT-induced PH值(136年]。最初,在体外机关文化研究使用MCT-induced PH鼠肺动脉显示弹性蛋白酶抑制剂抑制TN-C和诱导SMC细胞凋亡111年]。这个启动完成回归过分生长的血管壁由一个协调一致的细胞结构和ECM的损失。因此,在活的有机体内弹性蛋白酶抑制剂被证明反向先进MCT-induced PH值大鼠肺血管疾病(137年]。同样的,政府的丝氨酸弹性蛋白酶抑制剂(sc - 39026)大鼠低氧诱导的PH值降低(121年]。此外,转基因小鼠中的丝氨酸弹性蛋白酶抑制剂抑弹性酶蛋白过表达当暴露于慢性缺氧了减少丝氨酸弹性蛋白酶和MMP的活动比非转基因老鼠。重要的是,elafin-transgenic老鼠显示右心室压力降低,减少muscularisation和保存的数量远端血管与控制或非转基因小鼠相比40]。
此外,除了MMP抑制剂,一些药物化合物似乎回归建立了PH值函数通过调制MMP / TIMPs。Pullamsetti等。(One hundred.]表明,吸入联合选择性PDE3/4抑制剂(tolafentrine)表现出anti-proliferative anti-migratory和anti-remodelling效果,因此逆转MCT-induced PH值的老鼠。本研究表明,逆转重塑的影响tolafentrine甚至正常化的差别是由于对这些管制的几种基质金属蛋白酶和粘附分子来响应特定轴。Lercanidipine, vasoselective dihydropyridine钙通道阻滞剂,演示了有利影响患者的PH值减少循环MMP-9水平升高(93年,96年]。lercanidipine-induced效果会明显降低MMP-9活动而不影响proMMP-2活动和TIMP-1浓度除了减少氧化应激在酸碱和患者和糖尿病93年]。同样,管理第三代钙通道阻滞剂,氨氯地平,立即紧随其后的是特定治疗抑制MCT-induced MMP-2活动的增多,血小板激活,EC损伤和SMC增殖,从而抑制发展的PH值(133年]。fda批准的药物管理PH值,应用波生坦(双内皮素受体激动剂等A / B),也减毒MCT-induced upregulation MMP-2, TIMP-1,内皮没有合酶表达和MCT-induced PH值(104年]。MCT-induced PH值的改进氟西汀(选择性5 -羟色胺再摄取抑制剂)治疗与抑制MMP-2, MMP-9, TIMP-1 TIMP-2表达式(105年,106年]。在最近的一项研究中,卡托普利(血管紧张素转换酶抑制剂)和洛沙坦(血管紧张素ⅱ1型受体拮抗剂)政府减毒肺血管重塑,可能与调节MMP-2的表情,MMP-9和TIMP-1肺切除术+ MCT injection-induced严重多环芳烃(115年]。
MMP的药理研究抑制缺氧- PH值和MCT-induced实验模型已经证实可以双向选择性或pan-MMP抑制:变弱或加剧血管重塑和博士的微分结果部分建议的存在独特的机制涉及ECM积聚和MMP活动潜在的发展实验博士此外,临床研究执行在癌症、关节炎等疾病已经暗示可能dose-limiting pan-MMP抑制的副作用,如肌肉骨骼综合症,表现为疼痛和不动的肩膀关节,关节痛,挛缩手中和整体降低了患者的生活质量138年]。临床前研究使用选择性MMP抑制剂(表S2)可能缓解上述问题。
188bet体育备用网址当前的问题和未来的前景
管制表达式和活动的基质金属蛋白酶/ TIMPs已发现人类的多环芳烃和实验模型博士此外,一些药理研究MMP的抑制在不同PH值的实验动物模型已经证实,选择性或pan-MMP抑制可以减弱或增强血管重塑和博士因此,尚不清楚是否抑制基质金属蛋白酶是一个有用的策略治疗多环芳烃。几个问题需要解决在考虑MMP抑制作为临床治疗策略。
的冲突结果MMP的抑制研究缺氧和野百合碱模型PH值的回归肺血管病变显示MMP抑制介导的有利影响主要取决于侮辱,或抑制剂使用的类型。此外,这也可以大力支持发展的独特机制的存在低氧诱导酸度与MCT-induced相比及其微分对药物的反应。
MMP的表达和活动的详细描述在不同亚型缺乏人类的PH值,特别是肺血管和肺血管在不同的细胞类型。在动物上的数据推断人体酸碱的子组表明,慢性缺氧模型在啮齿动物认为关联组3,即。与缺氧相关的人类PH值(酸碱度与肺实质疾病有关,睡眠障碍性呼吸,严重的慢性阻塞性肺疾病和居住在高海拔),证明增加ECM营业额,即。增加elastolytic和gelatinolytic活动,以及积累的ECM蛋白质。然而,MCT-induced PH值被视为组1,多环芳烃由药物或毒素引起的。在这群ECM营业额的增加可能发挥主导作用。在这种背景下,获得更多信息的特定MMP / TIMP模式在不同的子组人类PH值是极端重要的。
此外,慢性缺氧和MCT-induced动物模型的PH值完全类似于人类复杂的情况。两种模型触发只有轻度到中度的PH值和不概括新生内膜增殖和丛状的病变,是严重的PH值的重要标志139年]。这使得它很难推断的结果MMP抑制人类在动物模型。然而,一个更加清晰的认识基质金属蛋白酶参与实验模型的血管内膜病变或plexogenic病变是必不可少的进一步探索治疗抑制MMP在PAH的可能性。
重要的是,广谱MMP抑制剂,如marimastat,没能证明其临床有效性由于严重的副作用。最常见的副作用与临床试验相关的MMP抑制剂是一个肌肉骨骼综合症(138年]。尽管如此,periostat是唯一MMP抑制剂已被FDA批准用于治疗牙周疾病。可能原因的低成功率MMP抑制剂在诊所包括不必要的副作用引起的他们缺乏选择性,可怜的口服生物利用度和降低效力在活的有机体内。绕过这是基本知道PH MMP的肺血管是有针对性的使用选择性MMP抑制剂和疾病的阶段,允许更好的选择性和功效。系统性毒性可能克服气溶胶交付的药物,允许地方政府和低剂量的化合物。
重要的是,缺乏选择性MMP抑制剂,与有限的知识的确切功能特定的MMP阻碍了临床应用。第三代MMP抑制剂是目前正在接受调查,并会增强逆转重塑过程中PH值,没有任何有害的脱靶效应。然而,这必须在适当的筛选在体外和在活的有机体内模型来评估疗效。
以前,大部分的在体外研究是由肺血管细胞的二维文化。然而,在活的有机体内血管细胞矩阵交互是复杂的,并且重申有必要采用三维培养系统或器官培养模型。此外,重要的是要确定基质/基因目标和基质金属蛋白酶之间的串扰和其他信号通路相关PH发病机理有待进一步调查。显然,选择性MMP抑制剂的临床前有效性评估严重PH值的动物模型是必要的。总之,基质金属蛋白酶在肺血管重塑中起着不可或缺的作用过程和可能是一个有吸引力的目标治疗PH值。
脚注
可以从本文的补充材料www.www.qdcxjkg.com
本系列之前的文章。1号:Loffek年代,先令O, Franzke北京市。生物基质金属蛋白酶的作用:一个关键的平衡。欧元和J2011;38:191 - 208。2号:Elkington PT, Ugarte-Gil CA, Friedland JS。基质金属蛋白酶在肺结核。欧元和J2011;38:456 - 464。3号:Gaggar,赫克托耳,Bratcher PE、等。基质金属蛋白酶的作用在囊性纤维化肺病。欧元和J2011;38:721 - 727。4号:戴维,McAuley DF, O 'Kane厘米。基质金属蛋白酶在急性肺损伤:损伤介质和司机的修复。欧元和J2011;38:959 - 970。5号:Vandenbroucke再保险,Dejonckheere R, Libert c .基质金属蛋白酶抑制剂在肺部疾病的治疗作用?欧元和J2011;38:1200 - 1214。6号博士:舞者RCA,木头,Thickett金属蛋白酶在特发性肺纤维化。欧元和J2011;38:1461 - 1467。7号周:Churg,年代,赖特杰。基质金属蛋白酶在慢性阻塞性肺病。欧元和J2012;39:197 - 209。8号:Dagouassat M, Lanone年代,Boczkowski j .交互与肺污染物基质金属蛋白酶。欧元和J2012;39:1021 - 1032。
感兴趣的语句
没有宣布。
- 收到了2011年11月30日。
- 接受2012年4月3日。
- ©2012人队











{kind=link}
{kind=link}
{kind=link}
{kind=link}